Abstract
Recent advances have greatly improved our understanding of the molecular mechanisms behind atherosclerosis pathogenesis. However, there is still a need to systematize this data from a general pathology perspective, particularly with regard to atherogenesis patterns in the context of both canonical and non-classical inflammation types. In this review, we analyze various typical phenomena and outcomes of cellular pro-inflammatory stress in atherosclerosis, as well as the role of endothelial dysfunction in local and systemic manifestations of low-grade inflammation. We also present the features of immune mechanisms in the development of productive inflammation in stable and unstable plaques, along with their similarities and differences compared to canonical inflammation. There are numerous factors that act as inducers of the inflammatory process in atherosclerosis, including vascular endothelium aging, metabolic dysfunctions, autoimmune, and in some cases, infectious damage factors. Life-critical complications of atherosclerosis, such as cardiogenic shock and severe strokes, are associated with the development of acute systemic hyperinflammation. Additionally, critical atherosclerotic ischemia of the lower extremities induces paracoagulation and the development of chronic systemic inflammation. Conversely, sepsis, other critical conditions, and severe systemic chronic diseases contribute to atherogenesis. In summary, atherosclerosis can be characterized as an independent form of inflammation, sharing similarities but also having fundamental differences from low-grade inflammation and various variants of canonical inflammation (classic vasculitis).
1. Introduction
The term ‘atherosclerosis’ comes from the Greek words ‘athero’, which means gruel or paste, and ‘sclerosis’, which means hardness. Atherosclerosis is a disease that is genetically intended for everyone who reaches a certain age, and its complications, primarily coronary heart disease and stroke are the main causes of death in developed countries [1,2]. There is a concerning trend of atherosclerosis development in young people [3,4]. Recently, significant advances have been made in understanding the molecular and cellular mechanisms of atherosclerosis. However, the systematization of these numerous data from the position of general pathology remains an urgent problem. Therefore, this review aims to study the main patterns of atherosclerosis in terms of the main typical pathological processes.
Aim and Objectives of the Review:
Aim: To elucidate the typical patterns of atherosclerosis pathogenesis as a unique variant of the general pathological process of inflammation, which exhibits distinctive features of both classical (canonical) and low-grade inflammation.
Objectives:
- Characterize atherosclerosis as a pressing issue in modern medicine and provide a general description of the various types of inflammation (Section 2);
- Demonstrate the pathogenetic role of cellular stress in atherosclerosis as an elementary functional component of general pathological processes (Section 3);
- Evaluate the importance of low-grade inflammation, endothelial dysfunction, and other associated pathogenesis links in atherosclerosis (Section 4);
- Systematize the primary typical immune mechanisms of inflammation development in stable and unstable atherosclerotic plaques (Section 5);
- Offer a comprehensive assessment of the inflammatory process in atherosclerosis (Section 6);
- Ascertain the relationship between atherosclerosis and systemic hyperinflammation (Section 7);
- Outline the main patterns of atherosclerosis pathogenesis from the perspective of general pathology and the theory of general pathological processes (Section 8).
2. General Characteristics of Atherosclerosis and Typical Inflammatory Processes Associated with Atherosclerosis
2.1. Definition
Atherosclerosis is a chronic inflammatory disease of elastic and musculoelastic arteries associated with the formation of atheromatous plaques (cholesterol) that cause stenosis or thrombosis (if the plaque is unstable).
2.2. General Patterns of Development of Atherosclerosis
The main pathogenetic factors of atherosclerosis are:
- Aging is associated with atherosclerosis, but it is not the only factor since early atherosclerosis and familial (genetic) variants of cholesterol metabolism disorders are known [5]. Pathoanatomical signs of the initial manifestations of atherosclerosis are also detected in children who died of other causes [6,7]. Therefore, preventing atherosclerosis in people aged 20–40 years is essential [8]. However, clinical manifestations of atherosclerosis are primarily observed in the elderly, associated with age-related changes in the metabolome, endotheliosis, immune dysfunction, shortening of telomeres, and other systemic changes in homeostasis [9,10]. Dyslipoproteinemia, dyslipidemia, hyperglycemia, and metabolic dysfunctions contribute not only to atherosclerosis but also to cell aging in a vicious circle [11];
- Atherosclerosis is linked to lipid metabolism disorders, mainly with the effect of atherogenic forms of lipoproteins on vessels, primarily modified (oxidized or methylated) low-density lipoproteins (LDL) [12];
- Atherosclerosis is associated with the immune system and inflammation [13,14,15]. However, “inflammation” is a broad concept uniting processes that are heterogeneous in their pathogenetic significance. This predetermines the need to specify “inflammation” in atherosclerosis from the standpoint of methodological approaches to general pathology.
2.3. Risk Factors for Atherosclerosis Development
Atherosclerosis is a complex multifactorial disease with various risk factors that contribute to its development. These risk factors can be broadly classified into several categories, including pro-inflammatory processes and metabolic dysfunctions, congenital factors, gender differences, lifestyle risk factors, disorders of intestinal microbiota, infections, and anatomical features.
- Pro-inflammatory processes and metabolic dysfunctions in atherosclerosis are closely associated with obesity, metabolic syndrome, and type 2 diabetes [16,17,18]. These pathologies are currently considered to be the result of low-grade chronic inflammation [19,20], which belongs to the category of nonclassical inflammation (meta-inflammation, para-inflammation, or quasi-inflammation) [21,22,23,24]. Low-grade chronic systemic inflammation is considered to be the basis of the pathogenesis of most age-related diseases, including hypertension and atherosclerosis [25,26,27];
- Congenital risk factors for atherosclerosis are associated with polymorphisms of polygenes that control the development of atherosclerosis, either directly or indirectly (e.g., through epigenetic mechanisms) [28,29,30,31,32]. The potential number of genes and genetic combinations that can serve as risk factors for atherosclerosis is very large [33]. In addition, there are familial variants of early atherosclerosis associated with specific genetic anomalies, primarily in cholesterol metabolism [34,35];
- Gender differences play a role in atherosclerosis development. Women under the age of about 70 are less susceptible to atherosclerosis than men, partly due to the effect of estrogen on lipid metabolism [36,37,38];
- Lifestyle risk factors such as smoking, unhealthy diet, alcohol abuse, chronic psychosocial stress, sleep deprivation, physical inactivity, and air pollution are considered risk factors for the disease [38,39,40,41,42];
- Disorders of intestinal microbiota and an increase in intestinal permeability for toxic products of microbial metabolism also contribute to the progression of atherosclerosis [43,44,45];
- Although atherosclerosis is not an infectious disease, infections, especially those that invade arteries, can serve as additional factors in the onset and progression of atherosclerosis [46,47,48];
- Anatomical features of the arteries, such as bifurcations, which exhibit turbulent blood flow compared to laminar flow, contribute to the progression of atherosclerosis [43]. The turbulent flow activates and repositions endotheliocytes, increasing their permeability to large molecules, including LDL. Atherosclerotic stenosis causes the transition to turbulence in the oscillatory flow of blood in the arteries, leading to greater hydrodynamic instability in the flow [49];
- Arterial hypertension shares common pathogenic mechanisms with atherosclerosis, primarily endothelial dysfunction, and is considered the most prevalent modifiable risk factor for atherosclerosis [50].
2.4. The Main Stages in the Development of Atherosclerosis
2.4.1. Morphology of the Normal Vascular Wall of the Arteries [51,52]
The morphology of the normal vascular wall of the arteries is well-established in the literature [51,52]. The inner wall of the arteries is composed of a monolayer of endothelial cells (ECs), which are connected with tight and gap junctions and are situated on a basement membrane. ECs are derived from mesenchymal cells and can be characterized as squamous epithelium. Their cytoplasm is thin, approximately 0.2–0.4 microns, and contains numerous transport vesicles that can form transendothelial channels. A dense layer of glycocalyx separates the ECs from the blood, and its thickness can reach several microns. Under normal physiological conditions, the endothelium is stable, slowly renewed, and does not have a significant amount of pro-inflammatory markers.
Beneath the endothelial lining is the intima, which is a layer of loose connective tissue primarily composed of collagen fibers, glycosaminoglycans of the main substance, and occasional stromal macrophages (Mf). The media is the next layer and is composed mainly of vascular smooth muscle cells (VSMCs). The outer layer of the arteries is called the adventitia, which is made up of fibrous structures and various connective tissue cells, including stromal Mf. The middle shell is sandwiched between the inner and outer elastic plates. The intima is located inside the inner elastic plate, and the adventitia is located outside the outer elastic plate. The microcirculatory network vasa vasorum provides oxygen and nutrients to the tunica media and tunica adventitia.
2.4.2. The Main Stages in the Development of Atherosclerosis
The development of atherosclerosis, a common disease in developed countries that significantly impacts life expectancy, involves a sequence of milestone events [15,53,54,55,56]. The events can be summarized as follows:
- ECs activation of results in the impairment of the barrier function of the glycocalyx and weakened contact between ECs, which increases the transport of LDL through the endothelial wall into the intima. This leads to the deposition of lipids in the intima in the form of “lipid bands.” Additionally, ECs produce chemokines and express adhesive molecules that facilitate the transendothelial migration of monocytes and other leukocytes;
- Monocytes migrate into the intima of the arteries, where they take up oxidized LDL (oxLDL). These monocytes differentiate into inflammatory Mf that accumulate cholesterol and become foam cells. Various forms of programmed necrosis, including incomplete apoptosis of foam cells, leading to the formation of a necrotic atheromatous nucleus consisting of cholesterol and cell degradation products. The release of necrotic cell products enhances the inflammatory response of cells surrounding the atheromatous nucleus;
- Activated VSMCs undergo transdifferentiating and become foam cells, fibroblasts, fibromyocytes, endothelial, adipocyte, and osteoblast-like cells, cells with a phenotype similar to mesenchymal stem cells, and cells of an intermediate phenotype that participate in the formation of foam cells (approximately up to 50%), fibrous plaque structures, and arterial calcification. Fibrous structures form the lid of an atherosclerotic plaque, and plaque sprouts microvessels from adventitia and media that provide nourishment to plaque cells and their periphery. If the plaque becomes unstable, its cap can rupture and form a blood clot inside the vessel. Rupture-prone atherosclerotic plaques often have large lipid cores covered by a thin fibrous sheath (<60 µm). Lesions with these characteristics are often referred to as “vulnerable plaques.” On the contrary, plaques with limited lipid accumulation and thicker fibrous caps are often referred to as “stable plaques.”
From a pathogenetic perspective, atherosclerosis is an inflammatory disease that appears to be an inevitable ontogenetic disease associated with local and systemic changes in homeostasis (allostasis), especially in the state of the endothelium, the immune system, and lipid metabolism. However, what is meant by “inflammation” in the context of general pathology in this case?
2.5. Classical and Non-Classical Variants of Inflammation, Their Possible Relationship with Atherosclerosis
The involvement of pro-inflammatory mechanisms at the tissue level, including the formation of a cytokine network, is a hallmark of inflammation as well as some physiological processes and diseases traditionally not classified as inflammatory, such as cancer and schizophrenia [57,58]. However, not all inflammatory processes can be characterized as canonical inflammation, including low-grade inflammation and systemic hyperinflammation [59,60].
2.5.1. Canonical Inflammation
Canonical inflammation is characterized by a focal reaction of microvessels with an exudative response and directed migration of leukocytes to the site of local damage. This reaction of the endothelium of postcapillary venules, known as endotheliitis, can only occur in vertebrates that have a network of lymphatic capillaries and blood microcirculatory units in various organs [61]. These processes are responsible for the five characteristic signs of inflammation known since ancient times, namely rubor, tumor, calor, dolore (Celsus) [62], and functio laesa (Galen) [63]. In contrast, invertebrates exhibit local inflammation of a nonclassical type, characterized by the accumulation of phagocytes in the damage zone but without a microvascular component.
The primary goal of local inflammation, whether classical or nonclassical, is to isolate and eliminate damaging agents. The systemic inflammatory response in classical inflammation performs a different task, primarily the resource provision of the focus of inflammation, and is characterized by a lower level of damaging factors and intensity of pro-inflammatory processes. Various variants of classical inflammation can develop in the focus of inflammation, depending on the severity of exudative vascular and cellular reactions. Productive inflammation, which is dominated by mononuclear leukocytes and inflammatory Mf formed from monocytes, is closest to atherosclerosis. However, atherosclerosis does not fully correspond to this variant of inflammation because the inflammatory role of the vasa vasorum is not clear, four local signs of inflammation formulated by Celsus are not present, and the reaction of the endothelium of the arteries can be defined as endotheliosis, which is a characteristic feature of low-grade inflammation.
2.5.2. Low-Grade Local and Systemic Inflammation [59,60]
Low-grade inflammation, also known as parainflammation, is a type of non-classical inflammation characterized by the long-term effects of damaging factors, the absence of characteristic signs of an inflammatory focus (such as an inflammatory reaction of microvessels), delocalization of the process, insufficiency of barrier mechanisms, and its interconnection with tissue aging processes, metabolic factors of damage, or systemic changes in the endothelium (endotheliosis) [59,60]. This type of inflammation is evident in conditions such as morbid obesity, metabolic syndrome, type 2 diabetes mellitus, and age-related sarcopenia [64,65]. While atherosclerosis is associated with these processes, it does not fully correspond to this variant of inflammation in terms of the severity of inflammatory mechanisms and other features of the plaque, such as local accumulation of immunocytes and pro-inflammatory changes.
2.5.3. High-Grade Systemic Inflammation (Hyperinflammation) [59,60]
This type of nonclassical inflammation is characterized by a severe systemic effect of damaging factors that lead to systemic microvascular endotheliitis, which is typical for a classical inflammatory focus. It also results in life-threatening microcirculatory disorders, intravascular activation of leukocytes, hemostasis, complements, and kallikrein-kinin systems. In chronic systemic inflammation, there may be a “cytokine storm” with latent microcirculatory disorders. The association of atherosclerosis with these variants of nonclassical inflammation is not well understood and requires further investigation. Section 7 briefly discusses this problem.
2.5.4. Cellular Stress as a Link between Various General Pathological Processes Associated with Inflammation
The fundamental functional unit of all types of inflammation, as well as several other pathological and even extreme physiological processes, is pro-inflammatory cellular stress [60]. One distinct attribute of cellular stress is the development of an inflammatory receptor and secretory phenotype, which ultimately leads to alterations at the organ and organism level (Figure 1).
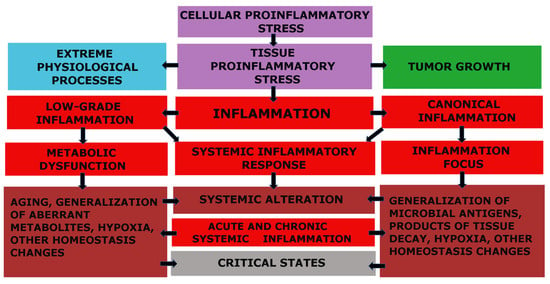
Figure 1.
Relationship between cellular and tissue stress and human pathology [66]. Note: The figure shows the relationship between cellular and tissue stress and human pathology. Cellular pro-inflammatory stress is a common response to various forms of damage to macromolecules and external stressors. This stress response is the basis for various physiological processes such as muscle contraction, tissue growth, mucosal function, and lymphocyte selection in primary lymphoid organs. However, it also underlies various pathological processes. Tumor cells and the tumor microenvironment are under stress, but tumor growth is not associated with inflammation. Systemic hyperinflammation, on the other hand, is a dysfunctional process that is a complication of other pathological processes that initiate systemic alteration.
3. Cellular Stress as an Elementary Functional Component of the Inflammatory Process in Atherosclerosis
3.1. General Pathological Patterns of Cellular Stress
3.1.1. Definition
Cellular pro-inflammatory stress (CS) can be defined as “a complex of interrelated universal and specific cell processes in response to the action of factors that cause actual or potential damage”.
3.1.2. Tissue Pro-Inflammatory Stress, Typical Pathological Processes of Inflammation, and Their Difference from Clinical Definitions
Models of general pathological (typical pathological) processes reflect the most fundamental patterns of human pathology. They are not clinical definitions but rather the theoretical basis for the formation of the latter. Inflammation is the most extensive general pathological process. Recent studies of the molecular mechanisms of the pathogenesis of various diseases have significantly expanded the concept of “inflammation”. Moreover, as noted earlier, various pro-inflammatory mechanisms (intracellular signaling pathways, cytokine networks, and other factors) are involved in many physiological processes [59]. This circumstance highlights the need to differentiate “inflammation” into various alternative general pathological processes, separating them from physiological processes and defining tissue stress as their common basis, with CS as an elementary component of this basis. However, CS must be differentiated from another common concept, namely “cell pathology”, which overlooks the cell’s response to damage and ignores the physiological significance of cell damage and controlled death (apoptosis and programmed necrosis).
3.2. Universal Links of CS in Atherosclerosis
The universal components of CS are depicted in Figure 2, where we can observe that changes in the genome, mitochondria, endoplasmic reticulum (ER), and other cellular compartments, as well as the activation effects of pro-inflammatory mediators, cause not only specific but also universal reactions in many types of cells, making CS an integral and typical phenomenon (Figure 2) [59,60,61,67,68,69].
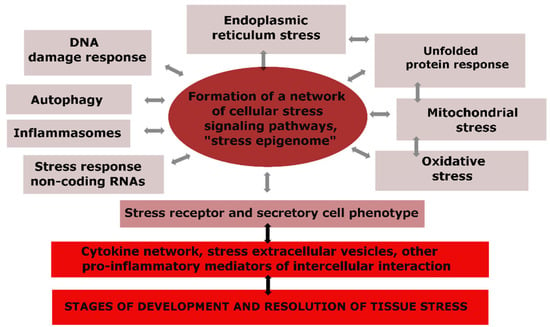
Figure 2.
The structure of typical cellular stress processes and their relationship with tissue stress.
The components of cellular senescence can be classified into several categories [60]:
- Oxidative stress involves the accumulation of reactive oxygen species (ROS) and free radicals in cells, leading to modifications in protein post-translational processes and the synthesis of biomolecules essential for all CS processes. Excessive ROS production, which primarily occurs in mitochondria, can damage macromolecules and lead to vascular damage [70,71]. Endogenous antioxidants serve as a checkpoint against these adverse effects, but an imbalance in pro-oxidant/antioxidant mechanisms can lead to a sustained state of oxidative stress. There is a consensus that oxidative stress is associated with cellular aging and the pathological activation of ECs, Mf, and other atherosclerotic plaque cells [72,73,74];
- DNA damage response (DDR) is another important component of CS that allows cells to control genotoxic stress and ensure the accurate transmission of genetic information to subsequent generations. DNA damage accumulation can result in various DDR outcomes, including cell cycle arrest, cell aging, malignancy, and apoptosis [75]. More than 1000 proteins, such as nuclear chaperones (ubiquitin, nucleophosmin, and SUMO protein), nuclear protein kinases (ATM, ATR, DNA-PKcs, and Chk1/2), nucleases, polymerases, ligases, DNA glycosylases, and transcription factors (TF), including p53, are involved in the DDR process in human cells [59,76,77]. DDR’s primary function is to stop the cell cycle, facilitate DNA repair, and ensure cell survival [78]. Conversely, apoptosis is an extreme variant of DDR that prevents the transmission of genetic abnormalities to daughter cells. Overall, DDR plays a protective role in vascular cell aging, promotes cell proliferation, and prevents the accumulation of cells with impaired differentiation in the focus of atherosclerosis [79,80,81];
- Mitochondrial stress is a complex process that involves the mitochondrial unfolded protein response reaction (UPR), associated with the inducible synthesis of heat shock proteins (HSP). Mitochondria are the main producers of ATP and ROS. There are approximately 1500 proteins that function in human mitochondria, of which only 13 are encoded by mitochondrial DNA (mtDNA) [82]. These are the most important proteins of mitochondrial respiratory complexes. The mitochondrial proteome is adjusted to the functional state of its cell and depends on the action of activating and damaging factors on the mitochondrion itself. The relationship between mitochondria and the cell nucleus is a well-established phenomenon that occurs in response to mitochondrial dysfunction. In this case, the UPR mt includes multidirectional changes in the biosynthesis of various mitochondrial proteins: a decrease in potentially toxic proteins but an increase in the production and transport into mitochondria of chaperones capable of restoring damaged mitochondrial proteins [83]. Integrative mitochondrial stress is primarily associated with the activation and production of HSP and several kinases by ATF4 that integrate mitochondria into the CS system [84]. Mitochondrial stress serves to reduce organelle damage and dysfunction, but mitochondrial factors also contribute to apoptosis, programmed necrosis, cellular senescence, and other outcomes of CS [60]. Currently, there are a significant number of facts that prove the key, but ambiguous, significance of mitochondrial stress in ECs, Mf, GMCS, and other cells during the development of various stages of atherosclerosis [85,86,87,88,89,90];
- ER stress occurs when there is a disruption of ER integrity or accumulation of misfolded proteins within it. This initiates UPR ER, primarily in the form of UPR ER [91]. The UPR ER process serves to restore altered ER homeostasis with the following goals: (1) arresting the synthesis and excretion of secretory proteins from the cell; (2) increasing transcription of chaperones and other proteins involved in protein folding and maturation; (3) utilizing denatured proteins via the ER-associated degradation complex (ERAD) [92]. Thus, the UPR ER provides a coordinated response that contributes to overcoming impaired ER proteostasis. The prolonged or critical intensity of UPR ER, in turn, induces apoptosis in several ways, including Ca2+ release into the cytoplasm of the ER [91,93]. However, since ER stress also activates antiapoptotic pathways (anti-apoptotic proteins of the Bcl-2 family), apoptosis is not the only outcome of ER stress. The development of ER stress is not an independent process of other components of CS [94,95]. In atherosclerosis, the development of ER stress in ECs, Mf, and other cells of focus of atheromatosis is closely associated with oxidative and mitochondrial stress, especially with cholesterol accumulation in foam cells [96,97,98,99,100,101];
- HSP response: The inducible synthesis of HSPs is a highly conserved and evolutionarily ancient molecular response that cells use to cope with disturbances in their proteostasis [102]. As mentioned earlier, HSPs play a crucial role in maintaining the UPR, and they are also involved in regulating several key cellular processes [103]. In fact, HSPs are involved in most of the major pathogenetic processes that lead to atherosclerosis, including autoimmune reactions [104]. However, the limited production of HSPs in the arteries during aging is a pathogenic mechanism that contributes to the development of atherosclerosis [105];
- Autophagy: Autophagy is a critical part of many physiological and pathological processes, and its severity increases significantly during starvation and severe CS with pronounced activation of catabolic processes [106]. In CS, the ubiquitin-proteasome pathway, which is responsible for the utilization of altered proteins, is overloaded, and this may act as an additional mechanism to activate autophagy [91,107]. Additionally, many long-lived proteins, large protein aggregates, and individual organelles can only be recycled through the autophagy process, which involves lysosomes and numerous accessory protein factors. Mitophagy, in particular, is the only mechanism for physiological mitochondrial recycling [108]. Therefore, autophagy plays a crucial role in maintaining the balance between protein biosynthesis, organelle biogenesis, and organelle degradation. Furthermore, autophagy can also prevent cell apoptosis or necrosis by removing damaged mitochondria, protein complexes, and intracellular parasites. Autophagy is generally classified into three main types and is regulated at various levels through CS mechanisms [109]. However, the regulation and implementation of autophagy can be altered as cells age [110]. In cells involved in the pathogenesis of atherosclerosis, the intensity of autophagy/mitophagy tends to increase, and the disruption of this process contributes to atheromatosis and its complications [111,112,113]. Adequate autophagy, proportional to the degree of damage, is an adaptive process in atherosclerosis. However, disproportionate autophagy itself contributes to cell damage and dysfunction in the focus of atheromatosis [114];
- Inflammasomes are multimeric cytosolic protein complexes with sensory molecules that recognize microbial and endogenous molecular patterns associated with damage. During the assembly of the protein complex, these receptors bind to procaspase-1, which is then converted into caspase-1. This induces the processing of IL-1β and IL-18 and, under certain conditions, leads to the development of pyroptosis (a variant of programmed necrosis) [115]. The formation of inflammasomes is indicative of a relatively pronounced cellular stress (CS). In the pathogenesis of atherosclerosis, the key role is played by NLRP3 inflammasomes, which are activated by a wide variety of factors, including cellular decay products, reactive oxygen species (ROS), lysosomal proteinases, cholesterol and calcium phosphate crystals, numerous exogenous stimuli, and the release of mitochondrial DNA into the cytoplasm [116,117,118]. Disruption of control over the formation and utilization of inflammasomes may inappropriately amplify the inflammatory response, particularly in macrophages, thereby exacerbating the progression of atherosclerosis [119,120];
- Noncoding RNAs, including microRNAs and long noncoding RNAs, have been shown to modulate gene expression and play a role in the development of atherosclerosis [121,122,123,124,125]. Noncoding RNAs can regulate cellular processes involved in proteostasis and autophagy, as well as intercellular communication, by being incorporated into extracellular vesicles [126]. Recent developments have focused on the potential of using miRNAs as a therapeutic approach for treating atherosclerosis [127];
- Formation of stress granules: In the post-transcriptional stage, RNA-binding proteins play a key role in stress-induced regulation of the fate and function of various RNAs [128]. Protein-RNA complexes can form large granules (approximately 0.1–1.0 μm) in the cytoplasm of stressed cells, whose function is presumably to protect and store mRNA [129]. Additionally, due to their non-membrane characteristics, these structures can serve as a center where various cellular signaling pathways converge and exchange components [130]. Meanwhile, CS can induce the formation of gel-like structures in cells, including those involving amyloid and prion-like proteins [131]. The formation of these structures is dynamic; they quickly condense or dissolve, making them ideal for participating in emergency cellular adaptation to stress. However, when autophagy function is insufficient and insoluble granules are formed from these proteins, there is a decompensated increase in chronic stress and cell damage [132]. Immunohistochemical analysis of atherosclerotic plaques in mice revealed an increase in stress-granule-specific markers in intimal Mf and VSMCs in direct relation to disease progression [130,133];
- Formation of a network of signaling pathways in cellular stress: At the cellular level, stress development is mediated by complex epigenetic control programs and the interweaving of signaling pathways, the protein elements of which are constantly subjected to multiple post-translational modifications [134,135]. Various extracellular and intracellular stress signals can activate common collector-type protein kinases (e.g., MAPK, STAT, Akt, PI3K, PKC, ATM, ATR, AMPK, PKA, PKR, mTOR) and key universal factors TF associated with cellular stress (NF-κB, p53, AP-1, HIF, HSF, NRF2, ATF4) in different cell types [136,137,138,139,140,141,142,143,144,145,146]. The same signaling molecules can be activated and participate in various multidirectional processes; however, several pleiotropic TFs may display functional preferences. Thus, HIF-1 (hypoxia-inducible factor-1) plays a key role in the development of cellular stress in hypoxia [147]; HSF1 (heat shock factor 1) is involved in the production of heat shock proteins [148]; NRF2 (nuclear factor erythroid 2-related factor 2) triggers the production of antioxidants by a negative feedback mechanism in oxidative stress [149]; ATF4 (activating transcription factor 4) plays a decisive role in the development of UPRmt [84].
The dynamic network of signaling pathways integrates various elements of cellular CS into a single entity, including the receptor and secretory phenotype of pro-inflammatory cells. To date, almost all of these universal mechanisms of CS have been shown to be involved in the pathogenesis of atherosclerosis [53,150,151,152,153]. It has been demonstrated that in areas of arterial blood flow with impaired (turbulent) blood flow, many key transcription factors (TFs) are activated in ECs, including NRF2, HIF-1α, NF-κB, AP-1, and others [151]. Additionally, activation of Runx2 (Runt-related transcription factor 2) in pathologically activated VSMCs promotes its transformation into osteoblast-like cells, which leads to the calcification of atherosclerotic plaques [154].
Several transcription factors, such as NRF2, p53, ATF3, ATF4, HIF1α, HSF1, and MAPK, and several other stress protein kinases, have been shown to play a multidirectional role in the development of ferroptosis (a variant of programmed necrosis) in Mf within atherosclerotic plaques [155]. Lipid-activated TFs, peroxisome proliferator-activated receptors (PPARs), play a crucial role in regulating lipid and lipoprotein metabolism, glucose homeostasis, and inflammation in various cells. Both animal models of atherosclerosis and clinical studies strongly support the antiatherosclerotic role of PPAR alpha and gamma in vivo [156]. Furthermore, inhibitors of the NF-κB family have been shown to slow down the development of atherosclerosis by inhibiting TF activity [157]. Overall, the pro-inflammatory role of NF-κB is crucial for many functions and dysfunctions, including those in cardiovascular diseases [158].
Thus, in various cells involved in the pathogenesis of atherosclerosis, pro-inflammatory CS develops as a holistic phenomenon that includes a network of signaling pathways. A separate component of CS is the pro-inflammatory secretory phenotype, which allows the formation of information networks (primarily cytokine) of tissue stress.
3.3. Outcomes of CS and Their Importance in the Pathogenesis of Atherosclerosis
The genetically programmed purpose of CS is to ensure the safety and survival of cells, with the priority task of maintaining the body’s homeostasis. Therefore, different forms of programmed cell death, such as apoptosis and necrosis, cannot be considered inherently pathological. Cell aging is closely linked to CS, but it also has its own adaptive components, such as an alternative to tumor cell transformation. The massive pro-inflammatory transformation of cells, primarily within the immune system, is the basis for the development of various types of inflammation during significant tissue damage. Figure 3 outlines the main routes for the implementation of CS, which are directly related to the pathogenesis of atherosclerosis, and where the final outcomes of CS are apoptosis and cell necrosis.
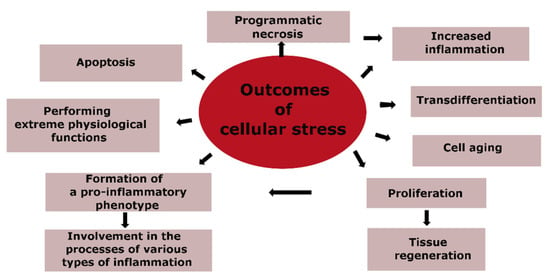
Figure 3.
Outcomes of cellular stress.
Apoptosis and cell necrosis are important biological processes with different implications for tissue homeostasis and disease progression. Apoptosis is a natural process of programmed cell death that is crucial for normal cell turnover, proper development and functioning of the immune system, and the resolution of inflammation [159]. Dead cells produced by apoptosis are rapidly cleared by Mf without significant generation of damage-associated molecular patterns (DAMPs), which are signals that can activate immune cells and promote inflammation. Therefore, apoptosis is a “delicate” variant of cell death that maintains tissue integrity and limits inflammatory responses. However, when apoptosis exceeds the regenerative capacity of the organ, it can contribute to tissue atrophy and subsequent fibrosis [160]. Apoptosis can be induced by various signaling pathways, which can be divided into extrinsic and intrinsic pathways, depending on their dependence on caspase activation. External pro-apoptotic signals act on TNF family cytokine receptors, primarily the Fas receptor (CD95). The main intrinsic pathway of apoptosis involves the release of pro-apoptotic molecules from mitochondria, such as cytochrome C, which activates caspase 9 and other pro-apoptotic caspases [161]. The mitochondrial response is controlled by a balance between pro-apoptotic and anti-apoptotic proteins of the Bcl-2 family, IAP, and other factors [162,163]. Therefore, the development of apoptosis is the result of a complex interplay between different signaling pathways that determine the balance between pro- and anti-apoptotic factors.
In the context of atherosclerosis, increased apoptosis can have both beneficial and detrimental effects, depending on the cell type and stage of the disease. For example, endothelial cell apoptosis can initiate atherosclerosis [164]. Apoptosis in Mf and foam cells is associated with ER stress, which can lead to an apoptotic response mediated by both Fas and mitochondrial pathways [165]. Apoptotic cell death is an important feature of atherosclerotic plaques, but its impact on plaque stability and inflammation is complex. In the late stages of atherosclerosis, a high level of VSMCs apoptosis can cause the fibrous cap to become smaller and thinner, leading to the formation of unstable plaques [166]. However, adequate uptake of apoptotic cells by Mf, also known as efferocytosis, is important to prevent the progression of atherosclerosis [167,168]. Efferocytosis is mainly carried out by functionally polarized Mf in the M2 direction, which has moderate pro-inflammatory activity and promotes tissue fibrosis, as opposed to the more pro-inflammatory M1 Mf [169,170]. When efferocytosis fails, incomplete apoptosis or secondary necrosis can occur, resulting in the release of DAMPs and the activation of inflammatory responses, including the Mf polarization toward M1 and the inhibition of efferocytosis by the CD47 receptor [169,171]. This creates a vicious cycle that perpetuates inflammation and incomplete apoptosis, making the atherosclerotic plaque unstable.
In addition to incomplete apoptosis in an unstable atherosclerotic plaque, other variants of programmed necrosis can occur, such as necroptosis (of Mf and VSMCs), pyroptosis (a final function of inflammasomes), ferroptosis (fatal iron-dependent lipid peroxidation), and autophagic death of foam cells (uncontrolled autophagy) [165]. It has become clear that neutrophils are involved in the pathological process in various stages of atherosclerosis (from the primary activation of the arterial endothelium and the formation of an unstable plaque) [172]. The neutrophil-specific cellular program (including the necrosis variant), characterized by the extracellular release of cobweb-like DNA structures called NET (NETosis), plays a significant role in tissue damage [172].
As noted in Section 2, transdifferentiation is a common consequence of the development of CS, particularly for VSMCs. These cells can differentiate into atypical cell populations with various functions that can have both adaptive and maladaptive significance.
The aging of cells is a result of the stochastic accumulation of damage in biomolecules such as the genome, transcriptome, and proteome, which are vital for proper cellular function. These changes can lead to the appearance of CS with the accumulation of ROS, cell cycle blockade, and the formation of cellular and tissue allostasis [173,174]. Cellular senescence is characterized by a state of proliferation arrest, in which cells remain metabolically active and secrete a range of pro-inflammatory and proteolytic factors and other components of the aging-associated secretory phenotype (SASP) [175]. Aging and pro-inflammatory SASP cells can form a vicious pathogenetic circle involved in the formation of tissue allostasis in aging tissue [176].
The aging of ECs and VSMCs is closely associated with vascular diseases such as hypertension and atherosclerosis [177,178]. Finally, aging Mf and other immunocytes that have accumulated genetic mutations can form populations of dysfunctional cells in atherosclerosis [9,179].
3.4. Triggers of Cellular Stress in Atherosclerosis
The development of atherosclerosis involves various factors that trigger cellular stress (CS). These factors can be classified into the following categories:
- Damage to macromolecules: Gradual accumulation of damage to the genome and proteome during aging, oxidative stress, and intracellular cholesterol accumulation during the formation of foam cells can cause damage to cells and extracellular matrix. This damage is recognized by CS sensors [180,181];
- Changes in homeostasis parameters: Several parameters of homeostasis can induce CS in atherosclerosis. These include an increase in osmotic [182,183] and hydrostatic pressure [183], changes in calcium concentrations in cells [184], and a decrease in ATP and oxygen concentrations in cells (hypoxia) [185,186];
- Lipotoxicity factors: Excess saturated free fatty acids (FFA), diacylglycerol, ceramides, modified carnitine, non-esterified cholesterol, and other hydrophobic molecules can damage mitochondria and other cellular structures [60,187,188,189];
- Recognition of PAMPs and DAMPs through PRRs: Pathogen-associated molecular patterns (PAMPs) and DAMPs recognized by pattern recognition receptors (PRRs) directly associate with inflammatory cells such as immunocytes, epitheliocytes, connective tissue cells, and endotheliocytes [190]. Cells can quickly enter a state of stress and realize their pro-inflammatory and immunocompetent functions. Toll-like receptors (TLRs) play a critical role in atherosclerosis [191]. TLRs contribute to the inflammatory Mf transformation and other cells in atheromatosis due to the release of significant amounts of DAMPs from necrotic cells [192,193]. TLRs can also be involved in earlier stages of atherosclerosis, including the development of endotheliosis, which is characteristic of low-grade inflammation and cardiovascular diseases [194,195]. Various stress cells can serve as a source of moderate amounts of DAMPs and HSP that interact with TLRs. Saturated FFA associated with blood plasma protein (FetA) may activate TLR4 [196], but the role of this mechanism in the pathogenesis of atherosclerosis is still unclear. TLR 4, expressed on various immunocytes, endotheliocytes and platelets, is the main receptor for bacterial endotoxin (LPS). Morbid obesity and systemic effects of lipotoxicity factors can lead to metabolic endotoxemia (an increase in LPS in the blood, approximately 1.5–2 times) as a result of impaired intestinal barrier function [197,198]. An increase in PAMPs in the blood can also be observed in infections indirectly associated with the development of atherosclerosis [199];
- Antibodies and T cell receptors (TCRs) recognize antigens, leading to T cell activation and interactions primarily with antigen-presenting cells. In atherosclerosis, these pathways of activation may be associated with the development of an auto-immune response to modified lipoproteins and other autoantigens [200,201];
- Excitotoxicity, in the narrow sense, refers to the toxic effect of high doses of glutamate on neurons, other neurotransmitters, and their catabolism products [202]. In the broad sense, it denotes the pathological hyperactivation of cells by various regulatory molecules, mainly pro-inflammatory cytokines such as TNF-α and IL-1β [59]. The phenomenon of cytokine-induced excitotoxicity contributes to the contagious development of chronic systemic inflammation. The formation of various variants of the local cytokine network in atherosclerosis is also an established fact [203,204];
- Scavenger receptors (SRs) are a family of molecular receptors that includes more than 30 members [170,205]. Although they do not share structural homology or genetic origin, they are united by common functional properties [206]. Structurally homologous receptors form separate SR classes (A-L) within this family. SRs are primarily located on the surface of stromal Mf but also on ECs and other cells, functioning at the intersection of metabolism, immunity, and inflammation. In particular, SRs are the primary sensors of modified LDLs. Another common feature of SRs is their ability to form functional clusters with Toll-like receptors (TLRs), integrins, and other receptors on cell membranes, modulating their activity in relation to the development of cardiovascular disease [206]. Due to this feature, SRs are involved not only in pathology but also in the implementation of physiological processes, providing clearance of abnormal metabolic products, relatively low concentrations of PAMPs and DAMPs, and apoptotic and abnormal cells. Moreover, in atherosclerosis, SRs can be involved in both adaptive and maladaptive processes.
Most SRs are involved in the processes of atherosclerosis. SR-E1 (LOX-1) is involved in endothelial cell activation and disruption of the endothelial barrier function, as well as in the activation of Mf and VSMCs [207,208]. SR-A1 (CD204) plays a key role in the uptake of modified LDL by Mf and VSMCs [209], while SR-B2 (CD36) plays a key role in the activation of lipoprotein-dependent Mf [210], and SR-J1 (RAGE) promotes negative activation of ECs and Mf [211]. At the same time, SR-J1 recognizes advanced glycation end products (AGEs), some DAMPs, and ROS-modified endogenous proteins (Table 1). SR-A1, SR-L1, and many other SRs on liver cells absorb modified LDLs, thus preventing their accumulation in the bloodstream and having an opposite effect on the development of atherosclerosis [170]. The nature of the course of atherosclerosis significantly depends on the morphological and functional features of the Mf formed and their ratio. Therefore, Mf with characteristic features of M2 (CD206 high, CD163 high/SR-E3, SR-I1) is mainly located in the periphery of atherosclerotic plaques in the annulus fibrosus zone [212].

Table 1.
Scavenger receptors that may be involved in the pathogenesis of atherosclerosis.
In contrast, M1 Mf-expressing SR-B2 and TLR, with a high pro-inflammatory and procoagulant potential, are more concentrated in the central region and shoulder of plaques directed into the lumen of the vessel [212]. It is with these types of Mf that the increase in pro-inflammatory mechanisms, the involvement of the blood coagulation system, the formation of atheromas, and other complications of atherosclerosis are associated [212,213]. Additionally, plaque core cells are more saturated with cholesterol than peripheral Mf while maintaining a relatively high level of expression of the main oxLDL scavenger, SR-A1 (CD204) [212].
The development of oxidative stress in the endothelium and M1 Mf contributes to an additional modification of LDL. In contrast, the predominance of M2 Mf promotes scarring of the focus of atherosclerosis and restoration of the endothelial lining of the vessel. At various stages of atherosclerosis, pro-inflammatory CD8+ and CD4+ T cells, as well as natural killer (NK) cells, can be involved in the process [263], and their migration can be facilitated by the soluble form of SR-G1 secreted by Mf (chemokine CXCL16) [264]. In turn, the function of SR-B1 in the development of atherosclerosis can be ambiguous. Therefore, on the one hand, SR-B1 promotes the transendothelial transport of LDL in the arteries [265], but on the other hand, it has anti-inflammatory and anti-atherogenic functions in the focus of atheromatosis [266].
As noted above, the mechanisms of the formation of an unstable plaque are largely associated with the failure of the efferocytosis process. Stromal and M2 Mf recognize apoptosis products primarily through SRs that bind spectrin and phosphatidylserine fixed on the cell membrane, HSP, complement fragment C1q, and some other markers of apoptosis (Table 1).
In general, numerous SRs actively manifest their role in atherosclerosis processes, from the transition from physiology to pathology to the development of various types of inflammation in vascular atheromas. Therefore, the orientation of these functions can have both adaptive and maladaptive significance, depending on the type of receptor, stage, and other characteristics of the pathological process.
3.5. Summary
Atherosclerosis is a disease characterized by low-grade inflammation, which is induced by pro-inflammatory CS. The accumulation of cells with a pro-inflammatory phenotype in atherosclerotic plaque leads to a process similar to canonical inflammation. Pro-inflammatory CS includes mitochondrial and ER stress, oxidative stress, formation of a pro-inflammatory secretory phenotype, inflammasomes, autophagy processes, and HSPs and noncoding RNAs. The main inducers of CS are the gradual accumulation of cellular damage during aging and the effect of aberrant metabolome factors, such as modified LDL, saturated FFA, and AGE, on cells. The direction and degree of inflammation in atheromas strongly depend on efferocytosis, cell necrosis, and DAMPs formation, which can lead to unstable plaque. The role of various types of SRs is crucial in controlling the development of CS.
4. Low-Grade Inflammation, Endothelial Dysfunction, and Associated Non-Immune Links in the Pathogenesis of Atherosclerosis
Typical pathological processes fall under the categories of general pathology and pathological physiology. These processes serve as the fundamental and regular manifestations of the pathogenesis of different diseases and provide the theoretical foundation for clinical definitions, including nosology and syndromes.
Atherosclerosis, one such disease, is characterized by two distinct pathological processes, low-grade inflammation and canonical inflammation. A common factor in the development of atherosclerosis is “inflamm-aging,” which can be considered low-grade systemic chronic inflammation in certain cases [9,267,268]. Common symptoms of low-grade systemic inflammation include morbid obesity, allostatic insulin resistance, endotheliosis (often with essential hypertension [269], relatively low manifestations of acute phase response and hypercytokinemia, hypercholesterolemia, hyperlipidemia, and increased blood concentrations of atherogenic forms of lipoproteins and AGE [59,64,270,271,272,273]. As atherosclerosis progresses, it can result in the development of metabolic syndrome (type 2 diabetes) alongside hepatosis and sarcopenia [274,275].
From this perspective, the initial stages of atherosclerosis are characterized by a localized increase in endotheliosis in the arterial wall and systemic manifestations of low-grade inflammation. This contributes to metabolic dysfunctions and lipotoxicity, which can be exacerbated by genetic predisposition, lifestyle risk factors, infections, increased intestinal permeability to PAMPs and toxins, chronic psychoemotional stress, environmental factors, and local turbulence of arterial blood flow. As atherogenesis progresses, local manifestations of low-grade inflammation can transform into classical inflammation (for instance, in the progression of nonalcoholic fatty liver disease and diabetic kidney disease) [276,277]. While renal glomerular pathology in diabetes may be complicated by fibrinous inflammation, which is a type of classical-type exudative destructive inflammation, a similar trend is typical for atherosclerosis, although the growing signs of arterial wall pathology do not fully align with the canons of classical inflammation.
4.1. Typical Conditions and Manifestations of the Initial (Clinically Latent) Phenomena of Atherosclerosis
The concept of ‘inflamm-aging’ [278] is quite broad and encompasses not only the pathology related to low-grade inflammation but also the manifestations of pro-inflammatory tissue stress, which are necessary for the adaptation of the body to age-related changes in homeostasis (allostasis). Consequently, it is not surprising that in aging, there is a natural tendency towards an acute phase response of the liver and hypercytokinemia [279].
Our data shows that 18% of individuals over the age of 65 years without signs of acute and severe chronic diseases, including metabolic syndrome and morbid obesity, have signs of a systemic inflammatory response [66]. However, the increase in the levels of C-reactive protein (CRP) and pro-inflammatory cytokines (TNF-α, IL-6, IL-8) in these patients does not exceed the upper value of the reference range by more than two-fold. In general, the accumulation of genomic mutations in cells, oxidative stress, mitochondrial dysfunction, and other manifestations of cellular and tissue stress are also typical of aging not complicated by low-grade inflammation and associated pathologies (morbid obesity, metabolic syndrome, type 2 diabetes). Low-grade inflammation should be considered a specific stage in the development of ‘inflamm-aging.’
An important issue to address is how to differentiate between low-grade inflammation, chronic systemic hyperinflammation, and manifestations of the systemic inflammatory response in classical types of chronic inflammation, such as autoimmune and infectious diseases. Pro-inflammatory tissue stress, as discussed in Section 3.3, is also characteristic of tumor diseases. Therefore, it is crucial to distinguish low-grade inflammation from other forms of inflammation to understand its role in the development of age-related pathologies.
The concept of ‘inflamm-aging’ involves both adaptive and maladaptive mechanisms, and age-related pro-inflammatory status can only be considered age-related pathology in specific cases. The transition from “age norm” to “age pathology” is not a discrete process and has a “grey” marginal zone of prepathological, latent changes. Local and systemic pro-atherogenic changes that occur before obvious signs of lipid deposition in the arterial intima represent the marginal stage of atherogenesis (pre-atherosclerosis).
In addition, altered hemodynamics in the arteries during the first decade of atherogenesis may also be an independent factor of mitochondrial and oxidative stress in ECs, even before the morphological manifestations of lipid deposition [280]. However, the presence of lipid bands in the arterial intima indicates the development of the initial (subclinical) stage of atherosclerosis.
The main characteristic of these and subsequent stages of atheromatosis is the increase in signs of endothelial dysfunction. This dysfunction, or endotheliosis, is characterized by less pronounced tissue stress in ECs, in contrast to microvascular reactions in the focus of canonical inflammation [60].
4.2. Endotheliosis as a Key Phenomenon of Age-Related Changes and Age-Related Vascular Pathologies
In healthy arteries, the endothelium is generally in an inactive state with regard to tissue stress, unlike other tissues, such as covering tissues, lymphoid organs, and working muscles [59]. However, internal and external damaging factors and changes in regulatory mechanisms within ECs can lead to the development of chronic CS and endothelial dysfunction, which is referred to as endotheliosis. This condition is a key factor in the pathogenesis of various cardiovascular diseases [74]. The development of atherosclerosis, starting from its pre-atherosclerotic stages, is closely linked to the pro-inflammatory activation of ECs, leading to vascular tone release, structural remodeling, inflammation, and atherogenesis regulation. The balance between these factors is critical for maintaining vascular homeostasis, and disturbances in this balance due to endotheliosis can lead to the development of vascular pathologies [74]. Several typical phenomena of endotheliosis can be identified, and it is important to note that normal interactions between ECs, blood plasma cells and proteins, other arterial cells, and the subendothelial extracellular matrix are inevitably disrupted in this condition. Therefore, the dysfunction of ECs cannot be isolated a priori when analyzing these phenomena [281].
4.2.1. Glycocalyx Pathology
The endothelial glycocalyx is a specialized extracellular matrix that covers the apical side of ECs and extends into the vascular lumen. Its composition, consisting of proteoglycans, glycosaminoglycans, and glycoproteins, has been extensively studied. The glycocalyx was once thought to be a passive physical barrier but is now known to be a multifunctional and dynamic structure involved in several vascular processes, such as vascular permeability, inflammation, thrombosis, mechanotransduction, and cytokine signaling [282,283].
Inflammation mediators such as cytokines and chemoattractants can cause degradation of the glycocalyx in different variants of inflammation, affecting arterioles, capillaries, and venules [284]. ROS, proteases, and glycosidases can directly damage the endothelial glycocalyx, which can also be produced by activated ECs. Moreover, ROS can enhance glycocalyx proteolysis by inactivating endogenous protease inhibitors [284,285,286]. Thus, a pathological feedback loop can occur between glycocalyx depletion and pro-inflammatory stress in ECs [286].
The barrier function of the endothelial glycocalyx deteriorates with age, preceding vascular dysfunction [287,288]. The degradation of the glycocalyx in various vessels is associated with negative local and systemic dynamics in the progression of atherosclerosis [289]. In oral submucosa capillaries, the degree of reduction in glycocalyx thickness correlates with the severity of coronary atherosclerosis [290]. Hypertension and arterial wall stiffness associated with aging can cause glycocalyx degradation in a pathological feedback loop, leading to increased endothelial dysfunction and vascular disease progression [291].
4.2.2. Changes in Nitric Oxide Production
The decrease in nitric oxide (NO) production is a characteristic difference between endotheliosis and microvascular endotheliitis in classical inflammation. This decrease is primarily due to a decrease in constitutive endothelial NO synthase (eNOS) activity and reduced NO bioavailability [59]. In endotheliosis, there are insufficient pro-inflammatory stimuli to activate inducible NO synthase (iNOS), which is responsible for NO hyperproduction. iNOS promotes vasodilation of the arterioles and the exudative response of the microvessels while simultaneously limiting pro-inflammatory processes due to the regulation of negative feedback [292,293]. Therefore, in endotheliosis, the limitation of NO bioavailability contributes to vasoconstriction, thrombinemia, and the maintenance of a moderate pro-inflammatory state of ECs, which is milder than classical inflammation. This reduction in NO production, along with the simultaneous hyperproduction of other ROS, characterizes the development of various stages of atherosclerosis [294,295,296].
Elevated oxLDL concentrations in the blood microcirculation zone, along with additional stimuli for inflammation development, lead to sustained activation of SR-E1 (LOX-1) and subsequent activation of NF-kB [297]. This, in turn, can activate iNOS, leading to more pronounced oxidative stress with the formation of more reactive peroxynitrite from NO and the accelerated apoptosis of ECs [297]. A similar mechanism may occur in atherosclerosis. However, more specific evidence is needed at the level of the endothelium of large arteries, not only microvessels and venules, to confirm this hypothesis.
4.2.3. Hyperproduction of Vascular Endothelial Growth Factors (VEGF)
One important characteristic of ECs and the cells surrounding them is their ability to produce growth factors, primarily from the VEGF family. These growth factors play a critical role in promoting vascular regeneration and the survival of ECs in extreme conditions. Most growth factors, including insulin, activate G-proteins and the PI3K/AKT/mTOR signaling pathway, which is associated with cell growth and anabolism. However, growth factors can also act through more specialized KC signaling pathways, which are unique to specific growth factors [59,298,299].
In particular, increased VEGF signaling has been shown to prevent age-related capillary loss, improve perfusion and organ function, and increase longevity in mice [300]. However, a chronic increase in VEGF-A production can lead to a more severe pro-inflammatory response, negatively affecting myocardial function in the aging heart [301]. Additionally, an increase in VEGF-A concentration can cause oxidative stress and multiple age-related eye diseases, such as cataracts, neovascular, and nonexudative pathologies [302].
In the development of atherosclerosis, the production of VEGFs can be induced by the TF family HIF, both as a result of hypoxia and hypoxia-independent signaling pathways such as TNF-α, LPS, and ROS/NF-κB/HIF/VEGFs [150,303]. This suggests that the strong inducers of VEGF and other growth factor production in ECs are mitochondrial and oxidative stress [304]. Additionally, oxLDL can activate NF-κB/HIF/VEGFs in ECs by acting on SR-E1 receptors (LOX-1) [233].
The effects of VEGFs on the development of atherosclerosis are complex and diverse. These effects have been described in detail in recent reviews, including [150,305,306,307]. VEGFs act on different cell types and are involved not only in angiogenesis and CS development but also in the regulation of lipid metabolism, glucose metabolism, and insulin sensitivity. Furthermore, the polymorphism of VEGF genes plays a significant role in predisposition to atherosclerosis and other cardiovascular diseases [308].
The VEGF family includes several members that play a role in atherosclerosis, with VEGF-A being a prominent marker [309]. VEGF-A has both beneficial and detrimental effects related to atherosclerosis. On the one hand, it protects ECs by stimulating the expression of antiapoptotic proteins and promoting NO synthesis by eNOS [310]. On the other hand, VEGF-A can prevent the repair of endothelial damage, contributing to atherogenesis and promoting monocyte adhesion, transendothelial migration, and activation [150].
Interestingly, the anti-atherogenic effects of VEGFs may be mediated through the regulation of arterial wall lymphatic drainage. Studies in mice suggest that a disruption of lymphatic function prior to the induction of atherosclerosis can contribute to the development of the disease, which is associated with a defect in the drainage function of the collecting lymphatic vessels. Systemic injections of VEGF-C can restore lymphatic transport in these mice [311].
4.2.4. The Role of Noncoding RNAs
Endothelial dysfunction is associated with various epigenetic mechanisms that are regulated by noncoding micro (mi) and long (lnc) RNAs [312]. It is worth noting that a single miRNA can inhibit the translation of hundreds of mRNAs, and collectively, noncoding RNAs can affect a wide range of processes, including angiogenesis, apoptosis, proliferation, and migration in vascular cells [313].
In this study, we will examine several examples of how noncoding RNAs affect the functions of ECs. For instance, the MEG3 lncRNA was found to suppress proliferation and induce apoptosis in vascular ECs [314]. Furthermore, the regulatory pathway of RNA MEG3/miR-223/NLRP3 limits the activation of the NLRP3 inflammasome and the development of pyroptosis in ECs [315]. Additionally, the pathogenic role of lncRNA MALAT1 in the development of endothelial dysfunction in diabetes and aging was described [316,317]. LncRNA H19 was shown to prevent the endothelial-mesenchymal transition in diabetic retinopathy [318]. The lncRNA TUG1 and miR-148b contribute to the expression of the IGF2 protein, which regulates proliferation and apoptosis in cells treated with oxLDL [319]. Overexpression of lncRNA SENCR regulates proliferation and migration in the HUVEC endothelial cell line [320]. The lncRNA GATA6-AS has been shown to be involved in the response of ECs to hypoxia [321]. Increased lncRNA GAS5 expression and decreased miR-26a expression have been reported in plasma from patients with atherosclerosis in oxLDL-activated human aortic ECs [322]. LncRNA and miR-320a regulate SR-E1 (LOX-1) expression in oxLDL-activated HUVEC cells [319]. MiR-638 has been shown to negatively regulate the AKT/mTOR signaling pathway and therefore inhibit proliferation and activate apoptosis of ECs treated with human aortic oxLDL [323]. Overexpression of lncRNA FENDRR was found to regulate VEGF-A and promote apoptosis by targeting miR-126 [324]. The lncRNA ROR enhances NF-κB activation and the JAK1/STAT3 signaling pathway in EC, promoting the survival of these cells [325]. Overexpression of miR-134 suppresses the ability of ECs to grow and migrate [326]. An increased expression of RNCR3 lncRNA has been found in human aortic atherosclerotic lesions [327]. In ECs, miR-5680 inhibits AMPK phosphorylation, thus regulating the development of KS [328]. The NORAD lncRNA is activated in response to DNA damage and maintains mitosis stability and genome integrity [329]. Overexpression of lncRNA p21 in ECs increases apoptosis and expression of SR-E1 (LOX-1) [330]. LncRNA HIF1A-AS1 stimulates apoptosis and decreases the proliferative activity of ECs [331]. A study by Song et al. showed that coronary atherosclerosis could be prevented by reducing lncRNA ANRIL expression [332]. Inhibition of the lncRNA XIST/miR-320/NOD2 regulatory pathway promotes the survival of oxLDL-activated ECs [314].
MiR-155 is an extensively researched miRNA that is linked to inflammation. It plays a crucial role in regulating eNOS levels and disrupting EC-dependent vasorelaxation [333]. Conversely, miR-103 promotes endothelial inflammation and increases the adhesion of monocytes to ECs [333]. Activated neutrophils can deliver miR-155 in microvesicles to endothelial cells (ECs) in arterial regions prone to atherosclerosis, thereby enhancing EC activation and monocyte adhesion to ECs [334].
During plaque formation in the aorta, ECs export miR-92 in extracellular vesicles to Mf. As a result, Mfs alter their phenotype to pro-atherosclerotic [335]. MiR-92 is, therefore, a potential target for regulating vascular disease. The complex interplay between miRNA, lncRNA, and other regulatory factors controls the development and resolution of pro-inflammatory CS, including ECs. However, prolonged exposure to damaging factors can stabilize pathological changes at the cellular, tissue, and systemic levels, leading to the formation of stable allostasis and promoting inflammation at the local level, such as in arteries prone to atherosclerosis.
4.2.5. Disruption of the Endothelial Barrier, Transendothelial Migration of Leukocytes in the Arteries
The process of cell migration across the endothelial barrier involves several fundamental mechanisms. First, there is ECs activation, followed by the adhesion of migrating cells to the endothelium and glycocalyx density reduction. Then, there is the dissociation of tight contacts between ECs, leading to the formation of gaps between them, which allows chemokines produced by ECs and their neighboring cells to direct the migration of leucocytes through the gaps formed via the paracellular pathway. Simultaneously, the paracellular pathway predominantly over the migration of leukocytes through the endothelial cell layer (transcellular pathway), which also necessitates prior adhesion of migrating cells to the endothelium [336,337].
The adhesion process includes several main steps. The first step is dynamic contact or “rolling”, which is determined by the inducible expression of E- and P-selectin receptors (CD62E/P) on ECs. The ligands for these receptors are leukocyte mucins, which are certain glycocalyx structures. P-selectins can be rapidly released from the Weibel-Palade body upon activation of ECs, which leads to the pre-activation of migrating leukocytes. Leukocyte (L) selectins (CD62L), which interact with endothelial mucins, also contribute to the rolling and pre-activation of migrating cells [336,337].
The second step is the formation of a sustained activation contact between cells or “arrest”, which involves ICAM-1 (CD54) and VCAM-1 (CD106) ECs. The main ligands of ICAM-1 are leucocyte β2-integrins (CD18), including LFA-1 (CD11a/CD18) and Mac-1 (CD11b/CD18). In turn, the main ligand of VCAM-1 is the β1-integrin (CD29), specifically VLA-4 (CD49b/CD29). These contacts are stimulated by several pro-inflammatory stimuli [336,338,339].
The third step is the disruption of contacts between ECs, which provides a paracellular pathway for leukocyte migration. A key mechanism of this process is the reduced expression of vascular endothelial (VE) cadherin (CD144) in the ECs contact area [340,341].
The fourth and final step is the remodeling of the actin cytoskeleton of ECs and migrating cells, which is a prerequisite for transendothelial leukocyte migration [341].
All the processes of exudative reaction and leukocyte migration to the inflammation focus are mainly observed in postcapillary venules, which are lined by type 2 ECs [59]. In contrast, the arterial part of the vascular network is not characterized by these processes, although in atherosclerosis, there may be a significant increase in the transendothelial migration of leukocytes in arteries [342,343]. For instance, treatment with oxLDL at a concentration of 40 µg/mL increased the expression of E- and P-selectins, VCAM-1, and ICAM-1 in a culture of human coronary artery ECs (HCAEC) mediated through LOX-1 [344]. In patients with diabetes, elevated levels of soluble VE-cadherin (sCD144) in plasma were identified as a significant risk factor for coronary heart disease compared to all other traditional risk factors [345,346,347].
In the elderly, problematic areas of the arteries for atherosclerosis are characterized by the presence of abnormal multinucleated ECs [347]. In this case, the basal endothelial membrane thickens, increasing its affinity to LDL, especially for modified LDL. This condition may result in the disruption of endothelial barrier function, either by the separation of ECs from the basal membrane or by the opening of dense contacts between ECs [348].
Chemokines are a group of small cytokines that are known for their ability to regulate leukocyte recruitment to sites of inflammation by inducing chemotaxis. This is achieved by binding chemokines to their receptors on white blood cells, leading to changes in cellular behavior, such as the rearrangement of actin and cell shape, allowing for leukocyte migration. In particular, the migration of neutrophils and monocytes across the endothelial barrier is regulated by CXCL8 (IL-8) and CXCL1, which can be secreted directly by ECs [349,350]. CXCL1, in particular, enhances the adhesive and pro-inflammatory properties of ECs in an autocrine manner, exerting an atherogenic effect. However, CXCL1 expression can be inhibited by miR-181b, which inhibits NF-κB in the atherosclerotic endothelium, exhibiting anti-atherogenic properties [351].
CXCL12 (SDF-1) is secreted by stromal cells [352]. It interacts with glycosaminoglycans in the endothelium, which are essential for its stability and presentation to leukocytes. CXCL12 acts through the CXCR4 receptor, which is widely expressed in various cell types associated with cardiovascular disease, including ECs, VSMCs, T cells, B cells, neutrophils, monocytes, and Mf [352]. While CXCL12 promotes the migration of these cells, it also accelerates the healing process of damaged vessels and supports ischemic neovascularization [353]. However, CXCL12 is considered to be a proatherogenic factor due to its involvement in dyslipidemia, angiogenesis, plaque destabilization, thrombus formation, neointimal hyperplasia, and pro-inflammatory effects against vascular endothelium [354]. Moreover, an increase in LDL levels is associated with the activation of CXCL12 expression, which is inversely proportional to HDL levels [355].
CXCL9, CXCL10 (IP-10), and CXCL11 are secreted by monocytes, ECs, and fibroblasts. In response to IFN-γ and TNF-α, they act through the CXCR3 receptors and promote the migration of monocytes, T lymphocytes, dendritic cells (DCs), and natural killer (NK) cells [356]. In patients with morbid obesity and metabolic syndrome, CXCL10 and CXCL11 are associated with insulin resistance and increased leukocyte adhesion on the endothelium [357]. The chemokine CCL20 (acting via the CCR6 receptor) is important for monocyte recruitment in atherosclerosis [358]. On the other hand, CCL5 (RANTES) is secreted by activated platelets and ECs and interacts with several receptors (CCR1, CCR3, CCR5), mediating monocyte/macrophage infiltration in atherosclerotic lesions [359].
The literature supports the importance of the CCL2 (MCP-1)—CCR2 axis in human atherosclerosis, highlighting its relevance and therapeutic potential [360]. CCL2, primarily secreted by monocytes and Mf, is bound to the endothelial glycocalyx and is considered the strongest chemoattractant for monocytes. Endotheliosis involves multiple interrelated processes that contribute to the development of arterial wall inflammation in atherosclerosis. In addition to ECs, other factors play a role in atherosclerotic inflammation.
4.3. Mast Cells in Atherogenesis and Aging
Adventitial mast cells (MCs) are located near the vasa vasorum of the aortic and coronary artery walls and in the intima, close to the lumen endothelium, as it lacks microvessels and is absent in normal conditions. MCs are powerful participants in the inflammatory response in various tissues, including both intimal and adventitial layers of atherosclerotic arteries [361], and contribute to the onset and progression of atherosclerosis [362]. Thus, activated MCs release histamine, heparin, neutral proteases (tryptase and chymase), ROS, and cytokines such as TNF-α, IL-6, CCL2, and CCL5 at the site of atherogenesis [361,363,364]. Due to their ability to release angiogenic compounds, histamine and proteases, MCs not only cause microvessel growth into the intima but also rupture new vessels, leading to intramembranous hemorrhage [365]. Immune complexes (including IgG-oxLDL), DAMPs, complement anaphylaxins (C3a and C5a), and pro-inflammatory cytokines act as MCs activators [362,366,367], while SR-B1 is involved in TC degranulation [367].
Several studies have shown a relationship between MCs and aging. In particular, an increase in the number and activity of MCs in the mesentery [368], heart, and kidneys were observed in aging rats [369]. In aging, MCs may also be involved in the pathogenesis of osteoporosis [370]. Aging causes basal activation of lymphoid MCs, which, in turn, limits the beneficial function of immune cells [371]. Taken together, these changes play an important role in the pathogenesis of inflammation and immunity associated with aging.
In humans, serum levels of tryptase, a marker of MCs activation and degranulation, have been found to be increased in aging, particularly in obesity [372,373]. However, some studies have reported a decrease in degranulation and an increase in MC numbers with aging [374]. In addition, activated MCs may contribute to the pathogenesis of neurodegenerative diseases in aging by disrupting the permeability of the blood-brain barrier, thereby allowing toxins and immune cells to enter the brain [375]. Overall, MCs are systemically involved in the formation of allostasis in the aging organism and thus participate in the pathogenesis of many aging-related chronic diseases, including atherosclerosis. In classical inflammation, particularly in immediate-type allergies, the role of mast cells is primarily to ensure local exudative and vascular reactions. However, in aging and low-grade inflammation, MCs’ function is less demonstrable and is associated with remodeling the microvascular network and modulating the function of different types of immune cells. At the same time, pathologically activated MCs are also capable of maladaptive effects in the tissues where they are localized.
4.4. Role of Platelets in the Pathogenesis of Atherogenesis
Platelets play a crucial role in hemostasis, preventing blood loss from injured blood vessels. Evolutionarily, platelets are thought to have originated from blood phagocytes, and their nuclear-free forms first appeared in mammals [59]. As a result, platelets share many functional similarities with phagocytic leukocytes, including the expression of receptors for TLR, SR, various complement factors, Fc receptors, and CD154 receptor (CD40 ligand on antigen-presenting cells). Platelets also express adhesion receptors such as P-selectin and integrins and are able to produce NO via eNOS and iNOS. Upon activation, platelets release various factors, including cationic proteins, proteases, cytokines (such as IL-1β, IL-6, IL-8, TNF-α, and various chemokines), and clotting factors [376,377,378,379,380,381,382,383,384,385,386].
Thus, platelets serve two primary functions: (1) participating in hemostasis to stop bleeding in case of vascular damage and (2) contributing to the development of various types of inflammation. In the latter case, the role of platelets and microthrombi in postcapillaries in the development of an exudative-vascular reaction in the focus of canonical inflammation is evident. At the system level, this process is one of the characteristic phenomena of systemic hyperinflammation and disseminated intravascular coagulation (DIC) [60].
However, platelets can also be involved in low-grade inflammation, specifically in the formation of moderate thrombophilia at the systemic level (not reaching the criteria for DIC) and, at the local level, in increased endotheliosis, endothelial damage, and leukocyte migration in arterial regions prone to atherosclerosis development [387].
The proatherogenic effects of activated platelets are related not only to their secretory function but also to their direct adhesion to EC, extracellular matrix structures, and their aggregation with monocytes and other leukocytes. Platelets can be activated by atherogenic lipoproteins, DAMP and PAMP, immune complexes, active hemostasis factors, some miRNAs, adhesive interactions, and complement [388,389,390,391].
In the presence of an intact and healthy endothelium and laminar blood flow, circulating platelets remain in a resting discoid state near the apical surface of ECs, without forming adhesive contacts. This is due to the antiadhesive properties of normal endothelium. After tissue injury, platelets are exposed to thrombogenic molecules present in the subendothelial tissue, such as collagen, von Willebrand factor (VWF), laminin, and thrombospondin, leading to their activation and adhesion to the damaged endothelium. Even with an abrupt change in blood flow shear stress or a change in the vascular microenvironment, circulating platelets can quickly perceive these signals and react with a subsequent interaction with activated ECs [389].
The formation of platelet-macrophage aggregates shifts Mf to a more pronounced pro-inflammatory phenotype [392], and in patients with atherosclerotic diseases, platelet activity correlates with Mf production of inflammatory cytokines [393]. Platelets express many surface receptors that interact directly (through receptors) or indirectly (through molecular adhesion cofactors) with collagen and other extracellular matrix structures of the basal endothelial membrane and atherosclerotic plaque [394,395]. Among these receptors, the integrin α2β1 (GPIa/IIa) and Ig-like receptor glycoprotein VI (GPVI) are the most important. Integrin α2β1 predominantly mediates adhesion, while GPVI is a strong platelet activator after contact with collagen. The presence of SR-B2 (CD36) on the surface of platelets has been shown to promote their hyperactivity upon contact with oxLDL [396].
Furthermore, platelets have been shown to play a role in modulating the differentiation of monocytes into Mf and influencing the ability of Mf to accumulate lipids and become foam cells [397]. When activated, platelets secrete numerous chemokines, including CXCL4 (PF4), CCL5 (RANTES), CXCL12 (SDF-1α), CXCL16, and others, which initiate or promote local inflammatory processes at sites of vascular injury. These processes are mainly mediated by the recruitment of circulating hematopoietic stem cells, neutrophils, monocytes, and lymphocytes to the vascular wall [398].
One of the key factors contributing to vascular complications in patients with type 2 diabetes mellitus (DM2) is increasing atherosclerosis. Hyperglycemia and insulin resistance can disrupt normal endothelial and platelet function, leading to the development of a prothrombotic state in DM2 patients [399]. These pro-atherogenic effects increase the risk of vascular complications in DM2 patients.
In summary, platelets act as inflammatory factors and may be involved in the pathogenesis of atherosclerosis, from the initial atherogenic manifestations of endotheliosis to their involvement in the inflammatory microenvironment of the atherosclerotic plaque.
4.5. Role of Lipoprotein Metabolism Dysfunction in the Pathogenesis of Atherosclerosis
The accumulation of plasma cholesterol and atherogenic lipoproteins (Lp), especially modified LDL, is a crucial metabolic condition for the development of atherosclerosis. All Lp have a hydrophobic lipid core surrounded by a hydrophilic envelope consisting of phospholipids and apolipoproteins (apo). Lp(a) has the highest atherogenic activity among native Lp and differs little from LDL in the composition of the lipid core and protein envelope [400,401]. However, Lp(a) contains not only the aroB-100 protein but also the apo (a) protein, which is structurally similar to plasminogen. As a result, Lp(a) has a high affinity for the extracellular matrix and rapidly accumulates in the vascular wall. Apo(a) undergoes oxidation and partial proteolysis by metalloproteinases, which contribute to the development of an atheromatous plaque. Recent review publications, such as those by Mehta (2022) [402], Lu (2022) [403], Di Giovanni (2023) [404], Ronda (2023) [405], Bellot (2023) [406], Guo (2023) [407], Močnik (2023) [408], Kim (2023) [409], and Poznyak (2023) [410], have detailed the pathogenetic role of impaired Lp metabolism in atherogenesis. It has been shown that all Lp-containing apoB-100 is associated with atherosclerosis. On the other hand, HDL helps remove cholesterol from Mf and VSMCs, thereby inhibiting the formation of foam cells. As mentioned in Section 4.2.3, effective arterial lymphatic drainage is important to allow HDL backtransport to the liver, where excess cholesterol undergoes oxidation and conversion to bile acids.
Initially, neutral fat (triacylglycerol) and cholesterol esters are synthesized in the liver, mainly from incoming blood glucose and FFA. These lipids then enter the bloodstream as very low-density lipoproteins (VLDL). Under the action of lipoprotein lipase, VLDL loses most of its neutral fat and becomes LDL, which transports cholesterol and esters to various cells by binding, via apolipoprotein B-100 (apoB-100), to the cellular receptor for LDL (LDLR).
ApoB-100 is the primary structural component of atherogenic lipoproteins, including VLDL, LDL, and Lp(a), while apoB-48 is specific to chylomicrons that are formed in the intestine to carry exogenous lipids to the liver and peripheral tissues. ApoA-I and apoA-II provide the framework for the lipidation of HDL, while apoC-II, apoC-III, and apoE are involved in the metabolism of triacylglycerol-rich lipoproteins. Apo(a) covalently binds to apoB-100 to form Lp(a). However, apoB-100 and apoB-48 are insoluble apolipoproteins and cannot remain in the blood in an Lp-free state. In contrast, other apolipoproteins are soluble and can exist as free proteins in plasma and be freely exchanged between lipoproteins.
After transendothelial transfer, amino acids of apoB-100 in LDL bind to proteoglycans of the intima, leading to LDL retention in the arterial wall. Some LDL undergoes multiple modifications, including oxidation, and then binds to SRs on Mf, leukocytes, ECs, and VSMCs, causing local inflammatory and immune responses, as well as the formation of foam cells. Foam cells mainly originate from Mf and transdifferentiated VSMCs and enhance the inflammatory cascade, promoting further migration into the intima, proliferation, and transdifferentiation of VSMCs, ultimately leading to the formation of atherosclerotic plaques. At the same time, VSMCs lose contractile function and acquire a pro-inflammatory secretory phenotype.
The accumulation of Lp in the blood is promoted by several factors, including excessive intake of glucose and FFA from the blood into the liver, impaired formation of phospholipids in the liver leading to dyslipoproteinemia, inadequate reverse HDL cholesterol transport, oxidative stress, hyperglycemia, and other modifications of atherogenic lipoproteins, as well as genetic features and defects in the formation of individual forms of apolipoproteins and lipoprotein cell receptors.
4.6. Role of Metabolic Dysfunction in the Pathogenesis of Atherosclerosis
Low-grade inflammation is a common factor in the pathogenesis of morbid obesity, metabolic syndrome, and DM2. This inflammation is directly linked to metabolic damage factors.
Several metabolites, including glucose, FFA, and many amino acids, have regulatory functions by acting on cellular receptors, allosteric centers of key enzymes in metabolic pathways, and various links of intracellular signaling pathways [411,412,413,414,415]. These metabolites are direct participants in forming network information-regulatory fields at the cell, tissue, and holistic levels of organisms. In pathology, these fields are distorted due to the accumulation of dysfunctional (dysregulatory) systems with the pathological character of forward and backward regulation.
At the organism level, these effects are largely associated with metabolic cycles, including the glucose fatty-acid cycle (Randle cycle) and glucose-alanine cycle. These cycles integrate metabolic processes in major facultatively glycolyzing organs, such as the liver, adipose tissue, and skeletal muscle, to regulate blood concentrations of the main energy metabolites, primarily glucose and FFA.
However, the functions of these cycles are not characterized by mobility and are complemented by neuroendocrine regulation. For example, the main insulin-dependent transmembrane glucose transporter, known as GLUT4, is mainly localized in adipocytes and muscle tissue [416,417]. On the other hand, the main glucose transporter in hepatocytes (GLUT2) functions depending on the ratio of intra- and extracellular glucose and the action of several factors on hepatocytes, including FFA and pro-inflammatory cytokines, that contribute to insulin resistance [418,419].
In individuals with obesity, there is a decrease in the sensitivity of GLUT4 to insulin. This leads to an excessive accumulation of FFA and glucose in the blood. The activation of gluconeogenesis in the liver and the dysregulatory effects of excess FFA contribute to this effect. In addition, pro-inflammatory tissue stress in the liver (hepatosis), which can be complicated by nonalcoholic fatty liver disease, exacerbates the process of forced glucose synthesis and dysfunction of lipoprotein formation in the liver [420,421,422]. The accumulation of lipotoxicity factors and pathological glycation products in the blood can act as inducers of chronic low-grade inflammation in the endothelium and other tissues [423].
Furthermore, lipolysis in adipocytes and increased FFA levels in the blood are correlated with hepatic Mf activity in patients with non-alcoholic fatty liver disease [424]. Recent studies have also shown a positive correlation between plasma levels of hydrophobic branched-chain amino acids, such as leucine, valine, and isoleucine, and the occurrence of atherogenic metabolic disorders [425].
On the other hand, an imbalance in histohormone production in the facultatively glycolyzing organs can also contribute to the development of low-grade inflammation in obesity. For instance, the development of low-grade inflammation in adipose tissue causes an increase in the number and pro-inflammatory activity of stromal Mf there [426,427,428]; aging intensifies this process [429]. The production of pro-inflammatory cytokines by Mf in adipose tissue can have not only local but also systemic effects, including increased gluconeogenesis and insulin resistance in the liver [430].
At the same time, an imbalance in lipokine production in adipocytes develops with a predominance of pro-inflammatory, atherogenic factors (leptin, chemerin, resistin, IL-6, and others) over anti-atherogenic factors (FGF-21, CTRP9, omentin, progranulin, adiponectin, and others) [431,432]. These dysfunctions contribute to endothelial dysfunction, hypertension, and the formation of pro-inflammatory allostasis in general.
Therefore, low-grade inflammation is associated with metabolic dysfunction and insulin resistance at the level of individual organs and the body as a whole. This variant of the inflammatory process can be considered a typical complication of aging, providing an additional stimulus for the development of atherosclerosis and other cardiovascular diseases.
4.7. Summary
Atherosclerosis is a complex pathology that involves metabolic, immune, and inflammatory disorders at both local and systemic levels. Endotheliosis and local low-grade inflammation within the arterial wall are closely linked to the development of atherosclerosis. Systemic low-grade inflammation and associated pathologies such as morbid obesity, metabolic syndrome, and DM2 are consequences of increased pro-inflammatory systemic tissue stress that accompanies aging, known as “inflamm-aging.” Low-grade inflammation is the underlying cause of negative dynamics in many pathologies, including atherosclerosis. Consequently, vascular complications arise due to the formation of unstable atherosclerotic plaques, which are directly related to increased local inflammation and the involvement of various immune mechanisms.
5. Immune Mechanisms of Atherosclerosis Pathogenesis
5.1. Macrophages
The immune system in the arteries в нoрме primarily consists of stromal Mf situated in the intima and adventitia. These Mf are an essential component of innate immunity and play a pivotal role in inflammation, wound healing, and defense against infection and other harmful factors. However, stromal Mf not only serve as immunocytes but also perform various specialized functions to maintain tissue homeostasis. As a result, stromal Mf acquire a tissue-specific phenotype, such as osteoclasts, chondroclasts, microglia, mesangial Mf, Kupffer cells (also known as stellate Mf of liver sinus capillaries), CD169+ Mf of secondary lymphoid organs, and more [433,434].
The majority of stromal Mf are of embryonic origin and maintain their numbers in tissues through proliferation [433,434]. However, in some organs, embryonic Mf are replaced by other Mf originating from bone marrow monocytes. This replacement primarily occurs in mucous membranes and lymphoid organs, as well as in the heart and arteries [433,434,435]. Mf of monocytic origin tend to have greater immune and pro-inflammatory activity, while embryonic cells exhibit more homeostatic activity. The number of stromal Mf of bone marrow origin increases with age, including in arteries and adipose tissue [436,437].
Under conditions of intimal lipidization, stromal Mf attempt to excrete excess lipids, but this process is quickly overwhelmed, turning Mf into “foam cells” [438]. Simultaneously, upon activation, similar to that occurring in hypercholesterolemia or at sites of impaired blood flow, monocytes are recruited into the subendothelial space and then differentiate into pro-inflammatory Mf, capable of forming foam cells [439]. This process is enhanced when Mf release inflammatory cytokines, primarily TNF-α and IL-1β, causing the expression of adhesion molecules and chemokine production in ECs [439].
At the same time, Mf themselves produce chemokines that attract monocytes and other leukocytes (from the bloodstream) and VSMCs (from the media) to the focus of atherogenesis. Mf can also be released from the plaque through interaction with chemokines CCR7, CCL19, and CCL21, which are produced by ECs (CCR7) and arterial stromal cells (CCL19, CCL21) [440].
The process of foam cell accumulation in the intima is realized already at the stage of lipoidosis during the formation of lipid spots and bands. With that, the main effectors of atherogenesis become inflammatory Mf of monocytic origin, which are replenished by monocyte migration and then by the proliferation of recruited Mf. At the stage of atherosclerotic plaque formation, transdifferentiated, modified VSMCs, primarily with a macrophage-like phenotype, are also actively involved in the formation of foamy cells [441]. During plaque formation and local hypoxia, vessels ingrown into the intima from the media, through which monocytes and other leukocytes can migrate [442]. The transport function of the vasa vasorum also increases with the increase in plaque size and accumulation of cells in it.
When cholesterol efflux is activated by HDL function, plaque Mf acquire a pro-resolving M2-like phenotype, releasing anti-inflammatory cytokines such as IL-10 and TGF-β, thereby promoting tissue repair or fibrosis [443]. In addition to M2 Mf, fibroblast-like VSMCs producing collagen and other extracellular matrix proteins contribute to fibrosis and stable plaque formation [444,445].
Conversely, the dominance of M1 Mf, macrophage-like, and osteoblast-like VSMCs is a prerequisite for unstable plaque formation [444,445]. M1 Mf are predominantly localized along the perimeter of the lipid-protein core and in the cap of the unstable plaque, while M2 Mf and myofibroblasts localize in the area of fibrous surroundings and intimal vascularization. M1 Mf not only inhibit the structural function of M2 but also actively secrete matrix metalloproteinases (MMP-1, MMP-3, MMP-10, MMP-12, MMP-4, and MMP-25) that degrade the fibrous membrane and contribute to tissue destruction zones from destroyed extracellular matrix structures, cellular necrosis, and unfinished apoptosis [442,446].
M1 cells are characterized by a pronounced pro-inflammatory phenotype [439,442,447,448], which includes:
- High expression of pro-inflammatory cytokines TNF-α, IL-1β, and IL-12;
- iNOS expression;
- High expression of TLRs, other PRRs, and SR-B2 (CD36), which is the most functionally related to TLRs of all SRs;
- High level of transcription factor (TF) expression: HIF-1, NF-kB, STAT.
- M1 cells are more active in secreting chemokines, such as CCL2, CCL3, and CCL5 than M2 cells;
- In some cases, M1 cells may actively form NLRP3 inflammasomes, which are associated with pro-inflammatory cellular stress, hyperproduction of IL-1 and IL-18, and pyroptosis.
The dual function of SR-B2 (CD36) on ECs, platelets, and various immunocytes, particularly M1 cells, should be considered. SR-B2 (CD36) acts as (1) a signal transducer from PAMPs, DAMPs, and oxLDL and (2) an FFA transporter. However, when FFA β-oxidation and aerobic phosphorylation function are deficient, it can increase lipotoxicity and contribute to mitochondrial dysfunction [449].
In contrast, M2 cells are characterized by a more pronounced expression of SRs in general, especially those involved in efferocytosis (Table 1) and the mannose receptor, SR-E3 (CD206), as well as arginase1, which is involved in the synthesis of extracellular matrix proteins. M2 cells also express TFs, such as STAT3, STAT6, PPAR, and anti-inflammatory cytokines (IL-10, TGF-β) [441,444,450]. M2 cells utilize the aerobic breakdown of glucose and FFA to meet their energy requirements, while M1 cells use glycolysis [448,450,451]. This difference is related to several factors, including the fact that M1 cells are typically located in a more hypoxic environment than M2 cells. Additionally, the absence of aerobic processes in M1 cells reduces the cell’s dependence on mitochondrial and oxidative stress, which are more pronounced in M1 cells. With glycolysis, there is no competition for oxygen between oxidative phosphorylation and ROS formation, which can be formed not only in mitochondria but also during microsomal oxidation. Moreover, M1 cells release the end product of glycolysis (lactate), further acidifying the extracellular environment, thereby enhancing local pro-inflammatory mechanisms.
In the bloodstream, at least two subpopulations of monocytes are distinguished, with the most significant of them (up to 90%) able to differentiate into M1, while the other can differentiate into M2 [438,441]. However, under the influence of cytokines and other environmental factors, a morphofunctional drift of Mf in the M1-M2 range is possible [75]. Thus, the division into classical (M1) and alternative (M2) polarization of Mf is, to some extent, conditional [436].
In vitro, the current classification of Mf formed from monocytes includes at least 10 distinct subpopulations [452]. This differentiation is probably even more complex in vivo [453,454,455,456]. Nevertheless, the various variants of Mf differentiation can be simplified into four conditional types: M1, M2a, M2b, and M2c, including atherosclerosis [436]. It is also possible to distinguish special variants of Mf differentiation, such as M4, as well as hemoglobin-stimulated Mf-M(Hb), which are also designated as Mhem or HA-Mac [448,456,457].
M(Hb) are characterized by antioxidant and anti-atherogenic activity and are located in the focus of atheromatosis near the vasa vasorum and associated with intrabladder hemorrhages [458,459]. These cells highly express SR-I1 (CD163) and a heme-dependent transcription factor (ATF1) that induces the expression of hemoxygenase 1 and LXR-β (liver X receptor). M(Hb) are also involved in hemoglobin clearance through erythrocyte phagocytosis and increases cholesterol efflux by expression of LXR-β-dependent genes [458,459]. In turn, M4 expresses genes responsible for the induction of extracellular matrix-destroying proteases such as MMP7, MMP8, and MMP12 [de Sousa, 2019] and genes facilitating foam cell formation [455].
Each Mf type cooperates with complementary CD4 T-helper (Th) types and other cells of the immune system during the development of immune inflammation, thereby forming certain immune response vectors (I) in the focus of inflammation: I1 (M1-Th1); I2 (M2a-Th2); I3 (M2b-Th17); I-reg (M2c-Treg) [75].
Regarding atherosclerosis, on the one hand, oxLDL uptake by Mf can be considered a protective mechanism since they remove cytotoxic elements from the intima. On the other hand, the increased migration of monocytes into the intima and their subsequent differentiation into inflammatory Mf and then foamy cells leads to the formation of an atherosclerotic plaque, which can become unstable and lead to severe hemodynamic disorders.
5.2. Cellular Immune Response Vectors in Atheromatous Inflammation
The process of atheromatosis during the formation of stable and unstable plaques resembles chronic classical inflammation with a fibrotic component. This type of inflammation involves complex intercellular interactions, particularly between Mf and T-helper cells. These interactions can be categorized into four vectors of immune response (I1, I2, I3, I-reg) that are active in the site of inflammation (see Table 2).
Table 2.
Vectors of immune response (I) [460,461,462,463,464,465,466,467,468,469,470].
It is important to consider that Th differentiation, as well as Mf, is characterized by plasticity. This means that certain spectrums of cytokines can cause transformations, such as Treg into Th17 or Th2, Th17 into Th1, or even the formation of functionally unstable T-cells with a mixed phenotype. For example, Th2 cells can transform into CD4+ T-cells that can simultaneously produce cytokines of competitive Th (Th2 and Th1), specifically IL-4 and IFN-γ [471,472,473]. Other cells with an intermediate Th1/Th17 phenotype simultaneously express TFs, such as T-bet and ROR, which allows for the simultaneous production of functionally different types of cytokines (IFN-γ, GM-CSF, and IL-17) [474]. Additionally, Th1, Th17, and Th2 can be divided into more specific subpopulations [475,476].
Furthermore, mature Treg cells can undergo epigenetic modifications, which can cause them to lose FOXP3 expression and transform from anti-inflammatory to pro-inflammatory cells [477].
Another group of cells to consider are γδ T cells, which sit at the border of adaptive and innate immunity and are also functionally plastic [478,479]. Studies have shown that γδ T cells in mice are predominantly found in the aortic root and contribute to the early formation of atherosclerotic lesions, plaque necrosis, and inflammation at this site [480]. However, in other studies, the immunological activity of total γδ T-cells in the blood of patients with coronary heart disease was significantly lower than that of healthy individuals [481]. The exact role of this subgroup of T cells in atherosclerosis is still unclear [482].
In general, epigenetic analysis has shown that the epigenome of cells involved in atherogenesis, such as ECs, fibroblasts, VSMCs, and various immunocytes, exhibits plasticity and heterogeneity [483]. The classification of pro-inflammatory immune responses (I1, I2, I3, I-reg) is conditional, and morphofunctional changes of immunocompetent cells occur within certain corridors. The boundaries and integration of these corridors are influenced by immune response triggers, immunocyte maturity, genetic and epigenetic interactions, inflammation activation factors, cytokine network features, and extracellular communication mechanisms like intercellular transvesicular exchange [484,485]. Additionally, even competing immune responses such as I1 and I2 have zones of overlap and functional cooperation, and the interaction between M1 and M2 Mf can lead to progressive interstitial fibrosis [486]. Overall, I1 factors promote atherosclerosis and unstable plaque formation by producing pro-inflammatory cytokines and chemokines. In contrast, M2c and Treg macrophages suppress inflammation and reduce plaque size, while M2a and Th2 promote fibrosis and plaque stability [436,439,440,442,447,448,487].
M2b Mf, like M1, produce pro-inflammatory cytokines such as IL-1, IL-6, TNF-α, and others, contributing to plaque destabilization [442]. Th17 cells not only activate M2b but also attract neutrophils to the focus of atherogenesis, which increases tissue destruction [171,488] (Figure 4). IL-17A signaling activates various downstream pathways, including the NF-κB pathway, intensifying inflammation and atherosclerosis in ECs, M2b Mf, and VSMCs [489,490]. The metabolic profiles of Th17 cells and Th1/M1 cells are similar, including the functions of glycolysis and hypoxia in the formation of these areas of the immune response [491,492]. However, some studies have also shown an anti-atherogenic role of IL-17 [428,493]. In addition to pro-inflammatory cytokines, M2b Mf also secrete significant amounts of IL-10, which is an IL-1 inhibitor and antiatherogenic factor [428,493]. The function of M2b Mf in unstable plaque formation is ambiguous, especially in I-reg deficiency.
Figure 4.
Involvement of Immune Response Vectors in Atherosclerotic Inflammation. Note: This figure illustrates the roles of various immune response vectors (I1, I2, I3, and I-reg) in the processes of atherosclerotic inflammation. (1) The formation of an atherosclerotic plaque and its transition to an unstable state depends on the balance of pro-inflammatory and specialized pro-resolvent mediators such as IL-10. (2) I2 dominance at the stable plaque stage promotes fibrosis and competitive inhibition of I1. However, I1 has a more pronounced pro-inflammatory potential and is an obvious mechanism of tissue destruction. (3) Cellular elements I-reg actively secrete anti-inflammatory mediators such as IL-10 to stabilize the atherosclerotic plaque. (4) Factors I3, along with I1, can be prominently activated in unstable plaque formation, where they promote tissue destruction. Nevertheless, M2b can actively secrete not only pro-inflammatory mediators but also IL-10, thereby limiting not only I1 but also I3, including Th17 and neutrophils [494].
B-cells may also be involved in the focus of atherogenesis and interact there with Mf and other lymphocytes. B-1 lymphocytes, which are on the edge of innate and adaptive immunity, exhibit their anti-atherogenic properties by secreting natural IgM antibodies that contribute to the removal of oxLDL from the bloodstream. However, antigen-specific B-2 cells stimulate Th1 cells and DCs to play a proatherogenic role [493].
5.3. Antigen-Specific Mechanisms of Atherosclerosis
The involvement of T cells, B cells, and DCs in atherogenic inflammation, as well as the presentation of antigenic oligopeptides to CD8 and CD4 T-lymphocytes via major histocompatibility complex (MHC, HLA) proteins, provides evidence for the presence of antigen-dependent mechanisms of atherosclerosis [495,496,497]. Upon encountering antigens, naive T cells transform into effector memory T cells, particularly Th1 cells that produce interferon-gamma and proatherogenic cytokines [497]. The accumulation of monoclonal and oligoclonal effector CD4 T cells and cytotoxic CD8 T cells, including those recognizing autoantigens such as oxLDL, in atheroma contributes to the formation of unstable plaques [498].
Mature, antigen-specific T cells interact with Mf expressing HLA-DR and cofactor receptors CD40 and B7 (CD80/CD86), which are necessary for their antinational interaction with T cells [499]. HLA-DRs are also expressed in atherosclerotic plaques and on VSMCs, which is not typical of normal arteries. Moreover, the involvement of B-2 lymphocytes in the pathogenesis of atherosclerosis indicates the participation of antigen-specific immunoglobulin (Ig) and follicular helper T cells (Tfh) in this process [500].
In advanced stages of atheromatosis development, T- and B-zone cell proliferations are present in arteries with active germinal centers, including DC and plasmocytes, while the formation of lymphatic follicles in the adventitia of atherosclerosis-affected arteries is designated as arterial tertiary lymphoid organs (ATLO) [501]. Analysis of ATLO immune cell subpopulations indicates antigen-specific T- and B-cell immune responses in the adventitia of atherosclerotic arteries. Additionally, ATLOs contain innate immune cells, including a large component of inflammatory Mf, B-1 cells, and an aberrant set of antigen-presenting cells, primarily DC. ATLOs are also characterized by neoangiogenesis, lymphangiogenesis, high endothelial venules (type 2 ECs), naive lymphocyte recruitment, and central and effector memory CD4 T cells [501,502].
Infectious antigens may contribute to the development of atherosclerosis, as seen in periodontal diseases [503]. Mayr et al. (2020) have shown a high reactivity of T cells against chlamydial antigens in atherosclerotic plaques [504]. Chronic viral infections [505] and COVID-19 [506] are also suggested to play a role in the development of cell aging and atherosclerosis in the literature. However, atherosclerosis is not generally considered an infectious disease, allowing us to focus on the autoimmune mechanisms of this pathology, which are primarily associated with I1 and I3 in atheroma (Table 2).
Multiple studies have demonstrated that several systemic autoimmune diseases are associated with accelerated development of atherosclerosis. Patients with antiphospholipid syndrome have been found to have high levels of oxLDL-binding antibodies detected in autoimmune-mediated atherothrombosis [507]. Additionally, autoantibodies to oxLDL are found in atherosclerosis, and these cross-react with antiphospholipid antibodies [508]. Furthermore, immunopathological problems such as systemic lupus erythematosus, antiphospholipid syndrome, and rheumatoid arthritis have been shown to accelerate atherogenesis [509,510,511,512,513].
In the context of atherogenesis, oxLDL interacts with CRP to form oxLDL-CRP complexes, which maintain not only vascular inflammation but also trigger autoimmune reactions [514]. Patients with atherosclerosis have significantly elevated autoantibodies against HSPs, and T-lymphocytes specifically react to HSPs as autoantigens in atherosclerotic plaques [515]. Additionally, IgG and IgM antibodies to oxLDL are present in animal and human plasma, and they form immune complexes with oxLDL in atherosclerotic lesions [515]. Furthermore, IgG antibodies to oxLDL and IgG antibodies against apoB-100 are positively associated with the presence of cardiovascular disease [516]. A proatherogenic role of IgG and IgG immune complexes has been revealed through the activation of Mf in atheromas via Fc receptors [517]. However, the specific influence of antibodies on atherosclerosis remains to be elucidated compared to other B-cell functions, such as cytokine production or the influence of B cells as antigen-presenting cells [518].
In summary, while infectious antigens can contribute to the development of atherosclerosis, autoimmune mechanisms are also important in this pathology. Multiple systemic autoimmune diseases are associated with accelerated atherogenesis, with autoantibodies against oxLDL and HSPs and IgG and IgG immune complexes playing a proatherogenic role. The specific role of antibodies in atherosclerosis compared to other B-cell functions requires further investigation.
Meanwhile, Fab-fragments of IgG or single-stranded variable fragments of antibodies against oxLDL can reduce the formation of foam cells [519], indicating that the neutralizing functions of antibodies against oxLDL may play an important protective role in atherosclerosis. Moreover, IgG against oxLDL can neutralize and facilitate the clearance of oxLDL by forming oxLDL-IgG complexes that are rapidly eliminated by Fc-receptor Mf in the liver and spleen [520]. Adoptive transfer of human IgG1 against modified apoB-100 can reduce the degree of atherosclerosis and autoimmune oxLDL epitopes in the blood of recipient mice [521]. Therefore, certain types of antibodies against modified LDL may have potential therapeutic value through the use of passive or active immunization [518]. However, further large-scale studies are required to definitively establish the correlation between different oxLDL-binding antibody isotypes and cardiovascular disease [522].
Additionally, it is necessary to consider the possibility of the formation of autoantigens and haptens (primarily phospholipids) not only as a result of the molecular modification of atherogenic lipoproteins but also the potential formation of other autoantigens due to a variety of reasons [496,523,524,525,526]. These reasons include molecular mimicry of microbial proteins during atheroma infection, “bystander activation”—the release of autoantigens from tissue damaged by inflammation, polyclonal activation of lymphocytes, “epitope spreading”—where the targets of autoimmune responses are expanded by including other epitopes on the same protein or in other proteins in the same tissue, systemic or local immune dysfunction, Treg deficiency, and increased mutagenesis (for example, as a result of oxidative cellular stress) and decreased efficiency of utilizing abnormal proteins. Another potential cause of immune dysfunction in atherosclerosis is increased stem cell proliferation, which can accelerate negative somatic evolution and proliferation of myeloid cell clones with driver mutations [527].
Thus, there is currently no doubt about the involvement of autoimmune mechanisms in the development of atherosclerosis, both as inducers of inflammation and as factors that facilitate the clearance of modified Lp from the bloodstream. Simultaneously, there is no convincing reason to deny the key role of scavenger receptor (SR)-dependent uptake of modified LDL by Mf and VSMCs in atherogenesis, which subsequently leads to foam cell formation.
5.4. Summary
In summary, the development of atherosclerosis is mediated by a complex interplay between innate and adaptive immune mechanisms, with inflammation primarily driven by inflammatory Mf and foam cells. Modified Lp and cholesterol deposition in the intercellular matrix and foam cells serve as major triggers of inflammation, with autoimmune and infectious factors also playing a role. The different immune reactivity vectors (I1, I2, I3, I-reg) are involved in the inflammatory process and may be localized in different layers of the atheroma, with the ratio of these immune responses affecting the severity of inflammation and stability of the atherosclerotic plaque. Despite the involvement of autoimmune mechanisms, the role of SR-dependent uptake of modified LDL by Mf and VSMCs with the subsequent formation of foam cells cannot be ignored in the development of atherosclerosis.
6. General Evaluation of the Inflammatory Process in Atherosclerosis
The development of atherosclerosis involves a local low-grade inflammation during the pre-atherosclerosis stage, characterized by manifestations of endotheliosis. In the initial subclinical stage of atherosclerosis, there is the formation of lipid spots and bands in the arterial intima (lipoidosis). This stage is associated with the appearance of frothy cells, namely accumulated cholesterol Mf and, in some cases, VSMCs, and deposition of lipids in the intercellular matrix of the intima. It is noteworthy that lipid spots and bands can be detected even in childhood, suggesting that lipoidosis can develop under conventionally physiological conditions without overt endotheliosis manifestations.
Lipoidosis can be considered a transitional stage to the development of the next stage of atherosclerosis, which is productive inflammation in the formation of atherosclerotic plaque (atheromatosis). The commonality of the molecular mechanisms of CS in atherosclerosis and low-grade inflammation suggests the proximity of epigenetic cellular changes in atherogenesis to other pro-inflammatory processes of the elderly, such as osteoporosis [528] and neurodegenerative diseases [529].
Abnormal states of autophagy, pyroptosis, and ferroptosis in vascular cells, including ECs, VSMCs, and Mf, associated with oxidative stress, pro-inflammatory cellular phenotype, and other manifestations of CS, play a key role in the development of atheromatosis [114]. The formation of an atherosclerotic plaque involves the destruction of the internal elastic lamina, large-scale migration of VSMCs into the focus of atherogenesis, and local vascularization of the media side intima. In addition, not only the media but also the adventitia is involved in the process, where tertiary lymphoid organs are formed. The peripheral nervous system uses the adventitia to regulate atherogenesis processes through the existence of neuroimmune interfaces in the arteries [282,530]. In these cases, the interactions between the nervous system and the immune system are bidirectional, which is especially important in the impact of psycho-emotional stress on human health [531]. Moreover, chronic psychosocial stress can contribute to the progression of atherosclerosis and its complications [532,533,534].
Perivascular adipose tissue, which produces adipokines and vasoactive substances such as leptin, adiponectin, resistin, and visfatin, cytokines, and growth factors that regulate vascular tone and homeostasis, is also involved in the process of atherogenesis [283,535,536].
The resolution of atherosclerosis can be hindered by an imbalance in the production of pro-inflammatory and specialized pro-resolvent mediators (SPMs), impaired clearance of dead cells, and functional changes in immune cells that contribute to ongoing inflammation [120]. The action of SPMs aims to limit the severity and duration of inflammation, activate regenerative processes, and prevent or reduce post-inflammatory tissue sclerosis. SPMs encompass various endogenous mediators, including non-classical eicosanoids (lipoxins), ω-3 polyunsaturated fatty acid derivatives (resolvins, protectins, and maresins), protein/peptide mediators such as annexin A1, IL-10, IL-37, and nucleotides like adenosine and inosine [120,535].
SPMs have diverse chemical structures and function in a receptor-dependent manner on a wide range of target cells. Most SPMs activate receptors associated with specific types of G proteins. An imbalance between SPMs and pro-inflammatory mediators is linked to several prevalent chronic inflammatory diseases in humans, including atherosclerosis [536]. Consequently, the utilization of SPMs and their chemical derivatives presents a pressing issue in the prevention and treatment of atherosclerosis [120,535].
A stable plaque is characterized by a thick fibrous cap, low inflammatory cell activity, and small necrotic nuclei, which includes foamy cells. It is considered atheroma with predominant I2-dependent productive inflammation with fibrous tissue overgrowth surrounding a stable, moderately sized lipid core. However, excessive fibrous tissue overgrowth, especially in the cap region, can lead to arterial stenosis [536]. Therefore, not all variants of stable plaque development are safe with regard to clinical complications of atherosclerosis.
On the other hand, an unstable plaque is characterized by a dynamic process in the core and the tissues adjacent to the core of the plaque, namely the presence of a necrotic process and incomplete apoptosis of foam cells, and an increased number of pro-inflammatory Mf, T-cells, neutrophils, and other leukocytes, which amplifies the I1 and I3 directions. This leads to an increased role of tissue destruction in the development of inflammation (DAMP formation, among others) but also to thinning of the fibrous capsule, a process that is especially dangerous in the cap region [537,538]. A vulnerable, unstable atherosclerotic plaque is typically characterized as a plaque with a high likelihood of forming a thrombogenic area. An unstable plaque or a plaque prone to ulceration and rupture has a cap less than 65 µm, infiltrated by Mf and T-lymphocytes, a large lipid core constituting more than 40% of the entire plaque area, and increased neovascularization and a large number of frothy cells surrounding the core [539].
Protective angiogenesis plays a crucial role in restoring vascular wall normoxia and resolving inflammation, leading to stabilization and partial regression of atherosclerosis. However, pathological, maladaptive angiogenesis exacerbates disease progression by increasing Mf infiltration and vascular wall thickness and perpetuating hypoxia and necrosis [540]. Additionally, thin-walled, fragile new vessels can rupture, resulting in intra-abdominal hemorrhage.
Several histological types of vulnerable unstable plaques are currently recognized [541,542], including atheroma with a thin fibrous cap (lipid type), plaques with increased inflammation leading to erosion and thrombosis (inflammatory-erosive type), and plaques with necrosis/calcinosis (dystrophic-necrotic type). Hence, among the different types of unstable plaques, there are three distinct types: lipid, inflammatory, and dystrophic-necrotic. Multiple recurrent episodes of rupture, thrombosis, and subsequent fibrosis can occur over several cycles in the same artery section, forming multiple “layers” of fibrous overgrowths.
In general, an unstable plaque’s characteristic feature is the presence of varying degrees of tissue destruction with possible foci of necrosis. The question arises as to the extent the immune system’s response in these cases corresponds to the canons of classical inflammation. The development of classical inflammation in such cases is manifested by several typical variants of exudative-destructive inflammation: purulent, gangrenous, curd-like, and fibrinous [59]. All these variants are characterized by a strong exudative reaction of microvessels, with varying degrees of severity of complement and neutrophil involvement in the process. In the acute course, these inflammation variants usually transform into productive inflammation with granulation formation, followed by potential fibrosis and scar formation. In chronic inflammation, these processes typically develop simultaneously, occupying different compartments of the inflammation focus. Aseptic inflammation, such as autoimmune inflammation, is characterized by the latter variant, related to massive fibrin deposition in the inflammation focus. In some cases, like diabetic kidney disease, fibrinous inflammation may arise as a result of the progression and transformation of local low-grade inflammation [275]. For tissue destruction in atheromatosis, these classical inflammation variants are not observed; rather, atherogenic inflammation, in this case, can be characterized as productive-destructive.
The closest variant of the classic destructive inflammation in atherosclerosis is the rapidly progressing atheroma, characterized by a thin fibrous cap infiltrated with Mf and lymphocytes, rare VSMCs, and underlying necrotic nuclei [543]. Intrabladder hemorrhages with subsequent fibrin deposition in the hemorrhagic zone are common in this type of atheromatosis. However, it cannot be fully attributed to fibrinous inflammation associated with a pronounced exudative reaction of microvessels. The probable reason for these differences is the insufficient inflammatory efficiency of secondary vasa vasorum, which may compensate for the insufficient efficiency of lymphatic drainage of the arterial wall.
Summary. The initial manifestations of atherogenesis (pre-atherosclerosis) are characterized by endotheliosis mechanisms typical of low-grade inflammation, while the subclinical stage of lipoidosis (lipid spots and bands) is a transition zone to productive inflammation and atheroma formation. Atherogenic inflammation at the stage of the formed plaque involves a large-scale involvement of the VSMCs as well as vasa vasorum in this process. The formation of a lipid core surrounded by inflammatory cells and pronounced fibrous deposits around this formation is a typical feature of this stage of atheromatosis.
Strengthening of primary and secondary (inflammation-related) alteration factors against the background of efferocytosis and SPM insufficiency leads to increased tissue destruction and unstable plaque formation. However, unlike classical variants of destructive inflammation in atherosclerosis, the vascular-exudative reaction is weakly expressed in this stage of atherogenesis. Therefore, this stage of atherogenesis can be characterized by the position of productive-destructive inflammation but not by the exudative-destructive inflammation of the classical type.
7. Relation of Atherosclerosis to Systemic Inflammation (Hyperinflammation)
Acute SI is a pathological process that underlies the development of critical conditions in patients in intensive care units [66,544]. In sepsis, SI is directly associated with rapidly progressing multiple organ dysfunction, which can be complicated by disseminated intravascular coagulation syndrome, acute respiratory distress syndrome, and septic shock [545]. SI, including the “cytokine storm” phenomenon, can also develop in COVID-19 [546]. To verify SI as a general pathological process, certain levels of hypercytokinemia [547] need to be established and compared to the severity of other SI phenomena [66]. The development of acute SI is also typical for the aseptic refractory shock [548] and severe polytrauma variants [509].
The dynamic development of SI is characterized by the alternation of hyperergic and hypoperergic phases, determined primarily by the intensity of its action at the systemic level rather than the nature of the damaging factor [66,549,550]. Chronic SI has less clear clinical equivalents, but its presence can reflect severe chronic diseases with high values of hypercytokinemia and signs of paracoagulation [66]. As applied to atherosclerosis, the relationship between SI and atherosclerosis can be considered from two perspectives: (1) the influence of atherosclerosis complications on the development of acute and chronic SI; and (2) the influence of acute and chronic SI on the development of atherosclerosis.
A study using a combination of existing murine models of atherosclerosis and sepsis found that the intra-abdominal sepsis model accelerated atheroma development [551]. However, the acceleration of atheroma development was associated with prolonged systemic, endothelial, and intimal inflammation and was not explained by ongoing infection. Clinical studies aimed at solving this problem are difficult, and the paucity of clinical data should not be surprising. A literature review by K. Laudanski showed a 3–9% increase in the risk of stroke or acute coronary event in sepsis survivors [552]. One possible explanation for this phenomenon is the persistence of lipid profile abnormalities caused by acute sepsis until recovery, which leads to accelerated atherosclerosis. The presence of an increased risk of cardiovascular disease, including myocardial infarction and stroke, in survivors of sepsis was demonstrated in a review by Merdji et al. [553]. The authors attribute these patterns to premature vascular aging after a critical condition.
In experimental mouse models, Wang et al. (2018) revealed the progression of atherosclerosis after craniocerebral trauma [554]. In humans, posttraumatic stress disorder syndrome has also been found to have an additional influence on atherosclerosis progression [555]. However, the association of this syndrome with SI has not yet been established.
Establishing the relationship between transferred SI and the subsequent progression of atherosclerosis is a significant challenge in acute critical conditions. In chronic diseases, the main challenge is differentiating between SI-dependent systemic inflammatory response and the typical systemic manifestations of classical inflammation and low-grade inflammation. For instance, in gout, classical inflammation (arthritis) develops [556,557], but Kimura et al. (2021) showed in their review that the systemic effects of hyperuricemia contribute to the development of atherosclerosis [558].
We demonstrated a high probability of SI development in systemic autoimmune diseases with established risks of atherosclerosis progression, such as systemic lupus erythematosus, rheumatoid arthritis, and primary antiphospholipid syndrome [66]. However, the influence of autoimmune mechanisms specific to these diseases on atherosclerosis must also be considered. Terminal renal failure, regardless of the features of the primary etiology of renal damage, is a well-known risk factor for the progression of atherosclerosis and its complications, as well as an obvious reason for the development of chronic SI [276].
In general, many diseases with risks of developing chronic SI can contribute to the development of atherosclerosis, such as chronic destructive pulmonary disease and chronic heart failure [558,559,560,561]. However, the problem of determining the relationship between SI and atherosclerosis will largely be determined by the existence of generally accepted and specified theoretical ideas about SI as a general pathological process and the availability of standard approaches to the verification of SI. Nonetheless, it can be concluded that the influence of SI and atherosclerosis on each other is mutual (Figure 5).
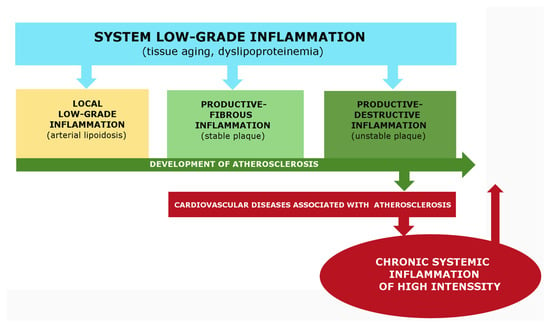
Figure 5.
Formation of a Vicious Pathogenetic Circle Integrating Atherogenesis and its Critical Complications. Note: This figure illustrates the development of a vicious pathogenetic circle integrating atherogenesis and its critical complications associated with acute and chronic systemic hyperinflammation. (1) The development of atherosclerosis is closely related to tissue aging, metabolic allostasis, and systemic and local low-grade inflammation. (2) Complications related to atherosclerosis can lead to systemic alteration, which is essential for the development of systemic hyperinflammation. (3) The development of acute or chronic systemic inflammation leads to more pronounced changes in homeostasis than low-grade inflammation, including in arteries that are problematic in relation to atherosclerosis, which in turn increases the likelihood and severity of critical complications. (4) In sepsis, severe trauma, various shock conditions, as well as in systemic autoimmune diseases, end-stage renal disease, and some other severe chronic diseases, systemic hyperinflammation may occur primarily in relation to progressive atherosclerosis. However, in this case, systemic inflammation will also provoke the accelerated development of atherosclerosis and the subsequent formation of a negative feedback loop.
The relationship between atherosclerosis and acute systemic inflammation (SI) is well established. Complications of atherosclerosis of specific arteries can lead to the development of acute SI, as demonstrated in the clinic and through data on the pathogenesis of cardiogenic shock [284,560,561,562,563]. Our studies have also shown that hemorrhagic stroke in elderly individuals complicated by severe coma and multiple organ failure is associated with the development of SI, including the phenomenon of paracoagulation (D-dimer > 500 ng/mL) [564]. In turn, we recorded the presence of chronic SI criteria, including paracoagulation, in patients with chronic limb-threatening ischemia caused by common femoral artery atherosclerotic lesions [66]. It should be noted that approximately 6% of adults worldwide have problems related to atherosclerotic ischemia of the lower extremities [565]. Furthermore, we detected signs of chronic SI in some patients over 70 years old who had New York Heart Association (NYHA) functional class II-IV heart failure [566]. While the causes of possible SI development, in this case, are polyvalent, ischemic changes in the heart and other organs related to atherosclerosis are undoubtedly contributing factors. Therefore, atherosclerosis is closely associated with chronic systemic and local low-grade inflammation, and critical complications of atherosclerotic arteries of the brain, heart, and lower extremities can provoke the development of acute and chronic variants of systemic hyperinflammation. In turn, acute and chronic SI can contribute to the progression of atherosclerosis in many diseases.
8. Conclusions
In modern medicine, a rational classification of pathologies is necessary not only based on the clinical principle but also on the main characteristics of pathogenesis manifested in various diseases, with their regularities reflecting abstract models of general pathological processes. A fundamental question remains regarding the definition of atherosclerosis as a clinical definition or as a general pathological process involving many universal mechanisms of pathologies while still having an independent meaning. ICD-11 demonstrates that atherosclerosis is a common platform for a large number of clinical definitions rather than a clinical nosology. Pathologies directly related to atherosclerosis occupy several sections in ICD-11, including cerebrovascular diseases, coronary heart disease, and atherosclerotic chronic arterial occlusive disease, all of which have arterial atheroma at the basis of pathogenesis or a typical pathological process leading to its formation.
Initially, atherosclerosis was not considered an inflammatory disease. However, many authors now view atherosclerosis as a low-grade chronic inflammation, which is not entirely accurate, as atheroma formation more closely resembles classical variants of productive inflammation, involving various immune response vectors in the process. Nevertheless, vascular atheroma does not fully align with canonical inflammation due to the low pro-inflammatory activity of vasa vasorum recruited into the damaged intima and the peculiarities of metabolic (cholesterol-dependent) damage. Moreover, despite the possible involvement of autoimmune mechanisms and infectious factors in atherosclerosis pathogenesis, leukocyte participation in productive inflammation is more limited compared to classical autoimmune and infectious diseases.
The primary participants in inflammatory atherogenesis are Mf interacting with cholesterol and modified VSMCs. The multivariate and large-scale transdifferentiation of VSMCs is also a characteristic feature of atherosclerosis. Possibly for these reasons, atherosclerosis is not defined in current literature as vasculitis or arteritis, as other vascular diseases with canonical inflammation are categorized. Instead, atherosclerosis corresponds more closely to noncanonical or quasi-inflammation, which differs from low-grade inflammation in several aspects. These features of atherosclerosis cannot be fully attributed to metabolic damage factors because, in diabetic kidney disease and nonalcoholic fatty liver disease, local low-grade chronic inflammation can transform into classical inflammation variants [59], but this is not entirely characteristic of atherosclerosis. Furthermore, arthritis in gout is characterized by classical inflammation variants, as uric acid functions as both a metabolite and a classical DAMP [539,566,567].
In general, the inflammatory process in atherosclerosis is closely related and shares many common mechanisms with low-grade inflammation (particularly at the initial stages of atherogenesis), as well as with productive inflammation of the classical type at the stage of formed atheroma. Simultaneously, the atherogenic process in unstable plaques can be characterized as productive-destructive inflammation, which is also atypical for destructive-exudative variants of classical inflammation.
Atherosclerosis complications, such as heart attacks and strokes, can lead to the development of acute systemic hyperinflammation. In contrast, complications like critical atherosclerotic ischemia of the lower extremities can result in chronic systemic inflammation. This relationship is bidirectional, as both acute and chronic variants of systemic inflammation can contribute to the progression of atherogenesis.
Therefore, while the atherogenic inflammatory process is related to other types of inflammation, it is not entirely identical to them. It’s important to note that atherosclerosis is a common platform for many significant diseases. Additionally, it is crucial to improve and implement new pathogenetic prevention and therapy methods for atherosclerosis in medical practice [14,568,569,570,571,572]. Advanced methods for studying atherosclerosis in vitro should also be introduced [572]. All of these factors lead us to characterize atherosclerosis as a special variant of the general pathological process.
If models of general pathological processes serve as pathogenetic platforms for many clinical definitions, is there a common platform for general pathological processes themselves? We propose that pro-inflammatory tissue stress is such a common platform, and its elementary functional unit is cellular stress. It’s important to note that the presence of alteration and cellular response to alteration is a condition for survival or programmed cell death not only in pathology but also in physiological conditions [59,60,61]. Moreover, typical cellular stress programs manifest themselves in pathologies not directly related to inflammation, such as tumor growth [59].
This fact highlights the presence of common molecular mechanisms, including epigenetic changes and backbone inducible signaling pathways, in processes that differ in nature and function. However, the context of these mechanisms at the cellular, tissue, and organismal levels gives rise to different functional systems that characterize the models of key general pathological processes (Figure 6).
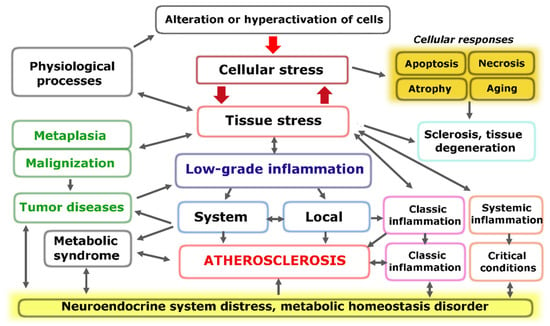
Figure 6.
Interrelation of the main general pathological processes on the basis of common mechanisms of cellular and tissue pro-inflammatory stress.
Section 3 illustrated the link between atherogenesis and all the main universal mechanisms of pro-inflammatory cellular stress. Additionally, atherosclerosis is characterized by typical response mechanisms of the immune system, connective tissue, and vascular endothelium, which are involved to varying degrees in the development of low-grade inflammation and inflammation of the classical type (Section 4 and Section 5). All of this not only differentiates various general pathological processes but also establishes their interrelation. Atherosclerosis may occupy a unique position within this system (Figure 6).
Thus, we believe there are several reasons to view the typical integrative regularities of atherosclerosis pathogenesis as a special variant of the general pathological inflammation process with distinctive features from both classical (canonical) inflammation and local low-grade inflammation.
Author Contributions
Conceptualization, E.G.; writing–original draft preparation, E.G. and A.S.; writing–review and editing, E.G. and A.S.; funding acquisition, E.G. and A.S. All authors have read and agreed to the published version of the manuscript.
Funding
The reported study was funded by the Government contract of the Institute of Immunology and Physiology (122020900136-4).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new experimental data were created.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| AGEs—advanced glycation end products |
| ATF4—activating transcription factor 4 |
| CRP—C-reactive protein |
| CS—cellular stress |
| DAMPs—danger-associated molecular patterns |
| DCs—dendritic cells |
| DDR—DNA damage response |
| DIC—disseminated intravascular coagulation |
| DM2—type 2 diabetes mellitus |
| ECs—endothelial cellsER—endoplasmic reticulum |
| FFA—free fatty acids |
| HDL—high-density lipoprotein |
| HIF-1—hypoxia-inducible factor-1 |
| HSF1—heat shock factor 1 |
| HSP—heat shock proteins |
| iNOS—inducible NO synthase |
| LDL—low-density lipoproteins |
| Lp—atherogenic lipoproteins |
| LPS—bacterial endo-toxin |
| MAPKs—mitogen-activated protein kinases |
| MCs—adventitial mast cells |
| Mf—macrophages |
| MMPs—matrix metalloproteinases |
| mtDNA—mitochondrial DNA |
| NF-κB—nuclear factor kappa-light-chain-enhancer of activated B cells, |
| NK—natural killer |
| NO—nitric oxide |
| NOS—NO synthase |
| NRF2—nuclear factor erythroid 2-related factor 2 |
| oxLDL—oxidized LDL |
| PAMPs—pathogen-associated molecular patterns |
| PRRs—pattern recognition receptors |
| ROS—reactive oxygen species |
| SI—systemic inflammation |
| SRs—scavenger receptors |
| TCRs—T cell receptors |
| TFs—transcription factors |
| Th—T-helper |
| TLRs—toll-like receptors |
| UPR—unfolded protein response reaction |
| VEGF—vascular endothelial growth factors |
| VSMCs—vascular smooth muscle cells |
References
- Barquera, S.; Pedroza-Tobías, A.; Medina, C.; Hernández-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global Overview of the Epidemiology of Atherosclerotic Cardiovascular Disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Sidelnikov, E.; Dornstauder, E.; Jacob, C.; Maas, C.; Pinto, L.; Leidl, R.; Ahrens, I. Healthcare resource utilization and costs of cardiovascular events in patients with atherosclerotic cardiovascular disease in Germany—Results of a claims database study. J. Med. Econ. 2022, 25, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kang, S.J.; Lee, C.W.; Han, S.; Park, D.W.; Lee, S.W.; Kim, Y.H.; Park, S.W.; Park, S.J.; Kim, J.J. Prevalence of coronary atherosclerosis in asymptomatic healthy subjects: An intravascular ultrasound study of donor hearts. J. Atheroscler. Thromb. 2013, 20, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Webber, B.J.; Seguin, P.G.; Burnett, D.G.; Clark, L.L.; Otto, J.L. Prevalence of and risk factors for autopsy-determined atherosclerosis among US service members, 2001–2011. JAMA 2012, 308, 2577–2583. [Google Scholar] [CrossRef]
- Santos, R.D.; Gidding, S.S.; Hegele, R.A.; Cuchel, M.A.; Barter, P.J.; Watts, G.F.; Baum, S.J.; Catapano, A.L.; Chapman, M.J.; Defesche, J.C.; et al. International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Defining severe familial hypercholesterolaemia and the implications for clinical management: A consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016, 4, 850–861. [Google Scholar] [CrossRef]
- Coakley, J.C. Lipids in Children and Links to Adult Vascular Disease. Clin. Biochem. Rev. 2018, 39, 65–76. [Google Scholar]
- Raitakari, O.; Pahkala, K.; Magnussen, C.G. Prevention of atherosclerosis from childhood. Nat. Rev. Cardiol. 2022, 19, 543–554. [Google Scholar] [CrossRef]
- Stone, N.J.; Smith, S.C., Jr.; Orringer, C.E.; Rigotti, N.A.; Navar, A.M.; Khan, S.S.; Jones, D.W.; Goldberg, R.; Mora, S.; Blaha, M.; et al. Managing Atherosclerotic Cardiovascular Risk in Young Adults: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 819–836. [Google Scholar] [CrossRef]
- Liberale, L.; Montecucco, F.; Tardif, J.C.; Libby, P.; Camici, G.G. Inflamm-ageing: The role of inflammation in age-dependent cardiovascular disease. J. Eur. Heart 2020, 41, 2974–2982. [Google Scholar] [CrossRef]
- Yegorov, Y.E.; Poznyak, A.V.; Nikiforov, N.G.; Starodubova, A.V.; Orekhov, A.N. Role of Telomeres Shortening in Atherogenesis: An Overview. Cells 2021, 10, 395. [Google Scholar] [CrossRef]
- Xiang, Q.; Tian, F.; Xu, J.; Du, X.; Zhang, S.; Liu, L. New insight into dyslipidemia-induced cellular senescence in atherosclerosis. Biol. Rev. Camb. Philos. Soc. 2022, 97, 1844–1867. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Roy, P.; Orecchioni, M.; Ley, K. How the immune system shapes atherosclerosis: Roles of innate and adaptive immunity. Nat. Rev. Immunol. 2022, 22, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Engelen, S.E.; Robinson, A.J.B.; Zurke, Y.X.; Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: How to proceed? Nat. Rev. Cardiol. 2022, 19, 522–542. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal. Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Yeh, W.C.; Tsai, Y.W.; Chen, J.Y. The Relationship between Atherogenic Index of Plasma and Obesity among Adults in Taiwan. Int. J. Environ. Res. Public Health 2022, 19, 14864. [Google Scholar] [CrossRef]
- López-Acosta, O.; Ruiz-Ramírez, A.; Barrios-Maya, M.Á.; Alarcon-Aguilar, J.; Alarcon-Enos, J.; Céspedes Acuña, C.L.; El-Hafidi, M. Lipotoxicity, glucotoxicity and some strategies to protect vascular smooth muscle cell against proliferative phenotype in metabolic syndrome. Food Chem. Toxicol. 2022, 172, 113546. [Google Scholar] [CrossRef]
- Zhao, Y.; Malik, S.; Criqui, M.H.; Allison, M.A.; Budoff, M.J.; Sandfort, V.; Wong, N.D. Coronary calcium density in relation to coronary heart disease and cardiovascular disease in adults with diabetes or metabolic syndrome: The Multi-ethnic Study of Atherosclerosis (MESA). BMC Cardiovasc. Disord. 2022, 22, 536. [Google Scholar] [CrossRef] [PubMed]
- Silveira Rossi, J.L.; Barbalho, S.M.; Reverete de Araujo, R.; Bechara, M.D.; Sloan, K.P.; Sloan, L.A. Metabolic syndrome and cardiovascular diseases: Going beyond traditional risk factors. Diabetes Metab. Res. Rev. 2022, 38, e3502. [Google Scholar] [CrossRef] [PubMed]
- Sharif, S.; Van der Graaf, Y.; Cramer, M.J.; Kapelle, L.J.; de Borst, G.J.; Visseren, F.L.J.; Westerink, J.; SMART study group. Low-grade inflammation as a risk factor for cardiovascular events and all-cause mortality in patients with type 2 diabetes. Cardiovasc. Diabetol. 2021, 20, 220. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front. Immunol. 2021, 12, 746151. [Google Scholar] [CrossRef]
- Wang, G.; Shen, G.; Jiang, X.; Chen, Z.; Yin, T. Assessment of para-inflammation in a wound healing model. Exp. Ther. Med. 2020, 20, 655–661. [Google Scholar] [CrossRef]
- Guarner, V.; Rubio-Ruiz, M.E. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 2015, 40, 99–106. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, T.; Larbi, A.; Witkowski, J.M. Human Inflammaging. Gerontology 2019, 65, 495–504. [Google Scholar] [CrossRef]
- Van Capelleveen, J.C.; Bochem, A.E.; Motazacker, M.M.; Hovingh, G.K.; Kastelein, J.J. Genetics of HDL-C: A causal link to atherosclerosis? Curr. Atheroscler. Rep. 2013, 15, 326. [Google Scholar] [CrossRef]
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis Is an Epigenetic Disease. Trends Endocrinol. Metab. 2018, 29, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zou, Y.F.; Xiao, Y.; Wang, T.X.; Wang, Y.N.; Dong, Z.C.; Huo, Y.H.; Yao, B.C.; Meng, L.B.; Du, S.X. Identification of Potential Hub Genes of Atherosclerosis Through Bioinformatic Analysis. J. Comput. Biol. 2021, 28, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Chyu, K.Y.; Dimayuga, P.C.; Shah, P.K. Immunogenetics of Atherosclerosis-Link between Lipids, Immunity, and Genes. Curr. Atheroscler. Rep. 2020, 22, 53. [Google Scholar] [CrossRef]
- Tada, H.; Takamura, M.; Kawashiri, M.A. What is the mechanism of genetic contributions to the development of atherosclerosis? Atherosclerosis 2020, 307, 72–74. [Google Scholar] [CrossRef]
- Nie, H.; Yan, C.; Zhou, W.; Li, T.S. Analysis of Immune and Inflammation Characteristics of Atherosclerosis from Different Sample Sources. Oxid. Med. Cell. Longev. 2022, 2022, 5491038. [Google Scholar] [CrossRef]
- Tokgozoglu, L.; Kayikcioglu, M. Familial Hypercholesterolemia: Global Burden and Approaches. Curr. Cardiol. Rep. 2021, 23, 151. [Google Scholar] [CrossRef] [PubMed]
- Di Taranto, M.D.; Giacobbe, C.; Fortunato, G. Familial hypercholesterolemia: A complex genetic disease with variable phenotypes. Eur. J. Med. Genet. 2020, 63, 103831. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; DiMario, S.; Philip, K. Gender Disparities in Health Resource Utilization in Patients with Atherosclerotic Cardiovascular Disease: A Retrospective Cross-Sectional Study. Adv. Ther. 2019, 36, 3424–3434. [Google Scholar] [CrossRef] [PubMed]
- Vakhtangadze, T.; Singh Tak, R.; Singh, U.; Baig, M.S.; Bezsonov, E. Gender Differences in Atherosclerotic Vascular Disease: From Lipids to Clinical Outcomes. Front. Cardiovasc. Med. 2021, 8, 707889. [Google Scholar] [CrossRef]
- Padro, T.; Muñoz-Garcia, N.; Peña, E.; Badimon, L. Sex Differences and Emerging New Risk Factors for Atherosclerosis and Its Thrombotic Complications. Curr. Pharm. Des. 2021, 27, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- German, C.A.; Baum, S.J.; Ferdinand, K.C.; Gulati, M.; Polonsky, T.S.; Toth, P.P.; Shapiro, M.D. Defining preventive cardiology: A clinical practice statement from the American Society for Preventive Cardiology. Am. J. Prev. Cardiol. 2022, 12, 100432. [Google Scholar] [CrossRef]
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Kränkel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle factors and high-risk atherosclerosis: Pathways and mechanisms beyond traditional risk factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406. [Google Scholar] [CrossRef]
- Yao, B.C.; Meng, L.B.; Hao, M.L.; Zhang, Y.M.; Gong, T.; Guo, Z.G. Chronic stress: A critical risk factor for atherosclerosis. J. Int. Med. Res. 2019, 47, 1429–1440. [Google Scholar] [CrossRef]
- Riccardi, G.; Giosuè, A.; Calabrese, I.; Vaccaro, O. Dietary recommendations for prevention of atherosclersis. Cardiovasc. Res. 2022, 118, 1188–1204. [Google Scholar] [CrossRef]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Qian, B.; Zhang, K.; Li, Y.; Sun, K. Update on gut microbiota in cardiovascular diseases. Front. Cell. Infect. Microbiol. 2022, 12, 1059349. [Google Scholar] [CrossRef]
- Kurilenko, N.; Fatkhullina, A.R.; Mazitova, A.; Koltsova, E.K. Act Locally, Act Globally-Microbiota, Barriers, and Cytokines in Atherosclerosis. Cells 2021, 10, 348. [Google Scholar] [CrossRef]
- Hidi, L.; Kovács, G.I.; Szabó, D.; Makra, N.; Pénzes, K.; Juhász, J.; Sótonyi, P.; Ostorházi, E. Human blood vessel microbiota in healthy adults based on common femoral arteries of brain-dead multi-organ donors. Front. Cell. Infect. Microbiol. 2022, 12, 1056319. [Google Scholar] [CrossRef]
- Li, C.; Yu, R.; Ding, Y. Association between Porphyromonas Gingivalis and systemic diseases: Focus on T cells-mediated adaptive immunity. Front. Cell. Infect. Microbiol. 2022, 12, 1026457. [Google Scholar] [CrossRef]
- Zou, Y.; Huang, Y.; Liu, S.; Yang, J.; Zheng, W.; Deng, Y.; Zhang, M.; Yan, Z.; Xie, H. Periodontopathic Microbiota and Atherosclerosis: Roles of TLR-Mediated Inflammation Response. Oxid. Med. Cell. Longev. 2022, 2022, 9611362. [Google Scholar] [CrossRef]
- Jain, K. The effect of varying degrees of stenosis on transition to turbulence in oscillatory flows. Biomech. Model. Mechanobiol. 2022, 21, 1029–1041. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Hypertension as a risk factor for atherosclerosis: Cardiovascular risk assessment. Front. Cardiovasc. Med. 2022, 9, 959285. [Google Scholar] [CrossRef]
- Mazurek, R.; Dave, J.M.; Chandran, R.R.; Misra, A.; Sheikh, A.Q.; Greif, D.M. Vascular Cells in Blood Vessel Wall Development and Disease. Adv. Pharmacol. 2017, 78, 323–350. [Google Scholar] [CrossRef]
- Majesky, M.W. Vascular Development. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e17–e24. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Tabas, I.; Bornfeldt, K.E. Intracellular and Intercellular Aspects of Macrophage Immunometabolism in Atherosclerosis. Circ. Res. 2020, 126, 1209–1227. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef]
- Upthegrove, R.; Khandaker, G.M. Cytokines, Oxidative Stress and Cellular Markers of Inflammation in Schizophrenia. Curr. Top. Behav. Neurosci. 2020, 44, 49–66. [Google Scholar] [CrossRef]
- Gusev, E.; Zhuravleva, Y. Inflammation: A New Look at an Old Problem. Int. J. Mol. Sci. 2022, 23, 4596. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Zotova, N.V. Cellular stress and general pathological processes. Curr. Pharm. Des. 2019, 25, 251–297. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Zhuravleva, Y.A.; Zotova, N.V. Correlation of the Evolution of Immunity and Inflammation in Vertebrates. Biol. Bull. Rev. 2019, 9, 358–372. [Google Scholar] [CrossRef]
- Celsus, A.C. De Medicina; Harvard College: Cambridge, MA, USA, 1904; Self published A.D. 25. [Google Scholar]
- Rather, L.J. Disturbance of function (functio laesa): The legendary fifth cardinal sign of inflammation, added by Galen to the four cardinal signs of Celsus. Bull. N. Y. Acad. Med. 1971, 47, 303–322. [Google Scholar]
- Tylutka, A.; Morawin, B.; Walas, Ł.; Michałek, M.; Gwara, A.; Zembron-Lacny, A. Assessment of metabolic syndrome predictors in relation to inflammation and visceral fat tissue in older adults. Sci. Rep. 2023, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Antuña, E.; Cachán-Vega, C.; Bermejo-Millo, J.C.; Potes, Y.; Caballero, B.; Vega-Naredo, I.; Coto-Montes, A.; Garcia-Gonzalez, C. Inflammaging: Implications in Sarcopenia. Int. J. Mol. Sci. 2022, 23, 15039. [Google Scholar] [CrossRef] [PubMed]
- Zotova, N.; Zhuravleva, Y.; Chereshnev, V.; Gusev, E. Acute and Chronic Systemic Inflammation: Features and Differences in the Pathogenesis, and Integral Criteria for Verification and Differentiation. Int. J. Mol. Sci. 2023, 24, 1144. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef]
- Morshed, S.A.; Davies, T.F. Understanding thyroid cell stress. J. Clin. Endocrinol. Metab. 2020, 105, e66–e69. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Angeles-Valencia, M.; Morales-González, Á.; Madrigal-Santillán, E.O.; Morales-Martínez, M.; Madrigal-Bujaidar, E.; Álvarez-González, I.; Gutiérrez-Salinas, J.; Esquivel-Chirino, C.; Chamorro-Cevallos, G.; et al. Oxidative stress, mitochondrial function and adaptation to exercise: New perspectives in nutrition. Life 2021, 11, 1269. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, X.; Xu, B.; Bao, X.; Jia, H.; Yu, B. Oxidative Stress-Mediated Programmed Cell Death: A Potential Therapy Target for Atherosclerosis. Cardiovasc. Drugs Ther. 2022. [Google Scholar] [CrossRef]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, C.K.; Yi, M.; Lui, K.O.; Huang, Y. Targeting endothelial dysfunction and inflammation. J. Mol. Cell. Cardiol. 2022, 168, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sławińska, N.; Krupa, R. Molecular aspects of senescence and organismal ageing-DNA damage response, telomeres, inflammation and chromatin. Int. J. Mol. Sci. 2021, 22, 590. [Google Scholar] [CrossRef]
- Hanawalt, P.; Sweasy, J. Mechanistic understanding of cellular responses to genomic stress. Environ. Mol. Mutagen. 2020, 61, 25–33. [Google Scholar] [CrossRef]
- Yoshida, K.; Fujita, M. DNA damage responses that enhance resilience to replication stress. Cell. Mol. Life Sci. 2021, 78, 6763–6773. [Google Scholar] [CrossRef]
- Xiong, H.; Hua, F.; Dong, Y.; Lin, Y.; Ying, J.; Liu, J.; Wang, X.; Zhang, L.; Zhang, J. DNA damage response and GATA4 signaling in cellular senescence and aging-related pathology. Front. Aging Neurosci. 2022, 14, 933015. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Finigan, A.; Figg, N.L.; Uryga, A.K.; Bennett, M.R. SIRT6 Protects Smooth Muscle Cells from Senescence and Reduces Atherosclerosis. Circ. Res. 2021, 128, 474–491. [Google Scholar] [CrossRef]
- Boström, K.I. DNA Damage Response, Runx2 (Runt-Related Transcription Factor 2), and Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1358–1359. [Google Scholar] [CrossRef]
- Duer, M.; Cobb, A.M.; Shanahan, C.M. DNA Damage Response: A Molecular Lynchpin in the Pathobiology of Arteriosclerotic Calcification. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e193–e202. [Google Scholar] [CrossRef] [PubMed]
- Priesnitz, C.; Becker, T. Pathways to balance mitochondrial translation and protein import. Genes Dev. 2018, 32, 1285–1296. [Google Scholar] [CrossRef]
- Eckl, E.-M.; Ziegemann, O.; Krumwiede, L.; Fessler, E.; Jae, L.T. Sensing, signaling and surviving mitochondrial stress. Cell. Mol. Life Sci. 2021, 78, 5925–5951. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.H.; Ramji, D.P. Investigation of Mitochondrial Bioenergetic Profile and Dysfunction in Atherosclerosis. Methods Mol. Biol. 2022, 2419, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zeng, Z.L.; Yang, S.; Li, A.; Zu, X.; Liu, J. Mitochondrial Stress in Metabolic Inflammation: Modest Benefits and Full Losses. Oxid. Med. Cell. Longev. 2022, 2022, 8803404. [Google Scholar] [CrossRef]
- Dorighello, G.G.; Rovani, J.C.; Paim, B.A.; Rentz, T.; Assis, L.H.P.; Vercesi, A.E.; Oliveira, H.C.F. Mild Mitochondrial Uncoupling Decreases Experimental Atherosclerosis, A Proof of Concept. J. Atheroscler. Thromb. 2022, 2, 825–838. [Google Scholar] [CrossRef]
- Coon, B.G.; Timalsina, S.; Astone, M.; Zhuang, Z.W.; Fang, J.; Han, J.; Themen, J.; Chung, M.; Yang-Klingler, Y.J.; Jain, M.; et al. A mitochondrial contribution to anti-inflammatory shear stress signaling in vascular endothelial cells. J. Cell Biol. 2022, 221, e202109144. [Google Scholar] [CrossRef]
- Zeng, Z.L.; Yuan, Q.; Zu, X.; Liu, J. Insights into the Role of Mitochondria in Vascular Calcification. Front. Cardiovasc. Med. 2022, 9, 879752. [Google Scholar] [CrossRef]
- Shemiakova, T.; Ivanova, E.; Wu, W.K.; Kirichenko, T.V.; Starodubova, A.V.; Orekhov, A.N. Atherosclerosis as Mitochondriopathy: Repositioning the Disease to Help Finding New Therapies. Front. Cardiovasc. Med. 2021, 8, 660473. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Leli, N.M.; Koumenis, C.; Amaravadi, R.K. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin. Cancer Biol. 2020, 66, 116–128. [Google Scholar] [CrossRef]
- Schröder, M. Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Yap, K.N.; Yamada, K.; Zikeli, S.; Kiaris, H.; Hood, W.R. Evaluating endoplasmic reticulum stress and unfolded protein response through the lens of ecology and evolution. Biol. Rev. Camb. Philos. Soc. 2021, 96, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, M.G.; Higuchi-Sanabria, R.; Garcia, G.; Tsui, C.K.; Dillin, A. Beyond the cell factory: Homeostatic regulation of and by the UPRER. Sci. Adv. 2020, 6, eabb9614. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, C.D.; Wu, R.F.; Terada, L.S. ROS signaling and ER stress in cardiovascular disease. Mol. Aspects Med. 2018, 63, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wu, M.; Li, X.; Zhao, R.; Zhao, Y.; Liu, L.; Wang, S. Role of Endoplasmic Reticulum Stress in Atherosclerosis and Its Potential as a Therapeutic Target. Oxid. Med. Cell. Longev. 2020, 2020, 9270107. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Orekhov, A.N. The Role of Endoplasmic Reticulum Stress and Unfolded Protein Response in Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 193. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Fernandes, C.; Liu, Y.; Wu, Y.; Wu, H.; Brophy, M.L.; Deng, L.; Song, K.; Wen, A.; Wong, S.; et al. Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diabetes Vasc. Dis. Res. 2017, 14, 14–23. [Google Scholar] [CrossRef]
- Wang, X.; Luo, D.; Wu, S. Molecular Dysfunctions of Mitochondria-Associated Endoplasmic Reticulum Contacts in Atherosclerosis. Oxid. Med. Cell. Longev. 2021, 2021, 2424509. [Google Scholar] [CrossRef]
- Morimoto, R.I. The heat shock response: Systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 91–99. [Google Scholar] [CrossRef]
- Lang, B.J.; Guerrero, M.E.; Prince, T.L.; Okusha, Y.; Bonorino, C.; Calderwood, S.K. The functions and regulation of heat shock proteins; key orchestrators of proteostasis and the heat shock response. Arch. Toxicol. 2021, 95, 1943–1970. [Google Scholar] [CrossRef]
- Cinoku, I.I.; Mavragani, C.P.; Moutsopoulos, H.M. Atherosclerosis: Beyond the lipid storage hypothesis. The role of autoimmunity. Eur. J. Clin. Investig. 2020, 50, e13195. [Google Scholar] [CrossRef]
- Bruxel, M.A.; Tavares, A.M.V.; Zavarize, N.L.D.; de Souza Borges, V.; Schroeder, H.T.; Bock, P.M.; Rodrigues, M.I.L.; Belló-Klein, A.; Homem de Bittencourt, P.I., Jr. Chronic whole-body heat treatment relieves atherosclerotic lesions, cardiovascular and metabolic abnormalities, and enhances survival time restoring the anti-inflammatory and anti-senescent heat shock response in mice. Biochimie 2019, 156, 33–46. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Liu, X.; Yamashita, T.; Shang, J.; Shi, X.; Morihara, R.; Huang, Y.; Sato, K.; Takemoto, M.; Hishikawa, N.; Ohta, Y.; et al. Molecular switching from ubiquitin–proteasome to autophagy pathways in mice stroke model. J. Cereb. Blood Flow Metab. 2020, 40, 214–224. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy—Unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; de Bruijn, J.; van Kuijk, K.; Riascos-Bernal, D.F.; Diaz, A.; Tasset, I.; Martín-Segura, A.; Gijbels, M.J.J.; Sander, B.; Kaushik, S.; et al. Protective role of chaperone-mediated autophagy against atherosclerosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2121133119. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, T.; Wang, X.; Zhang, J.; Ju, J.; Xu, H. Global research trends in atherosclerosis: A bibliometric and visualized study. Front. Cardiovasc. Med. 2022, 9, 956482. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Wu, W.K.; Kirichenko, T.V.; Orekhov, A.N. Autophagy and Mitophagy as Essential Components of Atherosclerosis. Cells 2021, 10, 443. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, M.X.; Zhang, L.; Zhang, D.; Li, C.; Li, Y.L. Autophagy, Pyroptosis, and Ferroptosis: New Regulatory Mechanisms for Atherosclerosis. Front. Cell Dev. Biol. 2022, 9, 809955. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Holley, C.L.; Schroder, K. The rOX-stars of inflammation: Links between the inflammasome and mitochondrial meltdown. Clin. Transl. Immunol. 2020, 9, e01109. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Savage, C.D.; Lopez-Castejon, G.; Denes, A.; Brough, D. NLRP3-inflammasome activating DAMPs stimulate an inflammatory response in glia in the absence of priming which contributes to brain inflammation after injury. Front. Immunol. 2012, 3, 288. [Google Scholar] [CrossRef]
- Takahashi, M. NLRP3 inflammasome as a key driver of vascular disease. Cardiovasc. Res. 2022, 118, 372–385. [Google Scholar] [CrossRef]
- Doran, A.C. Inflammation Resolution: Implications for Atherosclerosis. Circ. Res. 2022, 130, 130–148. [Google Scholar] [CrossRef]
- De Sousa, M.C.; Gjorgjieva, M.; Dolicka, D.; Sobolewski, C.; Foti, M. Deciphering miRNAs’ action through miRNA editing. Int. J. Mol. Sci. 2019, 20, 6249. [Google Scholar] [CrossRef]
- Fasolo, F.; Di Gregoli, K.; Maegdefessel, L.; Johnson, J.L. Non-coding, RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc. Res. 2019, 115, 1732–1756. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting epigenetics and non-coding RNAs in atherosclerosis: From mechanisms to therapeutics. Pharmacol. Ther. 2019, 196, 15–43. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Maleki, S.S.; Nazari-Jahantigh, M. Regulatory Non-coding RNAs in Atherosclerosis. Handb. Exp. Pharmacol. 2022, 270, 463–492. [Google Scholar] [CrossRef]
- Paloschi, V.; Winter, H.; Viola, J.; Soehnlein, O.; Maegdefessel, L. Mechanistic Links between Non-Coding RNAs and Myeloid Cell Inflammation in Atherosclerosis. Thromb. Haemost. 2019, 119, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.R.; Jovicic, N.; Djonov, V.; Arsenijevic, N.; Volarevic, V. Mesenchymal stem cell-derived exosomes and other extracellular vesicles as new remedies in the therapy of inflammatory diseases. Cells 2019, 8, 1605. [Google Scholar] [CrossRef]
- Vartak, T.; Kumaresan, S.; Brennan, E. Decoding microRNA drivers in atherosclerosis. Biosci. Rep. 2022, 42, BSR20212355. [Google Scholar] [CrossRef]
- Backlund, M.; Stein, F.; Rettel, M.; Schwarzl, T.; Perez-Perri, J.I.; Brosig, A.; Zhou, Y.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E. Plasticity of nuclear and cytoplasmic stress responses of RNA-binding proteins. Nucleic Acids Res. 2020, 48, 4725–4740. [Google Scholar] [CrossRef]
- Sidibé, H.; Vande Velde, C. Collective learnings of studies of stress granule assembly and composition. Methods Mol. Biol. 2022, 2428, 199–228. [Google Scholar] [CrossRef]
- Wang, J.; Gan, Y.; Cao, J.; Dong, X.; Ouyang, W. Pathophysiology of stress granules: An emerging link to diseases (Review). Int. J. Mol. Med. 2022, 49, 44. [Google Scholar] [CrossRef]
- Lau, Y.; Oamen, H.P.; Caudron, F. Protein phase separation during stress adaptation and cellular memory. Cells 2020, 9, 1302. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Fuxreiter, M. Protein condensation diseases: Therapeutic opportunities. Nat. Commun. 2022, 13, 5550. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.B.; Silva Afonso, M.; Kelemen, S.E.; Ray, M.; Vrakas, C.N.; Burke, A.C.; Scalia, R.G.; Moore, K.; Autieri, M.V. Regulation of Stress Granule Formation by Inflammation, Vascular Injury, and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2014–2027. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, L.; Zou, X.; Nie, Q.; Komarova, N.L. Mathematical models of specificity in cell signaling. Biophys. J. 2007, 92, 3425–3441. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Gundamaraju, R.; Vemuri, R.; Chong, W.C.; Geraghty, D.P.; Eri, R. Cell Stress Signaling Cascades Regulating Cell Fate. Curr. Pharm. Des. 2018, 24, 3176–3183. [Google Scholar] [CrossRef]
- Sadria, M.; Layton, A.T. Interactions among mTORC, AMPK and SIRT: A computational model for cell energy balance and metabolism. Cell Commun. Signal. 2021, 19, 57. [Google Scholar] [CrossRef]
- Dasgupta, I.; McCollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Shaw, P.; Chattopadhyay, A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83. [Google Scholar] [CrossRef]
- Ovens, A.J.; Scott, J.W.; Langendorf, C.G.; Kemp, B.E.; Oakhill, J.S.; Smiles, W.J. Post-Translational Modifications of the Energy Guardian AMP-Activated Protein Kinase. Int. J. Mol. Sci. 2021, 22, 1229. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Rein, T. Post-translational modifications and stress adaptation: The paradigm of FKBP51. Biochem. Soc. Trans. 2020, 48, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Pan, Z.; Ma, G.; Kong, L.; Du, G. Hypoxia-inducible factor-1: Regulatory mechanisms and drug development in stroke. Pharmacol. Res. 2021, 170, 105742. [Google Scholar] [CrossRef]
- Zhang, B.; Fan, Y.; Cao, P.; Tan, K. Multifaceted roles of HSF1 in cell death: A state-of-the-art review. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188591. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. Int. J. Mol. Sci. 2022, 23, 931. [Google Scholar] [CrossRef]
- Niu, N.; Xu, S.; Xu, Y.; Little, P.J.; Jin, Z.G. Targeting Mechanosensitive Transcription Factors in Atherosclerosis. Trends Pharmacol. Sci. 2019, 40, 253–266. [Google Scholar] [CrossRef]
- Wang, B.; Yang, X.; Sun, X.; Liu, J.; Fu, Y.; Liu, B.; Qiu, J.; Lian, J.; Zhou, J. ATF3 in atherosclerosis: A controversial transcription factor. J. Mol. Med. 2022, 100, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.; Charles, N.; Lehmann, U.; Hall, J.L. Transcription factor and kinase-mediated signaling in atherosclerosis and vascular injury. Curr. Atheroscler. Rep. 2006, 8, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, X.; Wu, H. Transcriptional Programming in Arteriosclerotic Disease: A Multifaceted Function of the Runx2 (Runt-Related Transcription Factor 2). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Ye, T.; Yang, L.; Shen, Y.; Li, H. Ferroptosis Signaling and Regulators in Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 809457. [Google Scholar] [CrossRef]
- Duval, C.; Chinetti, G.; Trottein, F.; Fruchart, J.C.; Staels, B. The role of PPARs in atherosclerosis. Trends Mol. Med. 2002, 8, 422–430. [Google Scholar] [CrossRef]
- Yang, L.; Chu, Y.; Wang, L.; Wang, Y.; Zhao, X.; He, W.; Zhang, P.; Yang, X.; Liu, X.; Tian, L.; et al. Overexpression of CRY1 protects against the development of atherosclerosis via the TLR/NF-κB pathway. Int. Immunopharmacol. 2015, 28, 525–530. [Google Scholar] [CrossRef]
- Gutiérrez, S.H.; Kuri, M.R.; del Castillo, E.R. Cardiac role of the transcription factor NF-kappaB. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 153–160. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Yu, S.; Gu, Y.; Wang, T.; Mu, L.; Wang, H.; Yan, S.; Wang, A.; Wang, J.; Liu, L.; Shen, H.; et al. Study of Neuronal Apoptosis ceRNA Network in Hippocampal Sclerosis of Human Temporal Lobe Epilepsy by RNA-Seq. Front. Neurosci. 2021, 15, 770627. [Google Scholar] [CrossRef]
- Arya, R.; White, K. Cell Death in Development: Signaling Pathways and Core Mechanisms. Semin. Cell Dev. Biol. 2015, 39, 12–19. [Google Scholar] [CrossRef]
- Rosa, N.; Ivanova, H.; Wagner, L.E., 2nd; Kale, J.; La Rovere, R.; Welkenhuyzen, K.; Louros, N.; Karamanou, S.; Shabardina, V.; Lemmens, I.; et al. Bcl-xL Acts as an Inhibitor of IP3R Channels, Thereby Antagonizing Ca2+-Driven Apoptosis. Cell Death Differ. 2021, 29, 788–805. [Google Scholar] [CrossRef] [PubMed]
- Bahatyrevich-Kharitonik, B.; Medina-Guzman, R.; Flores-Cortes, A.; García-Cruzado, M.; Kavanagh, E.; Burguillos, M.A. Cell Death Related Proteins Beyond Apoptosis in the CNS. Front. Cell Dev. Biol. 2022, 9, 825747. [Google Scholar] [CrossRef] [PubMed]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. 2019, 76, 1093–1106. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.W.; Fang, L.J.; Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022, 13, 467. [Google Scholar] [CrossRef]
- Rosner, D.; Stoneman, V.; Littlewood, T.; McCarthy, N.; Figg, N.; Wang, Y.; Tellides, G.; Bennett, M. Interferon-gamma induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-dependent mechanism. Am. J. Pathol. 2006, 168, 2054–2063. [Google Scholar] [CrossRef]
- Yurdagul, A., Jr. Metabolic Consequences of Efferocytosis and its Impact on Atherosclerosis. Immunometabolism 2021, 3, e210017. [Google Scholar] [CrossRef]
- Wang, Z.; Su, J.; Gong, F.; Xue, L.; Su, Z. The Impaired Mechanism and Facilitated Therapies of Efferocytosis in Atherosclerosis. J. Cardiovasc. Pharmacol. 2022, 80, 407–416. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, H.; Qu, P.; Qiao, X.; Guo, L.; Liu, L. Novel insight on the role of Macrophages in atherosclerosis: Focus on polarization, apoptosis and efferocytosis. Int. Immunopharmacol. 2022, 113 Pt A, 109260. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Zotova, N.V.; Zhuravleva, Y.A.; Chereshnev, V.A. Physiological and Pathogenic Role of Scavenger Receptors in Humans. Med. Immunol. 2020, 22, 7–48. [Google Scholar] [CrossRef]
- Dou, M.; Chen, Y.; Hu, J.; Ma, D.; Xing, Y. Recent Advancements in CD47 Signal Transduction Pathways Involved in Vascular Diseases. BioMed Res. Int. 2020, 2020, 4749135. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Olivares, L.; Soehnlein, O. Contemporary Lifestyle and Neutrophil Extracellular Traps: An Emerging Link in Atherosclerosis Disease. Cells 2021, 10, 1985. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. A common signature of cellular senescence; does it exist? Ageing Res. Rev. 2021, 71, 101458. [Google Scholar] [CrossRef] [PubMed]
- Uyar, B.; Palmer, D.; Kowald, A.; Murua Escobar, H.; Barrantes, I.; Möller, S.; Akalin, A.; Fuellen, G. Single-cell analyses of aging, inflammation and senescence. Ageing Res. Rev. 2020, 64, 101156. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Kirkland, J.L.; Schwartz, S. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging 2017, 9, 2411–2435. [Google Scholar] [CrossRef]
- Cui, X.Y.; Zhan, J.K.; Liu, Y.S. Roles and functions of antisense lncRNA in vascular aging. Ageing Res. Rev. 2021, 72, 101480. [Google Scholar] [CrossRef]
- Donato, A.J.; Morgan, R.G.; Walke, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89 Pt B, 122–135. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vascul. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Peng, Q.; Liu, H.; Luo, Z.; Zhao, H.; Wang, X.; Guan, X. Effect of autophagy on ferroptosis in foam cells via Nrf2. Mol. Cell. Biochem. 2022, 477, 1597–1606. [Google Scholar] [CrossRef]
- Gonzalez-Guerra, A.; Roche-Molina, M.; García-Quintáns, N.; Sánchez-Ramos, C.; Martín-Pérez, D.; Lytvyn, M.; de Nicolás-Hernández, J.; Rivera-Torres, J.; Arroyo, D.F.; Sanz-Rosa, D.; et al. Sustained Elevated Blood Pressure Accelerates Atherosclerosis Development in a Preclinical Model of Disease. Int. J. Mol. Sci. 2021, 22, 8448. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Jan, K.M.; Rumschitzki, D. Pre-atherosclerotic flow and oncotically active solute transport across the arterial endothelium. J. Theor. Biol. 2020, 499, 110275. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Kovanen, P.T.; Rezaee, M.; Banach, M.; Sahebkar, A. Ca2+ Flux: Searching for a Role in Efferocytosis of Apoptotic Cells in Atherosclerosis. J. Clin. Med. 2019, 8, 2047. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Nikolopoulou, E.A.; Xu, Q.; Qu, A. Hypoxia inducible factor as a therapeutic target for atherosclerosis. Pharmacol. Ther. 2018, 183, 22–33. [Google Scholar] [CrossRef]
- Farrell, G.C.; Haczeyni, F.; Chitturi, S. Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. Adv. Exp. Med. Biol. 2018, 1061, 19–44. [Google Scholar] [CrossRef]
- Ježek, P.; Jaburek, M.; Holendová, B.; Plecitá-Hlavatá, L. Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules 2018, 23, 1483. [Google Scholar] [CrossRef]
- Langlois, A.; Forterre, A.; Pinget, M.; Bouzakri, K. Impact of moderate exercise on fatty acid oxidation in pancreatic β-cells and skeletal muscle. J. Endocrinol. Investig. 2021, 44, 1815–1825. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal. Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Wicherska-Pawłowska, K.; Wróbel, T.; Rybka, J. Toll-Like Receptors (TLRs), NOD-Like Receptors (NLRs), and RIG-I-Like Receptors (RLRs) in Innate Immunity. TLRs, NLRs, and RLRs Ligands as Immunotherapeutic Agents for Hematopoietic Diseases. Int. J. Mol. Sci. 2021, 22, 13397. [Google Scholar] [CrossRef]
- Falck-Hansen, M.; Kassiteridi, C.; Monaco, C. Toll-like receptors in atherosclerosis. Int. J. Mol. Sci. 2013, 14, 14008–14023. [Google Scholar] [CrossRef] [PubMed]
- Jaén, R.; Val-Blasco, A.; Prieto, P.; Gil-Fernández, M.; Smani, T.; López-Sendón, J.L.; Delgado, C.; Boscá, L.; Fernández-Velasco, M. Innate Immune Receptors, Key Actors in Cardiovascular Diseases. JACC Basic Transl. Sci. 2020, 5, 735–749. [Google Scholar] [CrossRef]
- Bezhaeva, T.; Karper, J.; Quax, P.H.A.; de Vries, M.R. The Intriguing Role of TLR Accessory Molecules in Cardiovascular Health and Disease. Front. Cardiovasc. Med. 2022, 9, 820962. [Google Scholar] [CrossRef]
- Curtiss, L.K.; Tobias, P.S. Emerging role of Toll-like receptors in atherosclerosis. J. Lipid Res. 2009, 50 (Suppl.), S340–S345. [Google Scholar] [CrossRef]
- Heinrichsdorff, J.; Olefsky, J.M. Fetuin-A: The missing link in lipid-induced inflammation. Nat. Med. 2012, 18, 1182–1183. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Laugerette, F.; Féart, C. Metabolic Endotoxemia: A Potential Underlying Mechanism of the Relationship between Dietary Fat Intake and Risk for Cognitive Impairments in Humans? Nutrients 2019, 11, 1887. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2021, 11, 594150. [Google Scholar] [CrossRef]
- Li, B.; Xia, Y.; Hu, B. Infection and atherosclerosis: TLR-dependent pathways. Cell. Mol. Life Sci. 2020, 77, 2751–2769. [Google Scholar] [CrossRef]
- Kobiyama, K.; Ley, K. Atherosclerosis. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Michel, N.A.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic Autoimmunity in Atherosclerosis Evolves from Initially Protective Apolipoprotein B100-Reactive CD4+ T-Regulatory Cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Mannervik, B.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Muñoz, P. Astrocytes Protect Dopaminergic Neurons Against Aminochrome Neurotoxicity. Neural Regen. Res. 2022, 17, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Luo, J.; Sun, Y.; Zhao, Z. Cytokines associated with immune response in atherosclerosis. Am. J. Transl. Res. 2022, 14, 6424–6444. [Google Scholar]
- Libby, P. Inflammation in Atherosclerosis-No Longer a Theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef]
- Alquraini, A.; El Khoury, J. Scavenger receptors. Curr. Biol. 2020, 30, R790–R795. [Google Scholar] [CrossRef]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A consensus definitive classification of scavenger receptors and their roles in health and disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and Its Role in Atherosclerosis. Antioxidants 2019, 8, 218. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Kanuri, S.H.; Mehta, J.L. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 2019, 26, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2022, 118, 115–129. [Google Scholar] [CrossRef]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Baid, K.; Nellimarla, S.; Huynh, A.; Boulton, S.; Guarné, A.; Melacini, G.; Collins, S.E.; Mossman, K.L. Direct binding and internalization of diverse extracellular nucleic acid species through the collagenous domain of class A scavenger receptors. Immunol. Cell Biol. 2018, 96, 922–934. [Google Scholar] [CrossRef]
- Kelley, J.L.; Ozment, T.R.; Li, C.; Schweitzer, J.B.; Williams, D.L. Scavenger receptor-A (CD204): A two-edged sword in health and disease. Crit. Rev. Immunol. 2014, 34, 241–261. [Google Scholar] [CrossRef] [PubMed]
- Nellimarla, S.; Baid, K.; Loo, Y.M.; Gale, M., Jr.; Bowdish, D.M.; Mossman, K.L. Class A Scavenger Receptor-Mediated Double-Stranded RNA Internalization Is Independent of Innate Antiviral Signaling and Does Not Require Phosphatidylinositol 3-Kinase Activity. J. Immunol. 2015, 195, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Zheng, E.; Yu, B.; Zhang, Z.; Wang, Y.; Liu, Y.; He, Y. Recognition of lipoproteins by scavenger receptor class A members. J. Biol. Chem. 2021, 297, 100948. [Google Scholar] [CrossRef]
- Cheng, C.; Hu, Z.; Cao, L.; Peng, C.; He, Y. The scavenger receptor SCARA1 (CD204) recognizes dead cells through spectrin. J. Biol. Chem. 2019, 294, 18881–18897. [Google Scholar] [CrossRef]
- Yu, B.; Cheng, C.; Wu, Y.; Guo, L.; Kong, D.; Zhang, Z.; Wang, Y.; Zheng, E.; Liu, Y.; He, Y. Interactions of ferritin with scavenger receptor class A members. J. Biol. Chem. 2020, 295, 15727–15741. [Google Scholar] [CrossRef]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef]
- Jiang, Y.; Oliver, P.; Davies, K.E.; Platt, N. Identification and characterization of murine SCARA5, a novel class A scavenger receptor that is expressed by populations of epithelial cells. J. Biol. Chem. 2006, 281, 11834–11845. [Google Scholar] [CrossRef]
- Ley, K.; Pramod, A.B.; Croft, M.; Ravichandran, K.S.; Ting, J.P. How Mouse Macrophages Sense What Is Going On. Front. Immunol. 2016, 7, 204. [Google Scholar] [CrossRef]
- Stichling, N.; Suomalainen, M.; Flatt, J.W.; Schmid, M.; Pacesa, M.; Hemmi, S.; Jungraithmayr, W.; Maler, M.D.; Freudenberg, M.A.; Plückthun, A.; et al. Lung macrophage scavenger receptor SR-A6 (MARCO) is an adenovirus type-specific virus entry receptor. PLoS Pathog. 2018, 14, e1006914. [Google Scholar] [CrossRef] [PubMed]
- Whelan, F.J.; Meehan, C.J.; Golding, G.B.; McConkey, B.J.; Bowdish, D.M. The evolution of the class A scavenger receptors. BMC Evol. Biol. 2012, 12, 227. [Google Scholar] [CrossRef]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, L.K.; Sabnis, N.A.; Panchoo, M.; Nagarajan, B.; Lacko, A.G. Targeting the SR-B1 receptor as a gateway for cancer therapy and imaging. Front. Pharmacol. 2016, 7, 466. [Google Scholar] [CrossRef] [PubMed]
- Trigatti, B.L. SR-B1 and PDZK1: Partners in HDL regulation. Curr. Opin. Lipidol. 2017, 28, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Deitch, E.A.; Condon, M.; Feketeova, E.; Machiedo, G.W.; Mason, L.; Vinluan, G.M.; Alli, V.A.; Neal, M.D.; Tomaio, J.N.; Fishman, J.E.; et al. Trauma-hemorrhagic shock induces a CD36-dependent RBC endothelial-adhesive phenotype. Crit. Care Med. 2014, 42, e200–e2010. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Nabben, M.; Luiken, J.J.F.P. CD36 (SR-B2) as master regulator of cellular fatty acid homeostasis. Curr. Opin. Lipidol. 2022, 33, 103–111. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Investig. 2017, 97, 4–13. [Google Scholar] [CrossRef]
- Daub, K.; Siegel-Axel, D.; Schönberger, T.; Leder, C.; Seizer, P.; Müller, K.; Schaller, M.; Penz, S.; Menzel, D.; Büchele, B.; et al. Inhibition of foam cell formation using a soluble CD68-Fc fusion protein. J. Mol. Med. 2010, 88, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Stephen, S.L.; Freestone, K.; Dunn, S.; Twigg, M.W.; Homer-Vanniasinkam, S.; Walker, J.H.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger receptors and their potential as therapeutic targets in the treatment of cardiovascular disease. Int. J. Hypertens. 2010, 2010, 646929. [Google Scholar] [CrossRef] [PubMed]
- Murshid, A.; Borges, T.J.; Bonorino, C.; Lang, B.J.; Calderwood, S.K. Immunological Outcomes Mediated Upon Binding of Heat Shock Proteins to Scavenger Receptors SCARF1 and LOX-1, and Endocytosis by Mononuclear Phagocytes. Front. Immunol. 2020, 10, 3035. [Google Scholar] [CrossRef]
- Martinez-Pomares, L. The mannose receptor. J. Leukoc. Biol. 2012, 92, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Rødgaard-Hansen, S.; Rafique, A.; Christensen, P.A.; Maniecki, M.B.; Sandahl, T.D.; Nexø, E.; Møller, H.J. A soluble form of the macrophage-related mannose receptor (MR/CD206) is present in human serum and elevated in critical illness. Clin. Chem. Lab. Med. 2014, 52, 453–461. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Yang, S.; Vigerust, D.J.; Shepherd, V.L. Interaction of members of the heat shock protein-70 family with the macrophage mannose receptor. J. Leukoc. Biol. 2013, 93, 529–536. [Google Scholar] [CrossRef]
- Mastrotto, F.; Pirazzini, M.; Negro, S.; Salama, A.; Martinez-Pomares, L.; Mantovani, G. Sulfation at Glycopolymer Side Chains Switches Activity at the Macrophage Mannose Receptor (CD206) in vitro and in vivo. J. Am. Chem. Soc. 2022, 144, 23134–23147. [Google Scholar] [CrossRef]
- Patten, D.A. SCARF1: A multifaceted, yet largely understudied, scavenger receptor. Inflamm. Res. 2018, 67, 627–632. [Google Scholar] [CrossRef]
- Wicker-Planquart, C.; Dufour, S.; Tacnet-Delorme, P.; Bally, I.; Delneste, Y.; Frachet, P.; Housset, D.; Thielens, N.M. Molecular and Cellular Interactions of Scavenger Receptor SR-F1 With Complement C1q Provide Insights into Its Role in the Clearance of Apoptotic Cells. Front. Immunol. 2020, 11, 544. [Google Scholar] [CrossRef]
- Wicker-Planquart, C.; Tacnet-Delorme, P.; Preisser, L.; Dufour, S.; Delneste, Y.; Housset, D.; Frachet, P.; Thielens, N.M. Insights into the ligand binding specificity of SREC-II (scavenger receptor expressed by endothelial cells). FEBS Open Biol. 2021, 11, 2693–2704. [Google Scholar] [CrossRef] [PubMed]
- Elewa, U.; Sanchez-Niño, M.D.; Mahillo-Fernández, I.; Martin-Cleary, C.; Belen Sanz, A.; Perez-Gomez, M.V.; Fernandez-Fernandez, B.; Ortiz, A. Circulating CXCL16 in Diabetic Kidney Disease. Kidney Blood Press Res. 2016, 41, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Guan, T.; Emschermann, F.; Schories, C.; Groga-Bada, P.; Martus, P.; Borst, O.; Gawaz, M.; Geisler, T.; Rath, D.; Chatterjee, M. Platelet SR-PSOX/CXCL16-CXCR6 Axis Influences Thrombotic Propensity and Prognosis in Coronary Artery Disease. Int. J. Mol. Sci. 2022, 23, 11066. [Google Scholar] [CrossRef]
- Lee, W.; Park, S.Y.; Yoo, Y.; Kim, S.Y.; Kim, J.E.; Kim, S.W.; Seo, Y.K.; Park, E.K.; Kim, I.S.; Bae, J.S. Macrophagic Stabilin-1 Restored Disruption of Vascular Integrity Caused by Sepsis. Thromb. Haemost. 2018, 118, 1776–1789. [Google Scholar] [CrossRef] [PubMed]
- Gaus, H.; Miller, C.M.; Seth, P.P.; Harris, E.N. Structural Determinants for the Interactions of Chemically Modified Nucleic Acids with the Stabilin-2 Clearance Receptor. Biochemistry 2018, 57, 2061–2064. [Google Scholar] [CrossRef] [PubMed]
- Kzhyshkowska, J.; Gratchev, A.; Goerdt, S. Stabilin-1, a homeostatic scavenger receptor with multiple functions. J. Cell. Mol. Med. 2006, 10, 635–669. [Google Scholar] [CrossRef]
- Pandey, M.S.; Miller, C.M.; Harris, E.N.; Weigel, P.H. Activation of ERK and NF-κB during HARE-mediated heparin uptake require only one of the four endocytic motifs. PLoS ONE 2016, 11, e0154124. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moestrup, S.K. CD163 and inflammation: Biological, diagnostic, and therapeutic aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef]
- Schaer, D.J.; Alayash, A.I.; Buehler, P.W. Gating the radical hemoglobin to macrophages: The anti-inflammatory role of CD163, a scavenger receptor. Antioxid. Redox Signal. 2007, 9, 991–999. [Google Scholar] [CrossRef]
- Van Gorp, H.; Delputte, P.L.; Nauwynck, H.J. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol. Immunol. 2010, 47, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Skytthe, M.K.; Graversen, J.H.; Moestrup, S.K. Targeting of CD163+ Macrophages in Inflammatory and Malignant Diseases. Int. J. Mol. Sci. 2020, 21, 5497. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, J.; Girardi, C.S.; Somensi, N.; Ribeiro, C.T.; Moreira, J.C.F.; Michels, M.; Sonai, B.; Rocha, M.; Steckert, A.V.; Barichello, T.; et al. Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. J. Biol. Chem. 2018, 293, 226–244. [Google Scholar] [CrossRef]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Gröne, H.J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Knudson, C.B. Hyaluronan and CD44: Strategic players for cell-matrix interactions during chondrogenesis and matrix assembly. Birth Defects Res. C Embryo Today 2003, 69, 174–196. [Google Scholar] [CrossRef]
- Sackstein, R. Glycoengineering of HCELL, the human bone marrow homing receptor: Sweetly programming cell migration. Ann. Biomed. Eng. 2012, 40, 766–776. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef]
- Flütsch, A.; Henry, K.; Mantuano, E.; Lam, M.S.; Shibayama, M.; Takahashi, K.; Gonias, S.L.; Campana, W.M. Evidence that LDL receptor-related protein 1 acts as an early injury detection receptor and activates c-Jun in Schwann cells. Neuroreport 2016, 27, 1305–1311. [Google Scholar] [CrossRef]
- Lillis, A.P.; Van Duyn, L.B.; Murphy-Ullrich, J.E.; Strickland, D.K. LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 2008, 88, 887–918. [Google Scholar] [CrossRef]
- Gonias, S.L. Mechanisms by Which LRP1 (Low-Density Lipoprotein Receptor-Related Protein-1) Maintains Arterial Integrity. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2548–2549. [Google Scholar] [CrossRef]
- Kyaw, T.; Peter, K.; Li, Y.; Tipping, P.; Toh, B.H.; Bobik, A. Cytotoxic lymphocytes and atherosclerosis: Significance, mechanisms and therapeutic challenges. Br. J. Pharmacol. 2017, 174, 3956–3972. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, M.; Wu, J. CXCL16 may be a predisposing factor to atherosclerosis: An animal study. Mol. Med. Rep. 2021, 24, 716. [Google Scholar] [CrossRef]
- Huang, L.; Chambliss, K.L.; Gao, X.; Yuhanna, I.; Behling-Kelly, E.; Bergaya, S.; Ahmed, M.; Michaely, P.; Luby-Phelps, K.; Darehshouri, A.; et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019, 569, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yancey, P.G.; Blakemore, J.L.; Zhang, Y.; Ding, L.; Jerome, W.G.; Brown, J.D.; Vickers, K.C.; Linton, M.F. Macrophage SR-BI modulates autophagy via VPS34 complex and PPARα transcription of Tfeb in atherosclerosis. J. Clin. Investig. 2021, 131, e94229. [Google Scholar] [CrossRef]
- Songkiatisak, P.; Rahman, S.M.T.; Aqdas, M.; Sung, M.H. NF-κB, a culprit of both inflamm-ageing and declining immunity? Immun. Ageing 2022, 19, 20. [Google Scholar] [CrossRef]
- Cordero, M.D.; Ruiz-Cabello, J. Inflamm-ageing or inflammasom-ageing as independent events. Aging 2020, 12, 17759–17760. [Google Scholar] [CrossRef] [PubMed]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [CrossRef]
- Püschel, G.P.; Klauder, J.; Henkel, J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. J. Clin. Med. 2022, 11, 4358. [Google Scholar] [CrossRef]
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef]
- Pandey, A.; Chawla, S.; Guchhait, P. Type-2 diabetes: Current understanding and future perspectives. IUBMB Life 2015, 67, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Halim, M.; Halim, A. The effects of inflammation, aging and oxidative stress on the pathogenesis of diabetes mellitus (type 2 diabetes). Diabetes Metab. Syndr. 2019, 13, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Power Guerra, N.; Müller, L.; Pilz, K.; Glatzel, A.; Jenderny, D.; Janowitz, D.; Vollmar, B.; Kuhla, A. Dietary-Induced Low-Grade Inflammation in the Liver. Biomedicines 2020, 8, 587. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lin, H. Inflammation initiates a vicious cycle between obesity and nonalcoholic fatty liver disease. Immun. Inflamm. Dis. 2021, 9, 59–73. [Google Scholar] [CrossRef]
- Gusev, E.; Solomatina, L.; Zhuravleva, Y.; Sarapultsev, A. The pathogenesis of end-stage renal disease from the standpoint of the theory of general pathological processes of inflammation. Int. J. Mol. Sci. 2021, 22, 11453. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Cevenini, E.; Monti, D.; Franceschi, C. Inflamm-ageing. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 14–20. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Luo, J.Y.; Cheng, C.K.; He, L.; Pu, Y.; Zhang, Y.; Lin, X.; Xu, A.; Lau, C.W.; Tian, X.Y.; Ma, R.C.W.; et al. Endothelial UCP2 Is a Mechanosensitive Suppressor of Atherosclerosis. Circ. Res. 2022, 131, 424–441. [Google Scholar] [CrossRef]
- Xu, S.; Lyu, Q.R.; Ilyas, I.; Tian, X.Y.; Weng, J. Vascular homeostasis in atherosclerosis: A holistic overview. Front. Immunol. 2022, 13, 976722. [Google Scholar] [CrossRef]
- Foote, C.A.; Soares, R.N.; Ramirez-Perez, F.I.; Ghiarone, T.; Aroor, A.; Manrique-Acevedo, C.; Padilla, J.; Martinez-Lemus, L. Endothelial Glycocalyx. Compr. Physiol. 2022, 12, 3781–3811. [Google Scholar] [CrossRef]
- Moore, K.H.; Murphy, H.A.; George, E.M. The glycocalyx: A central regulator of vascular function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R508–R518. [Google Scholar] [CrossRef]
- Kolářová, H.; Ambrůzová, B.; Svihálková Šindlerová, L.; Klinke, A.; Kubala, L. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediat. Inflamm. 2014, 2014, 694312. [Google Scholar] [CrossRef]
- Van Golen, R.F.; van Gulik, T.M.; Heger, M. Mechanistic overview of reactive species-induced degradation of the endothelial glycocalyx during hepatic ischemia/reperfusion injury. Free Radic. Biol. Med. 2012, 52, 1382–1402. [Google Scholar] [CrossRef]
- Sieve, I.; Münster-Kühnel, A.K.; Hilfiker-Kleiner, D. Regulation and function of endothelial glycocalyx layer in vascular diseases. Vascul. Pharmacol. 2018, 100, 26–33. [Google Scholar] [CrossRef]
- Machin, D.R.; Bloom, S.I.; Campbell, R.A.; Phuong, T.T.T.; Gates, P.E.; Lesniewski, L.A.; Rondina, M.T.; Donato, A.J. Advanced age results in a diminished endothelial glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H531–H539. [Google Scholar] [CrossRef]
- Machin, D.R.; Phuong, T.T.; Donato, A.J. The role of the endothelial glycocalyx in advanced age and cardiovascular disease. Curr. Opin. Pharmacol. 2019, 45, 66–71. [Google Scholar] [CrossRef]
- Mitra, R.; O’Neil, G.L.; Harding, I.C.; Cheng, M.J.; Mensah, S.A.; Ebong, E.E. Glycocalyx in Atherosclerosis-Relevant Endothelium Function and as a Therapeutic Target. Curr. Atheroscler. Rep. 2017, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.J.; Jiang, Y.; Chen, L.; Chen, S.L. Relationship between the endothelial glycocalyx and the extent of coronary atherosclerosis. Microcirculation 2018, 25, e12504. [Google Scholar] [CrossRef] [PubMed]
- Weinbaum, S.; Cancel, L.M.; Fu, B.M.; Tarbell, J.M. The Glycocalyx and Its Role in Vascular Physiology and Vascular Related Diseases. Cardiovasc. Eng. Technol. 2021, 12, 37–71. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef] [PubMed]
- Minhas, R.; Bansal, Y.; Bansal, G. Inducible nitric oxide synthase inhibitors: A comprehensive update. Med. Res. Rev. 2020, 40, 823–855. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.F.; Liang, X.Y.; Liu, W.; Lv, S.; He, S.J.; Kuang, H.B.; Yang, S.L. Roles of eNOS in atherosclerosis treatment. Inflamm. Res. 2019, 68, 429–441. [Google Scholar] [CrossRef]
- Chen, J.Y.; Ye, Z.X.; Wang, X.F.; Chang, J.; Yang, M.W.; Zhong, H.H.; Hong, F.F.; Yang, S.L. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed. Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef]
- Karali, E.; Bellou, S.; Stellas, D.; Klinakis, A.; Murphy, C.; Fotsis, T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol. Cell 2014, 54, 559–572. [Google Scholar] [CrossRef]
- Jeong, J.H.; Ojha, U.; Lee, Y.M. Pathological angiogenesis and inflammation in tissues. Arch. Pharm. Res. 2021, 44, 1–15. [Google Scholar] [CrossRef]
- Grunewald, M.; Kumar, S.; Sharife, H.; Volinsky, E.; Gileles-Hillel, A.; Licht, T.; Permyakova, A.; Hinden, L.; Azar, S.; Friedmann, Y.; et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science 2021, 373, eabc8479. [Google Scholar] [CrossRef]
- Marneros, A.G. Effects of chronically increased VEGF-A on the aging heart. FASEB J. 2018, 32, 1550–1565. [Google Scholar] [CrossRef]
- Marneros, A.G. Increased VEGF-A promotes multiple distinct aging diseases of the eye through shared pathomechanisms. EMBO Mol. Med. 2016, 8, 208–231. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Zheng, X.; Yang, Y.; Yao, G.; Ye, L.; Doeppner, T.R.; Hermann, D.M.; Wang, H.; Dai, Y. Impairment of hypoxia-induced angiogenesis by LDL involves a HIF-centered signaling network linking inflammatory TNFα and angiogenic VEGF. Aging 2019, 11, 328–349. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef]
- Braile, M.; Marcella, S.; Cristinziano, L.; Galdiero, M.R.; Modestino, L.; Ferrara, A.L.; Varricchi, G.; Marone, G.; Loffredo, S. VEGF-A in Cardiomyocytes and Heart Diseases. Int. J. Mol. Sci. 2020, 21, 5294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, X.; Cui, H.; Shi, J.; Yuan, G.; Shi, S.; Hu, Y. The Role of the VEGF Family in Coronary Heart Disease. Front. Cardiovasc. Med. 2021, 8, 738325. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Lemnitzer, P.; Gall, J.; Schwager, S.; Toska, A.; Yvan-Charvet, L.; Detmar, M.; Soehnlein, O. Arterial Delivery of VEGF-C Stabilizes Atherosclerotic Lesions. Circ. Res. 2021, 128, 284–286. [Google Scholar] [CrossRef]
- Shimizu, Y.; Arima, K.; Noguchi, Y.; Yamanashi, H.; Kawashiri, S.Y.; Nobusue, K.; Nonaka, F.; Aoyagi, K.; Nagata, Y.; Maeda, T. Vascular endothelial growth factor (VEGF) polymorphism rs3025039 and atherosclerosis among older with hypertension. Sci. Rep. 2022, 12, 5564. [Google Scholar] [CrossRef]
- Yu, Z.M.; Deng, X.T.; Qi, R.M.; Xiao, L.Y.; Yang, C.Q.; Gong, T. Mechanism of Chronic Stress-induced Reduced Atherosclerotic Medial Area and Increased Plaque Instability in Rabbit Models of Chronic Stress. Chin. Med. J. 2018, 131, 161–170. [Google Scholar] [CrossRef]
- Kong, M.; Zhao, Y.; Chen, A.; Lin, A. The importance of physiologic ischemia training in preventing the development of atherosclerosis: The role of endothelial progenitor cells in atherosclerotic rabbits. Coron. Artery Dis. 2019, 30, 377–383. [Google Scholar] [CrossRef]
- Milasan, A.; Smaani, A.; Martel, C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr−/−mice. Atherosclerosis 2019, 283, 106–119. [Google Scholar] [CrossRef]
- Jayasuriya, R.; Ganesan, K.; Xu, B.; Ramkumar, K.M. Emerging role of long non-coding RNAs in endothelial dysfunction and their molecular mechanisms. Biomed. Pharmacother. 2022, 145, 112421. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, M.D.; McDonald, R.A.; Baker, A.H. lncRNA/MicroRNA interactions in the vasculature. Clin. Pharmacol. Ther. 2016, 99, 494–501. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Yang, W.; Yang, J.; Ding, J.; Li, S.; Wu, H.; Zhou, F.; Jiang, Y.; Teng, L.; Yang, J. Long Noncoding RNA MEG3 Negatively Regulates Proliferation and Angiogenesis in Vascular Endothelial Cells. DNA Cell Biol. 2017, 36, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.; Bai, X.; Lin, Y.; Li, Z.; Fu, J.; Li, M.; Zhao, T.; Yang, H.; Xu, R.; et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J. Pineal Res. 2018, 64, e12449. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Thomas, A.A.; Chen, S.; Aref-Eshghi, E.; Feng, B.; Gonder, J.; Sadikovic, B.; Chakrabarti, S. MALAT1: An Epigenetic Regulator of Inflammation in Diabetic Retinopathy. Sci. Rep. 2018, 8, 6526. [Google Scholar] [CrossRef]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef]
- Thomas, A.A.; Biswas, S.; Feng, B.; Chen, S.; Gonder, J.; Chakrabarti, S. lncRNA H19 prevents endothelial-mesenchymal transition in diabetic retinopathy. Diabetologia 2019, 62, 517–530. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, X.; Cheng, J.; Zhang, K.; Ma, C. LncRNA TUG1 regulates proliferation and apoptosis by regulating miR-148b/IGF2 axis in ox-LDL-stimulated VSMC and HUVEC. Life Sci. 2020, 243, 117287. [Google Scholar] [CrossRef]
- Sun, H.; Wang, S.; Song, M. Long non coding RNA SENCR alleviates the inhibitory effects of rapamycin on human umbilical vein endothelial cells. Mol. Med. Rep. 2018, 18, 1405–1414. [Google Scholar] [CrossRef]
- Neumann, P.; Jaé, N.; Knau, A.; Glaser, S.F.; Fouani, Y.; Rossbach, O.; Krüger, M.; John, D.; Bindereif, A.; Grote, P.; et al. The lncRNA GATA6-AS epigenetically regulates endothelial gene expression via interaction with LOXL2. Nat. Commun. 2018, 9, 237. [Google Scholar] [CrossRef]
- Liang, W.; Fan, T.; Liu, L.; Zhang, L. Knockdown of growth-arrest specific transcript 5 restores oxidized low-density lipoprotein-induced impaired autophagy flux via upregulating miR-26a in human endothelial cells. Eur. J. Pharmacol. 2019, 843, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guan, M.X.; Jiang, Q.H.; Li, S.; Zhang, H.Y.; Wu, Z.G.; Cong, H.L.; Qi, X.H. NEAT1 knockdown suppresses endothelial cell proliferation and induces apoptosis by regulating miR 638/AKT/mTOR signaling in atherosclerosis. Oncol. Rep. 2020, 44, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Zhou, B.; Sun, Z.; Huang, S.; Han, L.; Nie, H.; Chen, G.; Liu, S.; Zhang, Y.; Bao, N.; et al. LncRNA-FENDRR mediates VEGFA to promote the apoptosis of brain microvascular endothelial cells via regulating miR-126 in mice with hypertensive intracerebral hemorrhage. Microcirculation 2018, 25, e12499. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.W.; Xin, Z.L.; Wang, H.Q.; Wang, K.P.; Li, X.Y.; Wang, X. Inhibiting lncRNA ROR suppresses growth, migration and angiogenesis in microvascular endothelial cells by up-regulating miR-26. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7985–7993. [Google Scholar] [CrossRef]
- Miao, C.; Cao, H.; Zhang, Y.; Guo, X.; Wang, Z.; Wang, J. LncRNA DIGIT Accelerates Tube Formation of Vascular Endothelial Cells by Sponging miR-134. Int. Heart J. 2018, 59, 1086–1095. [Google Scholar] [CrossRef]
- Shan, K.; Jiang, Q.; Wang, X.Q.; Wang, Y.N.; Yang, H.; Yao, M.D.; Liu, C.; Li, X.M.; Yao, J.; Liu, B.; et al. Role of long non-coding RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell Death Dis. 2016, 7, e2248. [Google Scholar] [CrossRef]
- Zhao, X.; Su, L.; He, X.; Zhao, B.; Miao, J. Long noncoding RNA CA7-4 promotes autophagy and apoptosis via sponging MIR877-3P and MIR5680 in high glucose-induced vascular endothelial cells. Autophagy 2020, 16, 70–85. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, Y.; Lin, G.; Zhou, X.; Jiang, X.; Zhao, H. Noncoding RNA activated by DNA damage (NORAD): Biologic function and mechanisms in human cancers. Clin. Chim. Acta 2019, 489, 5–9. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, X. Long intergenic noncoding RNA p21 mediates oxidized LDL induced apoptosis and expression of LOX 1 in human coronary artery endothelial cells. Mol. Med. Rep. 2017, 16, 8513–8519. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Li, H.; Yang, J.; Gong, Z.; Wang, B.; Zhao, X. Clopidogrel reduces apoptosis and promotes proliferation of human vascular endothelial cells induced by palmitic acid via suppression of the long non-coding RNA HIF1A-AS1 in vitro. Mol. Cell. Biochem. 2015, 404, 203–210. [Google Scholar] [CrossRef]
- Song, C.L.; Wang, J.P.; Xue, X.; Liu, N.; Zhang, X.H.; Zhao, Z.; Liu, J.G.; Zhang, C.P.; Piao, Z.H.; Liu, Y.; et al. Effect of Circular ANRIL on the Inflammatory Response of Vascular Endothelial Cells in a Rat Model of Coronary Atherosclerosis. Cell. Physiol. Biochem. 2017, 42, 1202–1212. [Google Scholar] [CrossRef]
- Zhang, H.N.; Xu, Q.Q.; Thakur, A.; Alfred, M.O.; Chakraborty, M.; Ghosh, A.; Yu, X.B. Endothelial dysfunction in diabetes and hypertension: Role of microRNAs and long non-coding RNAs. Life Sci. 2018, 213, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; Ridger, V.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat. Commun. 2020, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Laura Francés, J.; Musolino, E.; Papait, R.; Pagiatakis, C. Non-Coding RNAs in Cell-to-Cell Communication: Exploiting Physiological Mechanisms as Therapeutic Targets in Cardiovascular Pathologies. Int. J. Mol. Sci. 2023, 24, 2205. [Google Scholar] [CrossRef]
- Schimmel, L.; Heemskerk, N.; van Buul, J.D. Leukocyte transendothelial migration: A local affair. Small GTPases 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.B.; Campos, O.A.; Criado-Hidalgo, E.; Chien, S.; Del Álamo, J.C.; Lasheras, J.C.; Yeh, Y.T. Elucidating the Biomechanics of Leukocyte Transendothelial Migration by Quantitative Imaging. Front. Cell Dev. Biol. 2021, 9, 635263. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.D. Neutrophil transendothelial migration: Updates and new perspectives. Blood 2019, 133, 2149–2158. [Google Scholar] [CrossRef]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef]
- Van Steen, A.C.I.; van der Meer, W.J.; Hoefer, I.E.; van Buul, J.D. Actin remodelling of the endothelium during transendothelial migration of leukocytes. Atherosclerosis 2020, 315, 102–110. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Mach, F. Leukocyte recruitment in atherosclerosis: Potential targets for therapeutic approaches? Cell. Mol. Life Sci. 2006, 63, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Matoba, K. Monitoring Leukocyte Migration During Atherosclerosis in vivo. Methods Mol. Biol. 2022, 2419, 475–479. [Google Scholar] [CrossRef]
- Li, D.; Chen, H.; Romeo, F.; Sawamura, T.; Saldeen, T.; Mehta, J.L. Statins modulate oxidized low-density lipoprotein-mediated adhesion molecule expression in human coronary artery endothelial cells: Role of LOX-1. J. Pharmacol. Exp. Ther. 2002, 302, 601–605. [Google Scholar] [CrossRef]
- Koga, H.; Sugiyama, S.; Kugiyama, K.; Watanabe, K.; Fukushima, H.; Tanaka, T.; Sakamoto, T.; Yoshimura, M.; Jinnouchi, H.; Ogawa, H. Elevated levels of VE-cadherin-positive endothelial microparticles in patients with type 2 diabetes mellitus and coronary artery disease. J. Am. Coll. Cardiol. 2005, 45, 1622–1630. [Google Scholar] [CrossRef]
- Bernard, S.; Loffroy, R.; Sérusclat, A.; Boussel, L.; Bonnefoy, E.; Thévenon, C.; Rabilloud, M.; Revel, D.; Moulin, P.; Douek, P. Increased levels of endothelial microparticles CD144 (VE-Cadherin) positives in type 2 diabetic patients with coronary noncalcified plaques evaluated by multidetector computed tomography (MDCT). Atherosclerosis 2009, 203, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Markin, A.M.; Sobenin, I.A.; Grechko, A.V.; Zhang, D.; Orekhov, A.N. Cellular Mechanisms of Human Atherogenesis: Focus on Chronification of Inflammation and Mitochondrial Mutations. Front. Pharmacol. 2020, 11, 642. [Google Scholar] [CrossRef]
- Mironov, A.A.; Beznoussenko, G.V. Opinion: On the Way towards the New Paradigm of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 2152. [Google Scholar] [CrossRef] [PubMed]
- Hedayati-Moghadam, M.; Hosseinian, S.; Paseban, M.; Shabgah, A.G.; Gholizadeh, J.; Jamialahmadi, T.; Sathyapalan, T.; Sahebkar, A. The Role of Chemokines in Cardiovascular Diseases and the Therapeutic Effect of Curcumin on CXCL8 and CCL2 as Pathological Chemokines in Atherosclerosis. Adv. Exp. Med. Biol. 2021, 1328, 155–170. [Google Scholar] [CrossRef]
- Girbl, T.; Lenn, T.; Perez, L.; Rolas, L.; Barkaway, A.; Thiriot, A.; Del Fresno, C.; Lynam, E.; Hub, E.; Thelen, M.; et al. Distinct Compartmentalization of the Chemokines CXCL1 and CXCL2 and the Atypical Receptor ACKR1 Determine Discrete Stages of Neutrophil Diapedesis. Immunity 2018, 49, 1062–1076. [Google Scholar] [CrossRef]
- Hartmann, P.; Schober, A.; Weber, C. Chemokines and microRNAs in atherosclerosis. Cell. Mol. Life Sci. 2015, 72, 3253–3266. [Google Scholar] [CrossRef]
- Murad, H.A.S.; Rafeeq, M.M.; Alqurashi, T.M.A. Role and implications of the CXCL12/CXCR4/CXCR7 axis in atherosclerosis: Still a debate. Ann. Med. 2021, 53, 1598–1612. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Kusano, K.F.; Masuo, O.; Kawamoto, A.; Silver, M.; Murasawa, S.; Bosch-Marce, M.; Masuda, H.; Losordo, D.W.; Isner, J.M.; et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation 2003, 107, 1322–1328. [Google Scholar] [CrossRef]
- Li, L.; Du, Z.; Rong, B.; Zhao, D.; Wang, A.; Xu, Y.; Zhang, H.; Bai, X.; Zhong, J. Foam cells promote atherosclerosis progression by releasing CXCL12. Biosci. Rep. 2020, 40, BSR20193267. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Liu, C.; Aviv, A.; Ho, J.E.; Courchesne, P.; Muntendam, P.; Larson, M.G.; Cheng, S.; Wang, T.J.; Mehta, N.N.; et al. Stromal cell-derived factor 1 as a biomarker of heart failure and mortality risk. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2100–2105. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Moreno, B.; Hueso, L.; Ortega, R.; Benito, E.; Martínez-Hervas, S.; Peiro, M.; Civera, M.; Sanz, M.J.; Piqueras, L.; Real, J.T. Association of chemokines IP-10/CXCL10 and I-TAC/CXCL11 with insulin resistance and enhance leukocyte endothelial arrest in obesity. Microvasc. Res. 2022, 139, 104254. [Google Scholar] [CrossRef]
- Zernecke, A.; Weber, C. Chemokines in atherosclerosis: Proceedings resumed. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Gencer, S.; Evans, B.R.; van der Vorst, E.P.C.; Döring, Y.; Weber, C. Inflammatory Chemokines in Atherosclerosis. Cells 2021, 10, 226. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Bernhagen, J.; Heitman, L.H.; Weber, C.; Dichgans, M. Targeting the CCL2-CCR2 axis for atheroprotection. Eur. Heart J. 2022, 43, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, P.T.; Bot, I. Mast cells in atherosclerotic cardiovascular disease—Activators and actions. Eur. J. Pharmacol. 2017, 816, 37–46. [Google Scholar] [CrossRef]
- Kovanen, P.T. Mast Cells as Potential Accelerators of Human Atherosclerosis-From Early to Late Lesions. Int. J. Mol. Sci. 2019, 20, 4479. [Google Scholar] [CrossRef]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef]
- Spinas, E.; Kritas, S.K.; Saggini, A.; Mobili, A.; Caraffa, A.; Antinolfi, P.; Pantalone, A.; Tei, M.; Speziali, A.; Saggini, R.; et al. Role of mast cells in atherosclerosis: A classical inflammatory disease. Int. J. Immunopathol. Pharmacol. 2014, 27, 517–521. [Google Scholar] [CrossRef]
- Bot, I.; Shi, G.P.; Kovanen, P.T. Mast cells as effectors in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 265–271. [Google Scholar] [CrossRef]
- Redegeld, F.A.; Yu, Y.; Kumari, S.; Charles, N.; Blank, U. Non-IgE mediated mast cell activation. Immunol. Rev. 2018, 282, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, N.B.; Persaud, I.; Brown, J.M. Silver Nanoparticle-Directed Mast Cell Degranulation Is Mediated through Calcium and PI3K Signaling Independent of the High Affinity IgE Receptor. PLoS ONE 2016, 11, e0167366. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, V.; Gashev, A.A. Aging-associated shifts in functional status of mast cells located by adult and aged mesenteric lymphatic vessels. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H693–H702. [Google Scholar] [CrossRef]
- Stamenov, N.; Kotov, G.; Iliev, A.; Landzhov, B.; Kirkov, V.; Stanchev, S. Mast cells and basic fibroblast growth factor in physiological aging of rat heart and kidney. Biotech. Histochem. 2022, 97, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Saxena, Y.; Routh, S.; Mukhopadhaya, A. Immunoporosis: Role of Innate Immune Cells in Osteoporosis. Front. Immunol. 2021, 12, 687037. [Google Scholar] [CrossRef]
- Pal, S.; Meininger, C.J.; Gashev, A.A. Aged Lymphatic Vessels and Mast Cells in Perilymphatic Tissues. Int. J. Mol. Sci. 2017, 18, 965. [Google Scholar] [CrossRef]
- Vos, B.J.; van der Veer, E.; van Voorst Vader, P.C.; Mulder, A.B.; van der Heide, S.; Arends, S.; Kluin-Nelemans, J.C.; de Monchy, J.G.; van Doormaal, J.J.; Oude Elberink, J.N. Diminished reliability of tryptase as risk indicator of mastocytosis in older overweight subjects. J. Allergy Clin. Immunol. 2015, 135, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Schliemann, S.; Seyfarth, F.; Hipler, U.C.; Elsner, P. Impact of age and heterophilic interference on the basal serum tryptase, a risk indication for anaphylaxis, in 1,092 dermatology patients. Acta Derm. Venereol. 2012, 92, 484–489. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Barron, M.J.; Watson, R.E.B.; Griffiths, C.E.M.; Bulfone-Paus, S. Aged human skin accumulates mast cells with altered functionality that localize to macrophages and vasoactive intestinal peptide-positive nerve fibres. Br. J. Dermatol. 2019, 180, 849–858. [Google Scholar] [CrossRef]
- Jones, M.K.; Nair, A.; Gupta, M. Mast Cells in Neurodegenerative Disease. Front. Cell. Neurosci. 2019, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Marjanovic, J.A.; Stojanovic, A.; Brovkovych, V.M.; Skidgel, R.A.; Du, X. Signaling-mediated functional activation of inducible nitric-oxide synthase and its role in stimulating platelet activation. J. Biol. Chem. 2008, 283, 28827–28834. [Google Scholar] [CrossRef]
- Hamad, M.A.; Krauel, K.; Schanze, N.; Gauchel, N.; Stachon, P.; Nuehrenberg, T.; Zurek, M.; Duerschmied, D. Platelet Subtypes in Inflammatory Settings. Front. Cardiovasc. Med. 2022, 9, 823549. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Duchez, A.C.; Audoux, E.; Ebermeyer, T.; Arthaud, C.A.; Prier, A.; Eyraud, M.A.; Mismetti, P.; Garraud, O.; Bertoletti, L.; et al. Platelets as Key Factors in Inflammation: Focus on CD40L/CD40. Front. Immunol. 2022, 13, 825892. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Linden, M.D. Platelet physiology. Methods Mol. Biol. 2013, 992, 13–30. [Google Scholar] [CrossRef]
- Schattner, M. Platelet TLR4 at the crossroads of thrombosis and the innate immune response. J. Leukoc. Biol. 2019, 105, 873–880. [Google Scholar] [CrossRef]
- Peerschke, E.I.; Yin, W.; Ghebrehiwet, B. Platelet mediated complement activation. Adv. Exp. Med. Biol. 2008, 632, 81–91. [Google Scholar] [CrossRef]
- Jurk, K.; Walter, U. New Insights into Platelet Signalling Pathways by Functional and Proteomic Approaches. Hamostaseologie 2019, 39, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Kerris, E.W.J.; Hoptay, C.; Calderon, T.; Freishtat, R.J. Platelets and platelet extracellular vesicles in hemostasis and sepsis. J. Investig. Med. 2020, 68, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Hilger, A.; Zarbock, A.; Rossaint, J. Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation. Cells 2022, 11, 1957. [Google Scholar] [CrossRef]
- Chen, Y.; Zhong, H.; Zhao, Y.; Luo, X.; Gao, W. Role of platelet biomarkers in inflammatory response. Biomark. Res. 2020, 8, 28. [Google Scholar] [CrossRef]
- Nording, H.; Baron, L.; Langer, H.F. Platelets as therapeutic targets to prevent atherosclerosis. Atherosclerosis 2020, 307, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Khodadi, E. Platelet Function in Cardiovascular Disease: Activation of Molecules and Activation by Molecules. Cardiovasc. Toxicol. 2020, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, C. Targeting Platelet in Atherosclerosis Plaque Formation: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 9760. [Google Scholar] [CrossRef]
- Lievens, D.; von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost. 2011, 106, 827–838. [Google Scholar] [CrossRef]
- Patzelt, J.; Verschoor, A.; Langer, H.F. Platelets and the complement cascade in atherosclerosis. Front. Physiol. 2015, 6, 49. [Google Scholar] [CrossRef]
- Finsterbusch, M.; Schrottmaier, W.C.; Kral-Pointner, J.B.; Salzmann, M.; Assinger, A. Measuring and interpreting platelet-leukocyte aggregates. Platelets 2018, 29, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.B. Pro-inflammatory atherogenic role of platelets. Nat. Rev. Cardiol. 2020, 17, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Huilcaman, R.; Venturini, W.; Fuenzalida, L.; Cayo, A.; Segovia, R.; Valenzuela, C.; Brown, N.; Moore-Carrasco, R. Platelets, a Key Cell in Inflammation and Atherosclerosis Progression. Cells 2022, 11, 1014. [Google Scholar] [CrossRef] [PubMed]
- Lebas, H.; Yahiaoui, K.; Martos, R.; Boulaftali, Y. Platelets Are at the Nexus of Vascular Diseases. Front. Cardiovasc. Med. 2019, 6, 132. [Google Scholar] [CrossRef]
- Massberg, S.; Schürzinger, K.; Lorenz, M.; Konrad, I.; Schulz, C.; Plesnila, N.; Kennerknecht, E.; Rudelius, M.; Sauer, S.; Braun, S.; et al. Platelet adhesion via glycoprotein IIb integrin is critical for atheroprogression and focal cerebral ischemia: An in vivo study in mice lacking glycoprotein IIb. Circulation 2005, 112, 1180–1188. [Google Scholar] [CrossRef]
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet signaling. Handb. Exp. Pharmacol. 2012, 210, 59–85. [Google Scholar] [CrossRef]
- Bakogiannis, C.; Sachse, M.; Stamatelopoulos, K.; Stellos, K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine 2019, 122, 154157. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Duarte Lau, F.; Giugliano, R.P. Lipoprotein(a) and its Significance in Cardiovascular Disease: A Review. JAMA Cardiol. 2022, 7, 760–769. [Google Scholar] [CrossRef]
- Lampsas, S.; Xenou, M.; Oikonomou, E.; Pantelidis, P.; Lysandrou, A.; Sarantos, S.; Goliopoulou, A.; Kalogeras, K.; Tsigkou, V.; Kalpis, A.; et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules 2023, 28, 969. [Google Scholar] [CrossRef]
- Mehta, A.; Shapiro, M.D. Apolipoproteins in vascular biology and atherosclerotic disease. Nat. Rev. Cardiol. 2022, 19, 168–179. [Google Scholar] [CrossRef]
- Lu, Y.; Cui, X.; Zhang, L.; Wang, X.; Xu, Y.; Qin, Z.; Liu, G.; Wang, Q.; Tian, K.; Lim, K.S.; et al. The Functional Role of Lipoproteins in Atherosclerosis: Novel Directions for Diagnosis and Targeting Therapy. Aging Dis. 2022, 13, 491–520. [Google Scholar] [CrossRef]
- Di Giovanni, G.; Kataoka, Y.; Bubb, K.; Nelson, A.J.; Nicholls, S.J. Impact of lipid lowering on coronary atherosclerosis moving from the lumen to the artery wall. Atherosclerosis 2023, 367, 8–14. [Google Scholar] [CrossRef]
- Ronda, N.; Zimetti, F.; Adorni, M.P.; Palumbo, M.; Karpouzas, G.A.; Bernini, F. Role of Lipoprotein Levels and Function in Atherosclerosis Associated with Autoimmune Rheumatic Diseases. Rheum. Dis. Clin. N. Am. 2023, 49, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Bellot, P.E.N.R.; Moia, M.N.; Reis, B.Z.; Pedrosa, L.F.C.; Tasic, L.; Barbosa, F., Jr.; Sena-Evangelista, K.C.M. Are Phosphatidylcholine and Lysophosphatidylcholine Body Levels Potentially Reliable Biomarkers in Obesity? A Review of Human Studies. Mol. Nutr. Food Res. 2023, e2200568. [Google Scholar] [CrossRef]
- Guo, X.; Ma, L. Inflammation in coronary artery disease-clinical implications of novel HDL-cholesterol-related inflammatory parameters as predictors. Coron. Artery Dis. 2023, 34, 66–77. [Google Scholar] [CrossRef]
- Močnik, M.; Marčun Varda, N. Lipid Biomarkers and Atherosclerosis-Old and New in Cardiovascular Risk in Childhood. Int. J. Mol. Sci. 2023, 24, 2237. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Park, H.J. New Therapeutic Approaches to the Treatment of Dyslipidemia 2: LDL-C and Lp(a). J. Lipid. Atheroscler. 2023, 12, 37–46. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sukhorukov, V.N.; Surkova, R.; Orekhov, N.A.; Orekhov, A.N. Glycation of LDL: AGEs, impact on lipoprotein function, and involvement in atherosclerosis. Front. Cardiovasc. Med. 2023, 10, 1094188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.F. Free fatty acid receptors in the endocrine regulation of glucose metabolism: Insight from gastrointestinal-pancreatic-adipose interactions. Front. Endocrinol. 2022, 13, 956277. [Google Scholar] [CrossRef]
- Milanesi, R.; Coccetti, P.; Tripodi, F. The Regulatory Role of Key Metabolites in the Control of Cell Signaling. Biomolecules 2020, 10, 862. [Google Scholar] [CrossRef]
- Yang, Q.; Vijayakumar, A.; Kahn, B.B. Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol. 2018, 19, 654–672. [Google Scholar] [CrossRef] [PubMed]
- Curi, R.; Newsholme, P.; Marzuca-Nassr, G.N.; Takahashi, H.K.; Hirabara, S.M.; Cruzat, V.; Krause, M.; de Bittencourt, P.I., Jr. Regulatory principles in metabolism-then and now. Biochem. J. 2016, 473, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T. Logic and mechanisms of metabolite signalling. Nat. Rev. Endocrinol. 2022, 18, 71–72. [Google Scholar] [CrossRef]
- Bryant, N.J.; Gould, G.W. Insulin stimulated GLUT4 translocation—Size is not everything! Curr. Opin. Cell Biol. 2020, 65, 28–34. [Google Scholar] [CrossRef]
- Govers, R. Molecular mechanisms of GLUT4 regulation in adipocytes. Diabetes Metab. 2014, 40, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflug. Arch. 2020, 472, 1273–1298. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, S.; Fan, P.; Li, L.; Feng, S.; Xiao, H.; Zhu, L. The Roles of Liver Inflammation and the Insulin Signaling Pathway in PM2.5 Instillation-Induced Insulin Resistance in Wistar Rats. Dis. Mark. 2021, 2021, 2821673. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017, 16, 203. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 11128. [Google Scholar] [CrossRef]
- Opazo-Ríos, L.; Soto-Catalán, M.; Lázaro, I.; Sala-Vila, A.; Jiménez-Castilla, L.; Orejudo, M.; Moreno, J.A.; Egido, J.; Mas-Fontao, S. Meta-Inflammation and De Novo Lipogenesis Markers Are Involved in Metabolic Associated Fatty Liver Disease Progression in BTBR ob/ob Mice. Int. J. Mol. Sci. 2022, 23, 3965. [Google Scholar] [CrossRef] [PubMed]
- Lipke, K.; Kubis-Kubiak, A.; Piwowar, A. Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge. Cells 2022, 11, 844. [Google Scholar] [CrossRef] [PubMed]
- Rosso, C.; Kazankov, K.; Younes, R.; Esmaili, S.; Marietti, M.; Sacco, M.; Carli, F.; Gaggini, M.; Salomone, F.; Møller, H.J.; et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease. J. Hepatol. 2019, 71, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Zaric, B.L.; Radovanovic, J.N.; Gluvic, Z.; Stewart, A.J.; Essack, M.; Motwalli, O.; Gojobori, T.; Isenovic, E.R. Atherosclerosis Linked to Aberrant Amino Acid Metabolism and Immunosuppressive Amino Acid Catabolizing Enzymes. Front. Immunol. 2020, 11, 551758. [Google Scholar] [CrossRef]
- Thomas, D.; Apovian, C. Macrophage functions in lean and obese adipose tissue. Metabolism 2017, 72, 120–143. [Google Scholar] [CrossRef]
- Li, B.; Sun, S.; Li, J.J.; Yuan, J.P.; Sun, S.R.; Wu, Q. Adipose tissue macrophages: Implications for obesity-associated cancer. Mil. Med. Res. 2023, 10, 1. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.Y.; Wu, L.L.; Jiang, L.Y.; Hu, Y.; Xiao, X.H.; Wang, Y.D. Paracrine Regulation of Adipose Tissue Macrophages by Their Neighbors in the Microenvironment of Obese Adipose Tissue. Endocrinology 2022, 163, bqac062. [Google Scholar] [CrossRef]
- Matacchione, G.; Perugini, J.; Di Mercurio, E.; Sabbatinelli, J.; Prattichizzo, F.; Senzacqua, M.; Storci, G.; Dani, C.; Lezoche, G.; Guerrieri, M.; et al. Senescent macrophages in the human adipose tissue as a source of inflammaging. Geroscience 2022, 44, 1941–1960. [Google Scholar] [CrossRef]
- Tao, Y.; Jiang, Q.; Wang, Q. Adipose tissue macrophages in remote modulation of hepatic glucose production. Front. Immunol. 2022, 13, 998947. [Google Scholar] [CrossRef]
- Liu, L.; Shi, Z.; Ji, X.; Zhang, W.; Luan, J.; Zahr, T.; Qiang, L. Adipokines, adiposity, and atherosclerosis. Cell. Mol. Life Sci. 2022, 79, 272. [Google Scholar] [CrossRef]
- Shi, J.; Fan, J.; Su, Q.; Yang, Z. Cytokines and Abnormal Glucose and Lipid Metabolism. Front. Endocrinol. 2019, 10, 703. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Ontogeny of Tissue-Resident Macrophages. Front. Immunol. 2015, 6, 486. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol. 2018, 330, 5–15. [Google Scholar] [CrossRef]
- Liu, Y.; Xia, Y.; Qiu, C.H. Functions of CD169 positive macrophages in human diseases (Review). Biomed. Rep. 2021, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Ji, H.H.; Li, Y.J.; Guo, S.D. Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosci. 2021, 8, 679797. [Google Scholar] [CrossRef]
- Trzebanski, S.; Jung, S. Plasticity of monocyte development and monocyte fates. Immunol. Lett. 2020, 227, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Susser, L.I.; Rayner, K.J. Through the layers: How macrophages drive atherosclerosis across the vessel wall. J. Clin. Investig. 2022, 132, e157011. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Li, X.; Xiong, K.; Song, Z.; Tian, J.; Wen, Y.; Sun, A.; Deng, X. The Entry and Egress of Monocytes in Atherosclerosis: A Biochemical and Biomechanical Driven Process. Cardiovasc. Ther. 2021, 2021, 6642927. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.; Mieremet, A.; de Vries, C.J.M.; Micha, D.; de Waard, V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2693–2707. [Google Scholar] [CrossRef]
- Yang, K.; Xiao, Q.; Niu, M.; Pan, X.; Zhu, X. Exosomes in atherosclerosis: Convergence on macrophages. Int. J. Biol. Sci. 2022, 18, 3266–3281. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Griendling, K.K. Mitochondrial Respiration and Atherosclerosis: R-E-S-P-I-R-E. Find Out What it Means to Mϕ (and VSMC). Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2229–2230. [Google Scholar] [CrossRef]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; McCluskey, K.; Fujii, K.; Wahl, L.M. Differential regulation of monocyte matrix metalloproteinase and TIMP-1 production by TNF-alpha, granulocyte-macrophage CSF, and IL-1 beta through prostaglandin-dependent and -independent mechanisms. J. Immunol. 1998, 161, 3071–3076. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, R.; Zhai, R.; Yang, S.; Peng, T.; Zheng, F.; Shen, Y.; Li, M.; Li, L. Matrix stiffness regulates macrophage polarization in atherosclerosis. Pharmacol. Res. 2022, 179, 106236. [Google Scholar] [CrossRef]
- Eshghjoo, S.; Kim, D.M.; Jayaraman, A.; Sun, Y.; Alaniz, R.C. Macrophage Polarization in Atherosclerosis. Genes 2022, 13, 756. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef]
- Bories, G.F.P.; Leitinger, N. Macrophage metabolism in atherosclerosis. FEBS Lett. 2017, 591, 3042–3060. [Google Scholar] [CrossRef]
- Dolfi, B.; Gallerand, A.; Haschemi, A.; Guinamard, R.R.; Ivanov, S. Macrophage metabolic regulation in atherosclerotic plaque. Atherosclerosis 2021, 334, 1–8. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Hashimoto, D.; Miller, J.; Merad, M. Dendritic cell and macrophage heterogeneity in vivo. Immunity 2011, 35, 323–335. [Google Scholar] [CrossRef]
- Jung, S.H.; Hwang, B.H.; Shin, S.; Park, E.H.; Park, S.H.; Kim, C.W.; Kim, E.; Choo, E.; Choi, I.J.; Swirski, F.K.; et al. Spatiotemporal dynamics of macrophage heterogeneity and a potential function of Trem2hi macrophages in infarcted hearts. Nat. Commun. 2022, 13, 4580. [Google Scholar] [CrossRef]
- De Sousa, J.R.; Da Costa Vasconcelos, P.F.; Quaresma, J.A.S. Functional aspects, phenotypic heterogeneity, and tissue immune response of macrophages in infectious diseases. Infect. Drug Resist. 2019, 12, 2589–2611. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Matuschik, L.; Riabov, V.; Schmuttermaier, C.; Sevastyanova, T.; Weiss, C.; Klüter, H.; Kzhyshkowska, J. Hyperglycemia Induces Inflammatory Response of Human Macrophages to CD163-Mediated Scavenging of Hemoglobin-Haptoglobin Complexes. Int. J. Mol. Sci. 2022, 23, 1385. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.J.; Johns, M.; Kampfer, T.; Nguyen, A.T.; Game, L.; Schaer, D.J.; Mason, J.C.; Haskard, D.O. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ. Res. 2012, 110, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Medbury, H.J.; Williams, H.; Fletcher, J.P. Clinical significance of macrophage phenotypes in cardiovascular disease. Clin. Transl. Med. 2014, 3, 63. [Google Scholar] [CrossRef]
- Zhu, J. T Helper cell differentiation, heterogeneity, and plasticity. Cold Spring Harb. Perspect. Biol. 2018, 10, a030338. [Google Scholar] [CrossRef]
- Wen, T.-H.; Tsai, K.-W.; Wu, Y.-J.; Liao, M.-T.; Lu, K.-C.; Hu, W.-C. The framework for human host immune responses to four types of parasitic infections and relevant key JAK/STAT signaling. Int. J. Mol. Sci. 2021, 22, 13310. [Google Scholar] [CrossRef] [PubMed]
- Castro, G.; Liu, X.; Ngo, K.; De Leon-Tabaldo, A.; Zhao, S.; Luna-Roman, R.; Yu, J.; Cao, T.; Kuhn, R.; Wilkinson, P.; et al. RORγt and RORα signature genes in human Th17 cells. PLoS ONE 2017, 12, e0181868. [Google Scholar] [CrossRef]
- Fujita, H.; Nograles, K.E.; Kikuchi, T.; Gonzalez, J.; Carucci, J.A.; Krueger, J.G. Human Langerhans cells induce distinct IL-22-producing CD4+ T cells lacking IL-17 production. Proc. Natl. Acad. Sci. USA 2009, 106, 21795–21800. [Google Scholar] [CrossRef] [PubMed]
- Paul, W.E.; Zhu, J. How are TH2-type immune responses initiated and amplified? Nat. Rev. Immunol. 2010, 10, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Wu, J.; Ma, M. Implications of macrophage polarization in corneal transplantation rejection. Transpl. Immunol. 2021, 64, 101353. [Google Scholar] [CrossRef]
- Loo, T.T.; Gao, Y.; Lazarevic, V. Transcriptional regulation of CD4+TH cells that mediate tissue inflammation. J. Leukoc. Biol. 2018, 104, 1069–1085. [Google Scholar] [CrossRef]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the heterogeneity in T-Cell mediated inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jiang, T.; Li, M.Q.; Zheng, X.L.; Zhao, G.J. Transcriptional Regulation of Macrophages Polarization by MicroRNAs. Front. Immunol. 2018, 9, 1175. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guan, L.; Tang, L.; Liu, S.; Zhou, Y.; Chen, C.; He, Z.; Xu, L. T Helper 9 cells: A new player in immune-related diseases. DNA Cell Biol. 2019, 38, 1040–1047. [Google Scholar] [CrossRef]
- Rasouli, J.; Casella, G.; Yoshimura, S.; Zhang, W.; Xiao, D.; Garifallou, J.; Gonzalez, M.V.; Wiedeman, A.; Kus, A.; Mari, E.R.; et al. A distinct GM-CSF+ T helper cell subset requires T-bet to adopt a TH1 phenotype and promote neuroinflammation. Sci. Immunol. 2020, 5, eaba9953. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef]
- Wang, Y.; Souabni, A.; Flavell, R.A.; Wan, Y.Y. An intrinsic mechanism predisposes Foxp3-expressing regulatory T cells to Th2 conversion in vivo. J. Immunol. 2010, 185, 5983–5992. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, A.; Maggi, L.; Liotta, F.; Cosmi, L.; Annunziato, F. Biological and clinical significance of T helper 17 cell plasticity. Immunology 2019, 158, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Caza, T.; Landas, S. Functional and Phenotypic Plasticity of CD4(+) T Cell Subsets. BioMed Res. Int. 2015, 2015, 521957. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, K.; Nakayama, T. CD4+T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Takai, K.; Fujiwara, N.; Arimitsu, N.; Ueda, Y.; Wakisaka, S.; Yoshikawa, H.; Kaneko, F.; Suzuki, T.; Suzuki, N. Excessive CD4+ T cells co-expressing interleukin-17 and interferon-γ in patients with behçet’s disease. Clin. Exp. Immunol. 2012, 168, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Malviya, V.; Yshii, L.; Junius, S.; Garg, A.D.; Humblet-Baron, S.; Schlenner, S.M. Regulatory T-cell stability and functional plasticity in health and disease. Immunol. Cell Biol. 2023, 101, 112–129. [Google Scholar] [CrossRef]
- Siegers, G.M.; Dutta, I.; Lai, R.; Postovit, L.M. Functional Plasticity of Gamma Delta T Cells and Breast Tumor Targets in Hypoxia. Front. Immunol. 2018, 9, 1367. [Google Scholar] [CrossRef]
- Giri, S.; Lal, G. Differentiation and functional plasticity of gamma-delta (γδ) T cells under homeostatic and disease conditions. Mol. Immunol. 2021, 136, 138–149. [Google Scholar] [CrossRef]
- Gil-Pulido, J.; Amézaga, N.; Jorgacevic, I.; Manthey, H.D.; Rösch, M.; Brand, T.; Cidlinsky, P.; Schäfer, S.; Beilhack, A.; Saliba, A.E.; et al. Interleukin-23 receptor expressing γδ T cells locally promote early atherosclerotic lesion formation and plaque necrosis in mice. Cardiovasc. Res. 2022, 118, 2932–2945. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, S.; Li, J.; Yin, M.; Yan, F.; Chen, Y.; Chen, Y.; Wu, T.; Cheng, M.; He, Y.; et al. Phenotypic Changes of Peripheral γδ T Cell and Its Subsets in Patients with Coronary Artery Disease. Front. Immunol. 2022, 13, 900334. [Google Scholar] [CrossRef]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef]
- Khan, A.W.; Paneni, F.; Jandeleit-Dahm, K.A.M. Cell-specific epigenetic changes in atherosclerosis. Clin. Sci. 2021, 135, 1165–1187. [Google Scholar] [CrossRef] [PubMed]
- Bart, V.M.T.; Pickering, R.J.; Taylor, P.R.; Ipseiz, N. Macrophage reprogramming for therapy. Immunology 2021, 163, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The good, the bad, and the gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Karim, M.R.; Izawa, T.; Kuwamura, M.; Yamate, J. Immunophenotypical characterization of M1/M2 macrophages and lymphocytes in cisplatin-induced rat progressive renal fibrosis. Cells 2021, 10, 257. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, J.; Hu, F.; Liu, J.; Li, M.; Zhao, L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019, 2019, 6724903. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Kashirskikh, D.A.; Khotina, V.A.; Grechko, A.V.; Orekhov, A.N. Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells. J. Clin. Med. 2019, 8, 1798. [Google Scholar] [CrossRef]
- He, X.; Liang, B.; Gu, N. Th17/Treg Imbalance and Atherosclerosis. Dis. Mark. 2020, 2020, 8821029. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Xu, D. Research progress on Th17 and T regulatory cells and their cytokines in regulating atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 929078. [Google Scholar] [CrossRef]
- Forteza, M.J.; Ketelhuth, D.F.J. Metabolism in atherosclerotic plaques: Immunoregulatory mechanisms in the arterial wall. Clin. Sci. 2022, 136, 435–454. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Sage, A.P.; Mallat, Z.; Tedgui, A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ. Res. 2014, 114, 1640–1660. [Google Scholar] [CrossRef]
- Wang, J.; Kang, Z.; Liu, Y.; Li, Z.; Liu, Y.; Liu, J. Identification of immune cell infiltration and diagnostic biomarkers in unstable atherosclerotic plaques by integrated bioinformatics analysis and machine learning. Front. Immunol. 2022, 13, 956078. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, J.; Zhang, W.; Xu, Y. A myriad of roles of dendritic cells in atherosclerosis. Clin. Exp. Immunol. 2021, 206, 12–27. [Google Scholar] [CrossRef]
- Bellini, R.; Bonacina, F.; Norata, G.D. Crosstalk between dendritic cells and T lymphocytes during atherogenesis: Focus on antigen presentation and break of tolerance. Front. Cardiovasc. Med. 2022, 9, 934314. [Google Scholar] [CrossRef] [PubMed]
- Razeghian-Jahromi, I.; Karimi Akhormeh, A.; Razmkhah, M.; Zibaeenezhad, M.J. Immune system and atherosclerosis: Hostile or friendly relationship. Int. J. Immunopathol. Pharmacol. 2022, 36, 3946320221092188. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bezsonov, E.E.; Popkova, T.V.; Starodubova, A.V.; Orekhov, A.N. Immunity in Atherosclerosis: Focusing on T and B Cells. Int. J. Mol. Sci. 2021, 22, 8379. [Google Scholar] [CrossRef]
- Jonasson, L.; Holm, J.; Skalli, O.; Gabbiani, G.; Hansson, G.K. Expression of class II transplantation antigen on vascular smooth muscle cells in human atherosclerosis. J. Clin. Investig. 1985, 76, 125–131. [Google Scholar] [CrossRef]
- Ghamar Talepoor, A.; Khosropanah, S.; Doroudchi, M. Functional subsets of circulating follicular helper T cells in patients with atherosclerosis. Physiol. Rep. 2020, 8, e14637. [Google Scholar] [CrossRef]
- Sima, P.; Vannucci, L.; Vetvicka, V. Atherosclerosis as autoimmune disease. Ann. Transl. Med. 2018, 6, 116. [Google Scholar] [CrossRef]
- Yin, C.; Mohanta, S.K.; Srikakulapu, P.; Weber, C.; Habenicht, A.J. Artery Tertiary Lymphoid Organs: Powerhouses of Atherosclerosis Immunity. Front. Immunol. 2016, 7, 387. [Google Scholar] [CrossRef] [PubMed]
- Czerniuk, M.R.; Surma, S.; Romańczyk, M.; Nowak, J.M.; Wojtowicz, A.; Filipiak, K.J. Unexpected Relationships: Periodontal Diseases: Atherosclerosis-Plaque Destabilization? From the Teeth to a Coronary Event. Biology 2022, 11, 272. [Google Scholar] [CrossRef]
- Mayr, M.; Kiechl, S.; Willeit, J.; Wick, G.; Xu, Q. Infections, immunity, and atherosclerosis: Associations of antibodies to Chlamydia pneumoniae, Helicobacter pylori, and cytomegalovirus with immune reactions to heat-shock protein 60 and carotid or femoral atherosclerosis. Circulation 2000, 102, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Ghamar Talepoor, A.; Doroudchi, M. Immunosenescence in atherosclerosis: A role for chronic viral infections. Front. Immunol. 2022, 13, 945016. [Google Scholar] [CrossRef]
- Das, D.; Podder, S. Unraveling the molecular crosstalk between Atherosclerosis and COVID-19 comorbidity. Comput. Biol. Med. 2021, 134, 104459. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, E.; Hughes, G.R.; Khamashta, M.A. Oxidation of LDL and its clinical implication. Autoimmun. Rev. 2008, 7, 558–566. [Google Scholar] [CrossRef]
- Suciu, C.F.; Prete, M.; Ruscitti, P.; Favoino, E.; Giacomelli, R.; Perosa, F. Oxidized low density lipoproteins: The bridge between atherosclerosis and autoimmunity. Possible implications in accelerated atherosclerosis and for immune intervention in autoimmune rheumatic disorders. Autoimmun. Rev. 2018, 17, 366–375. [Google Scholar] [CrossRef]
- Full, L.E.; Ruisanchez, C.; Monaco, C. The inextricable link between atherosclerosis and prototypical inflammatory diseases rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res. Ther. 2009, 11, 217. [Google Scholar] [CrossRef]
- Clancy, R.M.; Halushka, M.; Rasmussen, S.E.; Lhakhang, T.; Chang, M.; Buyon, J.P. Siglec-1 Macrophages and the Contribution of IFN to the Development of Autoimmune Congenital Heart Block. J. Immunol. 2019, 202, 48–55. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, H.; He, J. Role of dyslipidemia in accelerating inflammation, autoimmunity, and atherosclerosis in systemic lupus erythematosus and other autoimmune diseases. Discov. Med. 2020, 30, 49–56. [Google Scholar]
- Raj, R.; Thomas, S.; Gorantla, V. Accelerated atherosclerosis in rheumatoid arthritis: A systematic review. F1000research 2022, 11, 466. [Google Scholar] [CrossRef]
- Xing, H.; Pang, H.; Du, T.; Yang, X.; Zhang, J.; Li, M.; Zhang, S. Establishing a Risk Prediction Model for Atherosclerosis in Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 622216. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Mandal, K.; Jahangiri, M.; Xu, Q. Autoimmune mechanisms of atherosclerosis. Handb. Exp. Pharmacol. 2005, 170, 723–743. [Google Scholar] [CrossRef]
- Tsimikas, S.; Brilakis, E.S.; Lennon, R.J.; Miller, E.R.; Witztum, J.L.; McConnell, J.P.; Kornman, K.S.; Berger, P.B. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J. Lipid Res. 2007, 48, 425–433. [Google Scholar] [CrossRef]
- Hernández-Vargas, P.; Ortiz-Muñoz, G.; López-Franco, O.; Suzuki, Y.; Gallego-Delgado, J.; Sanjuán, G.; Lázaro, A.; López-Parra, V.; Ortega, L.; Egido, J.; et al. Fcgamma receptor deficiency confers protection against atherosclerosis in apolipoprotein E knockout mice. Circ. Res. 2006, 99, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Mussbacher, M.; Galkina, E.V. Functional Role of B Cells in Atherosclerosis. Cells 2021, 10, 270. [Google Scholar] [CrossRef]
- Tsimikas, S.; Miyanohara, A.; Hartvigsen, K.; Merki, E.; Shaw, P.X.; Chou, M.Y.; Pattison, J.; Torzewski, M.; Sollors, J.; Friedmann, T.; et al. Human oxidation-specific antibodies reduce foam cell formation and atherosclerosis progression. J. Am. Coll. Cardiol. 2011, 58, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Klimov, A.N.; Denisenko, A.D.; Popov, A.V.; Nagornev, V.A.; Pleskov, V.M.; Vinogradov, A.G.; Denisenko, T.V.; Magracheva, E.Y.; Kheifes, G.M.; Kuznetzov, A.S. Lipoprotein-antibody immune complexes. Their catabolism and role in foam cell formation. Atherosclerosis 1985, 58, 1–15. [Google Scholar] [CrossRef]
- Schiopu, A.; Bengtsson, J.; Söderberg, I.; Janciauskiene, S.; Lindgren, S.; Ares, M.P.; Shah, P.K.; Carlsson, R.; Nilsson, J.; Fredrikson, G.N. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation 2004, 110, 2047–2052. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bezsonov, E.E.; Popkova, T.V.; Starodubova, A.V.; Orekhov, A.N. Vaccination against Atherosclerosis: Is It Real? Int. J. Mol. Sci. 2022, 23, 2417. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.S.; Vanderlugt, C.J.; Hashimoto, T.; Nishikawa, T.; Zone, J.J.; Black, M.M.; Wojnarowska, F.; Stevens, S.R.; Chen, M.; Fairley, J.; et al. Epitope Spreading: Lessons from Autoimmune Skin Diseases. J. Investig. Dermatol. 1998, 110, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Tuohy, V.K.; Kinkel, R.P. Epitope Spreading: A Mechanism for Progression of Autoimmune Disease. Autoimmunity 2001, 48, 39–48. [Google Scholar]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716. [Google Scholar] [CrossRef]
- Heyde, A.; Rohde, D.; McAlpine, C.S.; Zhang, S.; Hoyer, F.F.; Gerold, J.M.; Cheek, D.; Iwamoto, Y.; Schloss, M.J.; Vandoorne, K.; et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 2021, 184, 1348–1361.e22. [Google Scholar] [CrossRef]
- Mo, L.; Ma, C.; Wang, Z.; Li, J.; He, W.; Niu, W.; Chen, Z.; Zhou, C.; Liu, Y. Integrated Bioinformatic Analysis of the Shared Molecular Mechanisms Between Osteoporosis and Atherosclerosis. Front. Endocrinol. 2022, 13, 950030. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Sazonova, M.D.; Kirichenko, T.V.; Khotina, V.A.; Khasanova, Z.B.; Doroschuk, N.A.; Karagodin, V.P.; Orekhov, A.N.; et al. Some Molecular and Cellular Stress Mechanisms Associated with Neurodegenerative Diseases and Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 699. [Google Scholar] [CrossRef]
- Mohanta, S.K.; Peng, L.; Li, Y.; Lu, S.; Sun, T.; Carnevale, L.; Perrotta, M.; Ma, Z.; Förstera, B.; Stanic, K.; et al. Neuroimmune cardiovascular interfaces control atherosclerosis. Nature 2022, 605, 152–159. [Google Scholar] [CrossRef]
- Glaser, R.; Kiecolt-Glaser, J.K. Stress-induced immune dysfunction: Implications for health. Nat. Rev. Immunol. 2005, 5, 243–251. [Google Scholar] [CrossRef]
- Hinterdobler, J.; Schunkert, H.; Kessler, T.; Sager, H.B. Impact of Acute and Chronic Psychosocial Stress on Vascular Inflammation. Antioxid. Redox Signal. 2021, 35, 1531–1550. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, T.; Darios, E.; Watts, S.W.; Roccabianca, S. Aortic stiffness is lower when PVAT is included: A novel ex vivo mechanics study. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H1003–H1013. [Google Scholar] [CrossRef] [PubMed]
- Hillock-Watling, C.; Gotlieb, A.I. The pathobiology of perivascular adipose tissue (PVAT), the fourth layer of the blood vessel wall. Cardiovasc. Pathol. 2022, 61, 107459. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Sadhu, S.; Rymut, N. Fine-tuning inflammation-resolution programs: Focus on atherosclerosis. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 117–123. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; de Winther, M.P.; Weber, C.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Atherosclerotic plaque destabilization: Mechanisms, models, and therapeutic strategies. Circ. Res. 2014, 114, 214–226. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Van Dijk, R.A.; Rijs, K.; Wezel, A.; Hamming, J.F.; Kolodgie, F.D.; Virmani, R.; Schaapherder, A.F.; Lindeman, J.H. Systematic Evaluation of the Cellular Innate Immune Response During the Process of Human Atherosclerosis. J. Am. Heart Assoc. 2016, 16, e002860. [Google Scholar] [CrossRef]
- Moreno, P.R.; Purushothaman, M.; Purushothaman, K.R. Plaque neovascularization: Defense mechanisms, betrayal, or a war in progress. Ann. N. Y. Acad. Sci. 2012, 1254, 7–17. [Google Scholar] [CrossRef]
- Waksman, R.; Seruys, P.W. Handbook of the Vulnerable Plaque; CRC Press: Boca Raton, FL, USA, 2007; pp. 1–48. [Google Scholar]
- Hellings, W.E.; Peeters, W.; Moll, F.L.; Pasterkamp, G. From vulnerable plaque to vulnerable patient: The search for biomarkers of plaque destabilization. Trends Cardiovasc. Med. 2007, 17, 162–171. [Google Scholar] [CrossRef]
- Kondakov, A.; Lelyuk, V. Clinical Molecular Imaging for Atherosclerotic Plaque. J. Imaging 2021, 7, 211. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Zotova, N.V.; Chereshnev, V.A.; Gusev, E.Y. Systemic inflammation: Methodological approaches to identification of the common pathological process. PLoS ONE 2016, 11, e0155138. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.Y.; Zotova, N.V.; Chereshnev, V.A. Sepsis-3: New edition—Old problems. Analysis from the perspective of general pathology. Russ. J. Infect. Immun. 2021, 11, 649–662. [Google Scholar] [CrossRef]
- Gusev, E.; Sarapultsev, A.; Hu, D.; Chereshnev, V. Problems of pathogenesis and pathogenetic therapy of COVID-19 from the perspective of the general theory of pathological systems (general pathological processes). Int. J. Mol. Sci. 2021, 22, 7582. [Google Scholar] [CrossRef] [PubMed]
- Zotova, N.V.; Zhuravleva, Y.A.; Zubova, T.E.; Gusev, E.Y. Integral estimation of systemic inflammatory response under sepsis. Gen. Physiol. Biophys. 2020, 39, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Brazhnikov, A.; Zotova, N.; Solomatina, L.; Sarapultsev, A.; Spirin, A.; Gusev, E. Shock-Associated Systemic Inflammation in Amniotic Fluid Embolism, Complicated by Clinical Death. Pathophysiology 2023, 30, 48–62. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Zotova, N.V. Pathogenesis and prediction of critical complications of polytrauma from the position of common pathological processes. Polytrauma 2021, 1, 97–105. [Google Scholar]
- Gusev, E.Y.; Chereshnev, V.A. Systemic inflammation: Theoretical and methodological approaches to description of general pathological process model. Part IV. A dynamics of the process. Patol. Fiziol. Eksp. Ter. 2014, 4, 4–16. [Google Scholar]
- Kaynar, A.M.; Yende, S.; Zhu, L.; Frederick, D.R.; Chambers, R.; Burton, C.L.; Carter, M.; Stolz, D.B.; Agostini, B.; Gregory, A.D.; et al. Effects of intra-abdominal sepsis on atherosclerosis in mice. Crit. Care 2014, 18, 469. [Google Scholar] [CrossRef]
- Laudanski, K. Persistence of Lipoproteins and Cholesterol Alterations after Sepsis: Implication for Atherosclerosis Progression. Int. J. Mol. Sci. 2021, 22, 10517. [Google Scholar] [CrossRef]
- Merdji, H.; Schini-Kerth, V.; Meziani, F.; Toti, F. Long-term cardiovascular complications following sepsis: Is senescence the missing link? Ann. Intensive Care 2021, 11, 166. [Google Scholar] [CrossRef]
- Wang, J.; Su, E.; Wang, H.; Guo, C.; Lawrence, D.A.; Eitzman, D.T. Traumatic Brain Injury Leads to Accelerated Atherosclerosis in Apolipoprotein E Deficient Mice. Sci. Rep. 2018, 8, 5639. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, C.; Klinger-König, J.; Frenzel, S.; Schminke, U.; Völzke, H.; Lübke, L.; Grabe, H.J. Association of traumatic stress and posttraumatic stress disorder with carotid atherosclerosis: Findings from the general population. Eur. J. Psychotraumatol. 2020, 11, 1815280. [Google Scholar] [CrossRef]
- Narang, R.K.; Dalbeth, N. Pathophysiology of Gout. Semin. Nephrol. 2020, 40, 550–563. [Google Scholar] [CrossRef]
- Ragab, G.; Elshahaly, M.; Bardin, T. Gout: An old disease in new perspective—A review. J. Adv. Res. 2017, 8, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Tsukui, D.; Kono, H. Uric Acid in Inflammation and the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 12394. [Google Scholar] [CrossRef] [PubMed]
- Brassington, K.; Selemidis, S.; Bozinovski, S.; Vlahos, R. Chronic obstructive pulmonary disease and atherosclerosis: Common mechanisms and novel therapeutics. Clin. Sci. 2022, 136, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Xanthopoulos, A.; Butler, J. Cardiovascular Aging and Heart Failure: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 804–813. [Google Scholar] [CrossRef]
- Shpektor, A. Cardiogenic shock: The role of inflammation. Acute Card. Care 2010, 12, 115–118. [Google Scholar] [CrossRef]
- Olarte, N.; Rivera, N.T.; Grazette, L. Evolving Presentation of Cardiogenic Shock: A Review of the Medical Literature and Current Practices. Cardiol. Ther. 2022, 11, 369–384. [Google Scholar] [CrossRef]
- Bertini, P.; Guarracino, F. Pathophysiology of cardiogenic shock. Curr. Opin. Crit. Care 2021, 27, 409–415. [Google Scholar] [CrossRef]
- Bochkarev, P.Y.; Berdyugina, O.V.; Zhidkova, V.S.; Zubova, T.E.; Gusev, E.Y. The role of systemic inflammation in the pathogenesis of hemorrhagic stroke in the presence or absence of effective brain blood flow. Z. Nevrol. Psikhiatr. Imeni SS Korsakova 2020, 120, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J. Update on the pathophysiology and medical treatment of peripheral artery disease. Nat. Rev. Cardiol. 2022, 19, 456–474. [Google Scholar] [CrossRef] [PubMed]
- Shaker, M.E. The contribution of sterile inflammation to the fatty liver disease and the potential therapies. Biomed. Pharmacother. 2022, 148, 112789. [Google Scholar] [CrossRef] [PubMed]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Tan, J.; Wei, L.; Wu, D.; Gao, S.; Weng, Y.; Chen, J. Recent advance in treatment of atherosclerosis: Key targets and plaque-positioned delivery strategies. J. Tissue Eng. 2022, 13, 20417314221088509. [Google Scholar] [CrossRef]
- Kotlyarov, S.; Kotlyarova, A. Molecular Pharmacology of Inflammation Resolution in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 4808. [Google Scholar] [CrossRef]
- Gan, J.; Guo, L.; Zhang, X.; Yu, Q.; Yang, Q.; Zhang, Y.; Zeng, W.; Jiang, X.; Guo, M. Anti-inflammatory therapy of atherosclerosis: Focusing on IKKβ. J. Inflamm. 2023, 20, 8. [Google Scholar] [CrossRef]
- Chen, J.; Xiang, X.; Nie, L.; Guo, X.; Zhang, F.; Wen, C.; Xia, Y.; Mao, L. The emerging role of Th1 cells in atherosclerosis and its implications for therapy. Front. Immunol. 2023, 13, 1079668. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Millican, R.; Lynd, T.; Gangasani, M.; Malhotra, S.; Sherwood, J.; Hwang, P.T.; Cho, Y.; Brott, B.C.; et al. Recent Progress in in vitro Models for Atherosclerosis Studies. Front. Cardiovasc. Med. 2022, 8, 790529. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).