
Molecular Factors and Pathways of Hepatotoxicity Associated with HIV/SARS-CoV-2 Protease Inhibitors

Abstract
:1. Overview of Antiviral Protease Inhibitors and Side Effects on the Liver
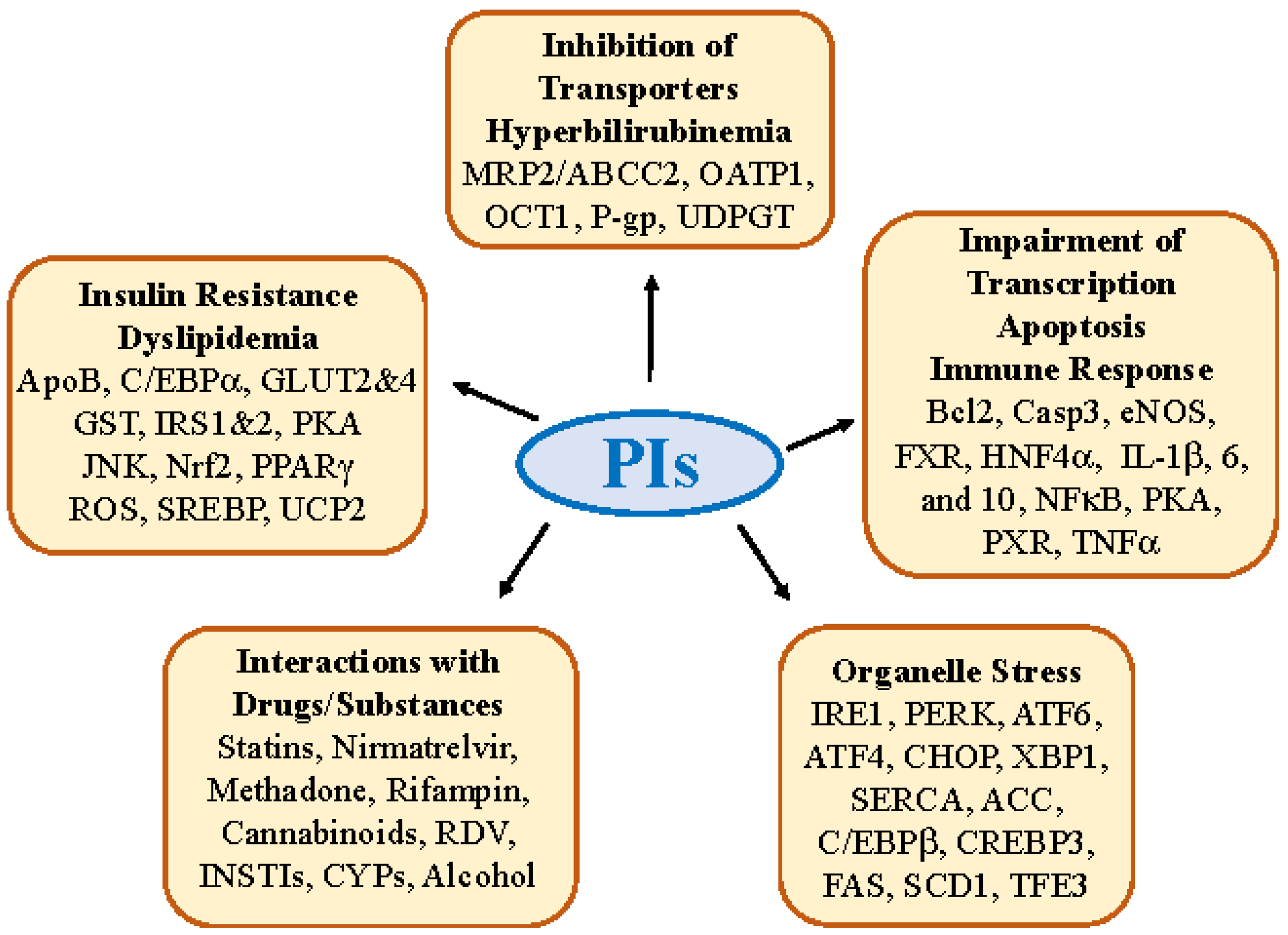
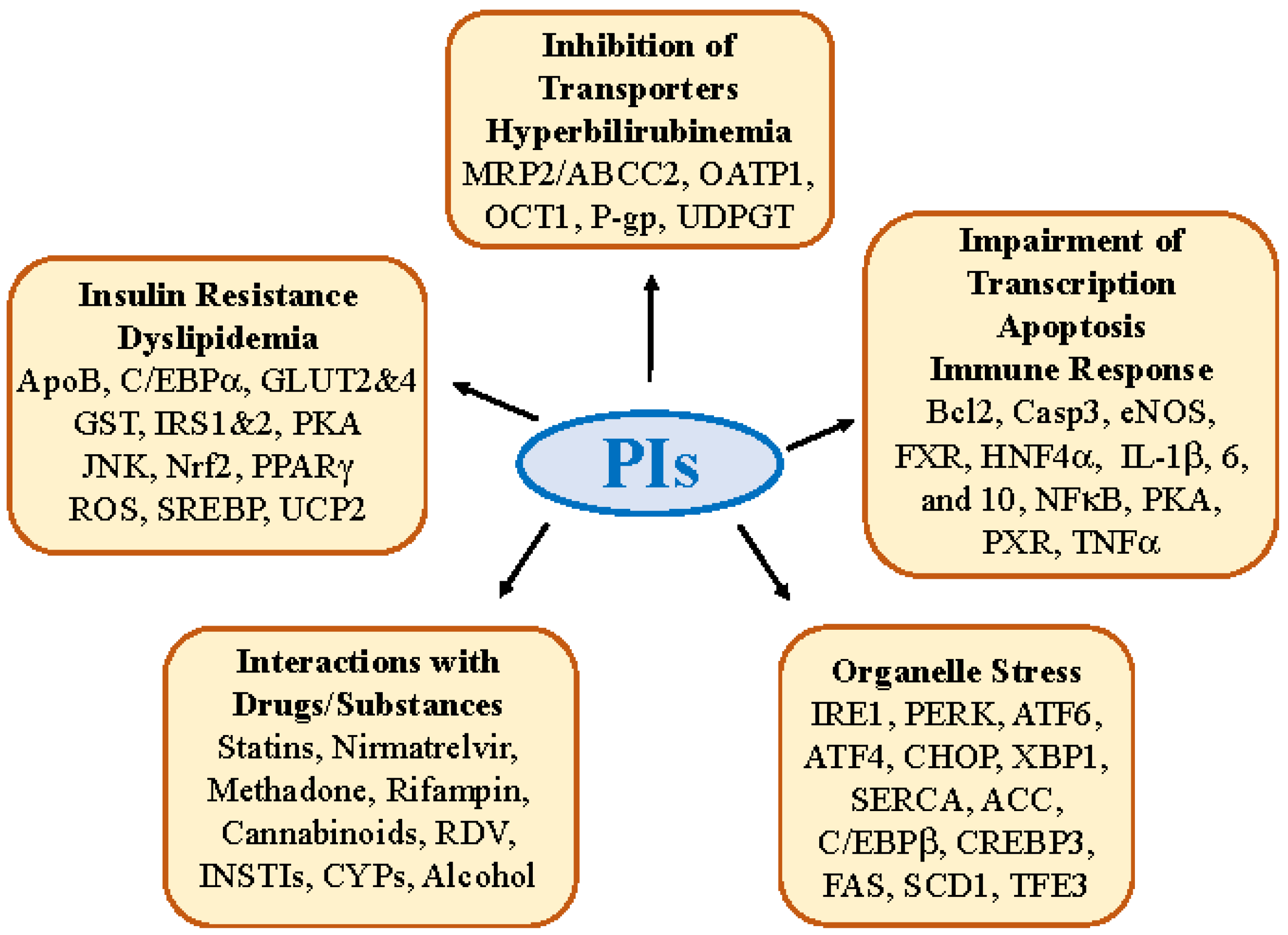
2. Molecular Factors and Pathways Involved in PI-induced Hepatotoxicity
2.1. Insulin Resistance, Dyslipidemia and Lipodystrophy
2.2. Hepatic Transporter Inhibition and Hyperbilirubinemia
2.3. Impairment in Hepatic Transcription, Apoptosis, and Immune Response
2.4. Molecular Interactions of PIs with Other Drugs/Substances
3. Antiviral PI-Induced Organelle Stress Pathways and Specific Off-Targets
3.1. Hepatic ER and Golgi Stress Response
3.2. PI-induced Organelle Stress Response
3.3. Specific Off-Targets of PIs Linking to ER-Golgi Trafficking and Dyslipidemia
4. Potential Therapeutic/Pharmaceutical Solutions for Antiviral PIs Associated with Liver Injury
4.1. Targeting Insulin Resistance, Cellular Stress and Dyslipidemia
4.2. Improving Drug Delivery and Bioavailability
4.3. Designing Safer PIs
5. Conclusive Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- HIV.gov. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. 2019. Available online: https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv/initiation-antiretroviral-therapy (accessed on 15 January 2023).
- Gandhi, R.T.; Bedimo, R.; Hoy, J.F.; Landovitz, R.J.; Smith, D.M.; Eaton, E.F.; Lehmann, C.; Springer, S.A.; Sax, P.E.; Thompson, M.A.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2022 Recommendations of the International Antiviral Society-USA Panel. JAMA 2023, 329, 63–84. [Google Scholar] [CrossRef]
- Otto, A.O.; Rivera, C.G.; Zeuli, J.D.; Temesgen, Z. Hepatotoxicity of Contemporary Antiretroviral Drugs: A Review and Evaluation of Published Clinical Data. Cells 2021, 10, 1263. [Google Scholar] [CrossRef]
- Flexner, C. HIV-protease inhibitors. N. Engl. J. Med. 1998, 338, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Marzi, M.; Vakil, M.K.; Bahmanyar, M.; Zarenezhad, E. Paxlovid: Mechanism of Action, Synthesis, and In Silico Study. Biomed. Res. Int. 2022, 2022, 7341493. [Google Scholar] [CrossRef] [PubMed]
- Casalini, G.; Giacomelli, A.; Antinori, S. Liver tests abnormalities with licensed antiviral drugs for COVID-19: A narrative review. Expert Opin. Drug Saf. 2022, 21, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Lory, P.; Combret, S.; Michot, J.; Veyrac, G.; Chouchana, L.; Grandvuillemin, A. Safety profile of the lopinavir/ritonavir combination before and during the SARS-CoV-2 pandemic. Therapie, 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Grandvuillemin, A.; Rocher, F.; Valnet-Rabier, M.B.; Drici, M.D.; Dautriche, A. French Pharmacovigilance Network. Pharmacovigilance follow-up of patients in the context of the COVID-19 pandemic. Therapie, 2023; in press. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H.; Björnsson, E.S. Drug-Induced Liver Injury—Types and Phenotypes. N. Engl. J. Med. 2019, 381, 264–273. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS Auckl. 2015, 7, 95–104. [Google Scholar] [PubMed]
- Voshavar, C. Protease Inhibitors for the Treatment of HIV/AIDS: Recent Advances and Future Challenges. Curr. Top. Med. Chem. 2019, 19, 1571–1598. [Google Scholar] [CrossRef]
- Rivera, C.G.; Otto, A.O.; Zeuli, J.D.; Temesgen, Z. Hepatotoxicity of contemporary antiretroviral drugs. Curr. Opin. HIV AIDS 2021, 16, 279–285. [Google Scholar] [CrossRef]
- Gervasoni, C.; Cattaneo, D.; Filice, C.; Galli, M. “Gruppo Italiano Studio-NASH in malattie infettive”. Drug-induced liver steatosis in patients with HIV infection. Pharmacol. Res. 2019, 145, 104267. [Google Scholar] [CrossRef]
- Kumar, S.; Rao, P.S.; Earla, R.; Kumar, A. Drug-drug interactions between anti-retroviral therapies and drugs of abuse in HIV systems. Expert Opin. Drug Metab. Toxicol. 2015, 11, 343–355. [Google Scholar] [CrossRef]
- Makarov, E.; Wang, W.; Kharbanda, K.K.; Kidambi, S.; Poluektova, L.Y.; Osna, N.A. Alcohol Metabolism Potentiates HIV-Induced Hepatotoxicity: Contribution to End-Stage Liver Disease. Biomolecules 2019, 9, 851. [Google Scholar]
- Ganesan, M.; Poluektova, L.Y.; Kharbanda, K.K.; Osna, N.A. Liver as a target of human immunodeficiency virus infection. World J. Gastroenterol. 2018, 24, 4728–4737. [Google Scholar] [CrossRef]
- van der Valk, M.; Bisschop, P.H.; Romijn, J.A.; Ackermans, M.T.; Lange, J.M.; Endert, E.; Reiss, P.; Sauerwein, H.P. Lipodystrophy in HIV-1-positive patients is associated with insulin resistance in multiple metabolic pathways. Aids 2001, 15, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Hadigan, C.; Borgonha, S.; Rabe, J.; Young, V.; Grinspoon, S. Increased rates of lipolysis among human immunodeficiency virus-infected men receiving highly active antiretroviral therapy. Metabolism 2002, 51, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Lagathu, C.; Béréziat, V.; Gorwood, J.; Fellahi, S.; Bastard, J.P.; Vigouroux, C.; Boccara, F.; Capeau, J. Metabolic complications affecting adipose tissue, lipid and glucose metabolism associated with HIV antiretroviral treatment. Expert Opin. Drug Saf. 2019, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.R.; McCandless, L.; Harrigan, P.R.; Hogg, R.S.; Bondy, G.; Iloeje, U.H.; Mukherjee, J.; Montaner, J.S. Changes in lipids over twelve months after initiating protease inhibitor therapy among persons treated for HIV/AIDS. Lipids Health Dis. 2005, 4, 4. [Google Scholar] [CrossRef]
- Koethe, J.R. Adipose Tissue in HIV Infection. Compr. Physiol. 2017, 7, 1339–1357. [Google Scholar] [PubMed]
- Mira, J.A.; Macías, J.; Girón-González, J.A.; Merino, D.; González-Serrano, M.; Jiménez-Mejías, M.E.; Caballero-Granado, F.J.; Torre-Cisneros, J.; Terrón, A.; Becker, M.I.; et al. Grupo Andaluz Para el Estudio de las Enfermedades Infecciosas (GAEI). Incidence of and risk factors for severe hepatotoxicity of nelfinavir-containing regimens among HIV-infected patients with chronic hepatitis C. J. Antimicrob. Chemother. 2006, 58, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; Behrens, G.M. HIV-therapy associated lipodystrophy: Experimental and clinical evidence for the pathogenesis and treatment. Endocr. Metab. Immune Disord. Drug Targets 2007, 7, 237–249. [Google Scholar] [CrossRef]
- Katz, E.B.; Stenbit, A.E.; Hatton, K.; DePinho, R.; Charron, M.J. Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature 1995, 377, 151–155. [Google Scholar] [CrossRef]
- Murata, H.; Hruz, P.W.; Mueckler, M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J. Biol. Chem. 2000, 275, 20251–20254. [Google Scholar] [CrossRef]
- Murata, H.; Hruz, P.W.; Mueckler, M. Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. Aids 2002, 16, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.Y. Effects of HIV protease inhibitor therapy on lipid metabolism. Prog. Lipid Res. 2003, 42, 81–92. [Google Scholar] [CrossRef]
- Hruz, P.W.; Murata, H.; Qiu, H.; Mueckler, M. Indinavir induces acute and reversible peripheral insulin resistance in rats. Diabetes 2002, 51, 937–942. [Google Scholar] [CrossRef]
- Noor, M.A.; Lo, J.C.; Mulligan, K.; Schwarz, J.M.; Halvorsen, R.A.; Schambelan, M.; Grunfeld, C. Metabolic effects of indinavir in healthy HIV-seronegative men. Aids 2001, 15, F11–F18. [Google Scholar] [CrossRef]
- Kotani, K.; Peroni, O.D.; Minokoshi, Y.; Boss, O.; Kahn, B.B. GLUT4 glucose transporter deficiency increases hepatic lipid production and peripheral lipid utilization. J. Clin. Investig. 2004, 114, 1666–1675. [Google Scholar] [CrossRef]
- Vyas, A.K.; Koster, J.C.; Tzekov, A.; Hruz, P.W. Effects of the HIV protease inhibitor ritonavir on GLUT4 knock-out mice. J. Biol. Chem. 2010, 285, 36395–36400. [Google Scholar] [CrossRef] [PubMed]
- Buettner, C.; Camacho, R.C. Hypothalamic control of hepatic glucose production and its potential role in insulin resistance. Endocrinol. Metab. Clin. N. Am. 2008, 37, 825–840. [Google Scholar] [CrossRef]
- Schütt, M.; Zhou, J.; Meier, M.; Klein, H.H. Long-term effects of HIV-1 protease inhibitors on insulin secretion and insulin signaling in INS-1 beta cells. J. Endocrinol. 2004, 183, 445–454. [Google Scholar] [CrossRef]
- Schütt, M.; Meier, M.; Meyer, M.; Klein, J.; Aries, S.P.; Klein, H.H. The HIV-1 protease inhibitor indinavir impairs insulin signalling in HepG2 hepatoma cells. Diabetologia 2000, 43, 1145–1148. [Google Scholar] [CrossRef]
- Hresko, R.C.; Hruz, P.W. HIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PLoS ONE 2011, 6, e25237. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R.; Dolci, W.; Thorens, B. Glucose sensing by the hepatoportal sensor is GLUT2-dependent: In vivo analysis in GLUT2-null mice. Diabetes 2000, 49, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef]
- Schütt, M.; Meier, M.; Jost, M.M.; Klein, H.H. The HIV protease inhibitor indinavir impairs glycogen synthesis in HepG2 hepatoma cells. Exp. Clin. Endocrinol. Diabetes 2003, 111, 16–20. [Google Scholar] [CrossRef]
- Liang, J.S.; Distler, O.; Cooper, D.A.; Jamil, H.; Deckelbaum, R.J.; Ginsberg, H.N.; Sturley, S.L. HIV protease inhibitors protect apolipoprotein B from degradation by the proteasome: A potential mechanism for protease inhibitor-induced hyperlipidemia. Nat. Med. 2001, 7, 1327–1331. [Google Scholar] [CrossRef]
- Riddle, T.M.; Kuhel, D.G.; Woollett, L.A.; Fichtenbaum, C.J.; Hui, D.Y. HIV protease inhibitor induces fatty acid and sterol biosynthesis in liver and adipose tissues due to the accumulation of activated sterol regulatory element-binding proteins in the nucleus. J. Biol. Chem. 2001, 276, 37514–37519. [Google Scholar] [CrossRef]
- Bastard, J.P.; Caron, M.; Vidal, H.; Jan, V.; Auclair, M.; Vigouroux, C.; Luboinski, J.; Laville, M.; Maachi, M.; Girard, P.M.; et al. Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet 2002, 359, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Caron-Debarle, M.; Le Dour, C.; Magré, J.; Capeau, J. Molecular mechanisms of human lipodystrophies: From adipocyte lipid droplet to oxidative stress and lipotoxicity. Int. J. Biochem. Cell Biol. 2011, 43, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Ngondi, J.L.; Oben, J.; Forkah, D.M.; Etame, L.H.; Mbanya, D. The effect of different combination therapies on oxidative stress markers in HIV infected patients in Cameroon. AIDS Res. Ther. 2006, 22, 19. [Google Scholar] [CrossRef]
- Chandra, S.; Mondal, D.; Agrawal, K.C. HIV-1 protease inhibitor induced oxidative stress suppresses glucose stimulated insulin release: Protection with thymoquinone. Exp. Biol. Med. 2009, 234, 442–453. [Google Scholar] [CrossRef]
- Kobuchi, S.; Fukushima, K.; Aoyama, H.; Ito, Y.; Sugioka, N.; Takada, K. Effects of oxidative stress on the pharmacokinetics and hepatic metabolism of atazanavir in rats. Free Radic. Res. 2013, 47, 291–300. [Google Scholar] [CrossRef]
- Ganta, K.K.; Chaubey, B. Endoplasmic reticulum stress leads to mitochondria-mediated apoptosis in cells treated with anti-HIV protease inhibitor ritonavir. Cell Biol. Toxicol. 2019, 35, 189–204. [Google Scholar] [CrossRef]
- ElZohary, L.; Weglicki, W.B.; Chmielinska, J.J.; Kramer, J.H.; Mak, I.T. Mg-supplementation attenuated lipogenic and oxidative/nitrosative gene expression caused by Combination Antiretroviral Therapy (cART) in HIV-1-transgenic rats. PLoS ONE 2019, 14, e0210107. [Google Scholar] [CrossRef]
- Wang, Y.; van der Tuin, S.; Tjeerdema, N.; van Dam, A.D.; Rensen, S.S.; Hendrikx, T.; Berbée, J.F.; Atanasovska, B.; Fu, J.; Hoekstra, M.; et al. Plasma cholesteryl ester transfer protein is predominantly derived from Kupffer cells. Hepatology 2015, 62, 1710–1722. [Google Scholar] [CrossRef]
- Wang, X.; Mu, H.; Chai, H.; Liao, D.; Yao, Q.; Chen, C. Human immunodeficiency virus protease inhibitor ritonavir inhibits cholesterol efflux from human macrophage-derived foam cells. Am. J. Pathol. 2007, 171, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Blas-Garcia, A.; Apostolova, N.V.; Esplugues, J. Oxidative Stress and Mitochondrial Impairment After Treatment with Anti-HIV Drugs: Clinical Implications. Curr. Pharm. Des. 2012, 17, 4076–4086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gorset, W.; Washington, C.B.; Blaschke, T.F.; Kroetz, D.L.; Giacomini, K.M. Interactions of HIV protease inhibitors with a human organic cation transporter in a mammalian expression system. Drug Metab. Dispos. 2000, 28, 329–334. [Google Scholar]
- Ye, Z.W.; Camus, S.; Augustijns, P.; Annaert, P. Interaction of eight HIV protease inhibitors with the canalicular efflux transporter ABCC2 (MRP2) in sandwich-cultured rat and human hepatocytes. Biopharm. Drug Dispos. 2010, 31, 178–188. [Google Scholar] [CrossRef]
- Holmstock, N.; Oorts, M.; Snoeys, J.; Annaert, P. MRP2 Inhibition by HIV Protease Inhibitors in Rat and Human Hepatocytes: A Quantitative Confocal Microscopy Study. Drug Metab. Dispos. 2018, 46, 697–703. [Google Scholar] [CrossRef] [PubMed]
- McRae, M.P.; Lowe, C.M.; Tian, X.; Bourdet, D.L.; Ho, R.H.; Leake, B.F.; Kim, R.B.; Brouwer, K.L.; Kashuba, A.D. Ritonavir, saquinavir, and efavirenz, but not nevirapine, inhibit bile acid transport in human and rat hepatocytes. J. Pharmacol. Exp. Ther. 2006, 318, 1068–1075. [Google Scholar] [CrossRef]
- De Bruyn, T.; Stieger, B.; Augustijns, P.F.; Annaert, P.P. Clearance Prediction of HIV Protease Inhibitors in Man: Role of Hepatic Uptake. J. Pharm. Sci. 2016, 105, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Annaert, P.; Ye, Z.W.; Stieger, B.; Augustijns, P. Interaction of HIV protease inhibitors with OATP1B1, 1B3, and 2B1. Xenobiotica 2010, 40, 163–176. [Google Scholar] [CrossRef]
- Shitara, Y. Clinical importance of OATP1B1 and OATP1B3 in drug-drug interactions. Drug Metab. Pharmacokinet. 2011, 26, 220–227. [Google Scholar] [CrossRef]
- Tátrai, P.; Krajcsi, P. Prediction of Drug-Induced Hyperbilirubinemia by In Vitro Testing. Pharmaceutics 2020, 12, 755. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.D.; Qin, X.; Rouster, S.D.; Yu, F.; Green, R.M.; Keshavan, P.; Feinberg, J.; Sherman, K.E. Mechanism of indinavir-induced hyperbilirubinemia. Proc. Natl. Acad. Sci. USA 2001, 98, 12671–12676. [Google Scholar] [CrossRef] [PubMed]
- Torti, C.; Lapadula, G.; Antinori, A.; Quirino, T.; Maserati, R.; Castelnuovo, F.; Maggiolo, F.; De Luca, A.; Paraninfo, G.; Antonucci, F.; et al. Hyperbilirubinemia during atazanavir treatment in 2404 patients in the Italian atazanavir expanded access program and MASTER Cohorts. Infection 2009, 37, 244–249. [Google Scholar] [CrossRef]
- Culley, C.L.; Kiang, T.K.; Gilchrist, S.E.; Ensom, M.H. Effect of the UGT1A1*28 allele on unconjugated hyperbilirubinemia in HIV-positive patients receiving Atazanavir: A systematic review. Ann. Pharmacother. 2013, 47, 561–572. [Google Scholar] [CrossRef]
- Panagopoulos, P.; Maltezos, E.; Hatzakis, A.; Paraskevis, D. Hyperbilirubinemia in atazanavir treated HIV-infected patients: The impact of the UGT1A1*28 allele. Pharmgenomics Pers. Med. 2017, 10, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Wang, A.; Ma, Y.; Li, X. Association between the UGT1A1*28 allele and hyperbilirubinemia in HIV-positive patients receiving atazanavir: A meta-analysis. Biosci. Rep. 2019, 39, BSR20182105. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chan, W.W.; Zucker, S.D. Association Between Atazanavir-Induced Hyperbilirubinemia and Cardiovascular Disease in Patients Infected with HIV. J. Am. Heart Assoc. 2020, 9, e016310. [Google Scholar] [CrossRef] [PubMed]
- Lankisch, T.O.; Behrens, G.; Ehmer, U.; Möbius, U.; Rockstroh, J.; Wehmeier, M.; Kalthoff, S.; Freiberg, N.; Manns, M.P.; Schmidt, R.E.; et al. Gilbert’s syndrome and hyperbilirubinemia in protease inhibitor therapy—An extended haplotype of genetic variants increases risk in indinavir treatment. J. Hepatol. 2009, 50, 1010–1018. [Google Scholar] [CrossRef]
- Mencarelli, A.; Cipriani, S.; Renga, B.; Francisci, D.; Palladino, G.; Distrutti, E.; Baldelli, F.; Fiorucci, S. The bile acid sensor FXR protects against dyslipidemia and aortic plaques development induced by the HIV protease inhibitor ritonavir in mice. PLoS ONE 2010, 5, e13238. [Google Scholar] [CrossRef]
- Shehu, A.I.; Lu, J.; Wang, P.; Zhu, J.; Wang, Y.; Yang, D.; McMahon, D.; Xie, W.; Gonzalez, F.J.; Ma, X. Pregnane X receptor activation potentiates ritonavir hepatotoxicity. J. Clin. Investig. 2019, 129, 2898–2903. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, L.; Kong, W.; Xie, Q.; Li, P.; Liu, X.; Zhang, J.; Liu, M.; Wang, Z.; Zhu, L.; et al. PXR activation impairs hepatic glucose metabolism partly via inhibiting the HNF4α-GLUT2 pathway. Acta Pharm. Sin. B 2022, 12, 2391–2405. [Google Scholar] [CrossRef]
- Wang, X.; Yu, Y.; Wang, P.; Yang, K.; Wang, Y.; Yan, L.; Zhong, X.B.; Zhang, L. Long Noncoding RNAs Hepatocyte Nuclear Factor 4A Antisense RNA 1 and Hepatocyte Nuclear Factor 1A Antisense RNA 1 Are Involved in Ritonavir-Induced Cytotoxicity in Hepatoma Cells. Drug Metab. Dispos. 2022, 50, 704–715. [Google Scholar] [CrossRef]
- Kuang, C.C.; Wang, Y.; Hu, P.C.; Gao, F.F.; Bu, L.; Wen, X.M.; Xiang, Q.M.; Song, H.; Li, Q.; Wei, L.; et al. Ritonavir-induced hepatotoxicity and ultrastructural changes of hepatocytes. Ultrastruct. Pathol. 2014, 38, 329–334. [Google Scholar] [CrossRef]
- Sun, L.; Niu, L.; Zhu, X.; Hao, J.; Wang, P.; Wang, H. Antitumour effects of a protease inhibitor, nelfinavir, in hepatocellular carcinoma cancer cells. J. Chemother. 2012, 24, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.A.; Malik, M.Y.; Azmi, L.; Shukla, I.; Naseem, Z.; Rao, C.; Agarwal, N.K. Formononetin and biochanin A protects against ritonavir induced hepatotoxicity via modulation of NfκB/pAkt signaling molecules. Life Sci. 2018, 213, 174–182. [Google Scholar]
- Gruevska, A.; Moragrega, Á.B.; Cossarizza, A.; Esplugues, J.V.; Blas-García, A.; Apostolova, N. Apoptosis of Hepatocytes: Relevance for HIV-Infected Patients under Treatment. Cells 2021, 10, 410. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jarujaron, S.; Wu, X.; Sun, L.; Zha, W.; Liang, G.; Wang, X.; Gurley, E.C.; Studer, E.J.; Hylemon, P.B.; et al. HIV protease inhibitor lopinavir-induced TNF-alpha and IL-6 expression is coupled to the unfolded protein response and ERK signaling pathways in macrophages. Biochem. Pharmacol. 2009, 78, 70–77. [Google Scholar] [CrossRef]
- Gallego-Escuredo, J.M.; Gutierrez, M.D.M.; Diaz-Delfin, J.; Domingo, J.C.; Mateo, M.G.; Domingo, P.; Giralt, M.; Villarroya, F. Differential effects of efavirenz and lopinavir/ritonavir on human adipocyte differentiation, gene expression and release of adipokines and pro-inflammatory cytokines. Curr. HIV Res. 2010, 8, 545–553. [Google Scholar] [CrossRef]
- Sulkowski, M.S. Drug-induced liver injury associated with antiretroviral therapy that includes HIV-1 protease inhibitors. Clin. Infect. Dis. 2004, 38 (Suppl. 2), S90–S97. [Google Scholar] [CrossRef]
- Dick, T.B.; Lindberg, L.S.; Ramirez, D.D.; Charlton, M.R. A clinician’s guide to drug-drug interactions with direct-acting antiviral agents for the treatment of hepatitis C viral infection. Hepatology 2016, 63, 634–643. [Google Scholar] [CrossRef]
- Kosel, B.W.; Aweeka, F.T.; Benowitz, N.L.; Shade, S.B.; Hilton, J.F.; Lizak, P.S.; Abrams, D.I. The effects of cannabinoids on the pharmacokinetics of indinavir and nelfinavir. Aids 2002, 16, 543–550. [Google Scholar] [CrossRef]
- McCain, J.D.; Chascsa, D.M. Special Considerations in the Management of HIV and Viral Hepatitis Coinfections in Liver Transplantation. Hepatic Med. 2022, 14, 27–36. [Google Scholar] [CrossRef]
- Hodge, D.; Hodel, E.M.; Hughes, E.; Hazenberg, P.; Grañana Castillo, S.; Gibbons, S.; Wang, D.; Marra, F.; Marzolini, C.; Back, D.; et al. Prevalence of Potentially Clinically Significant Drug-Drug Interactions With Antiretrovirals Against HIV Over Three Decades: A Systematic Review of the Literature. JAIDS J. Acquir. Immune Defic. Syndr. 2023, 92, 97–105. [Google Scholar] [CrossRef]
- Basoulis, D.; Mastrogianni, E.; Voutsinas, P.M.; Psichogiou, M. HIV and COVID-19 Co-Infection: Epidemiology, Clinical Characteristics, and Treatment. Viruses 2023, 15, 577. [Google Scholar] [CrossRef]
- Jülg, B.; Bogner, J.R.; Goebel, F.D. Severe hepatotoxicity associated with the combination of enfuvirtide and tipranavir/ritonavir: Case report. Aids 2006, 20, 1563. [Google Scholar] [CrossRef]
- Chan-Tack, K.M.; Struble, K.A.; Birnkrant, D.B. Intracranial hemorrhage and liver-associated deaths associated with tipranavir/ritonavir: Review of cases from the FDA’s Adverse Event Reporting System. AIDS Patient Care STDS 2008, 22, 843–850. [Google Scholar] [CrossRef]
- Li, F.; Wang, L.; Guo, G.L.; Ma, X. Metabolism-mediated drug interactions associated with ritonavir-boosted tipranavir in mice. Drug Metab. Dispos. 2010, 38, 871–878. [Google Scholar] [CrossRef]
- Hulskotte, E.G.; Feng, H.P.; Xuan, F.; van Zutven, M.G.; Treitel, M.A.; Hughes, E.A.; O’Mara, E.; Youngberg, S.P.; Wagner, J.A.; Butterton, J.R. Pharmacokinetic interactions between the hepatitis C virus protease inhibitor boceprevir and ritonavir-boosted HIV-1 protease inhibitors atazanavir, darunavir, and lopinavir. Clin. Infect. Dis. 2013, 56, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Vadlapatla, R.K.; Patel, M.; Paturi, D.K.; Pal, D.; Mitra, A.K. Clinically relevant drug-drug interactions between antiretrovirals and antifungals. Expert Opin. Drug Metab. Toxicol. 2014, 10, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Yew, W.W. Clinically significant interactions with drugs used in the treatment of tuberculosis. Drug Saf. 2002, 25, 111–133. [Google Scholar] [CrossRef]
- Niemi, M.; Backman, J.T.; Fromm, M.F.; Neuvonen, P.J.; Kivistö, K.T. Pharmacokinetic interactions with rifampicin: Clinical relevance. Clin. Pharmacokinet. 2003, 42, 819–850. [Google Scholar] [CrossRef]
- Schmitt, C.; Riek, M.; Winters, K.; Schutz, M.; Grange, S. Unexpected Hepatotoxicity of Rifampin and Saquinavir/Ritonavir in Healthy Male Volunteers. Arch. Drug Inf. 2009, 2, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.M.; Pithavala, Y.K.; Vourvahis, M.; Chen, J. A literature review of liver function test elevations in rifampin drug-drug interaction studies. Clin. Transl. Sci. 2022, 15, 1561–1580. [Google Scholar] [CrossRef]
- Janneh, O.; Anwar, T.; Jungbauer, C.; Kopp, S.; Khoo, S.H.; Back, D.J.; Chiba, P. P-glycoprotein, multidrug resistance-associated proteins and human organic anion transporting polypeptide influence the intracellular accumulation of atazanavir. Antivir Ther. 2009, 14, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Elsby, R.; Coghlan, H.; Edgerton, J.; Hodgson, D.; Outteridge, S.; Atkinson, H. Mechanistic in vitro studies indicate that the clinical drug-drug interactions between protease inhibitors and rosuvastatin are driven by inhibition of intestinal BCRP and hepatic OATP1B1 with minimal contribution from OATP1B3, NTCP and OAT3. Pharmacol. Res. Perspect. 2023, 11, e01060. [Google Scholar] [CrossRef] [PubMed]
- Bachmeier, C.J.; Spitzenberger, T.J.; Elmquist, W.F.; Miller, D.W. Quantitative assessment of HIV-1 protease inhibitor interactions with drug efflux transporters in the blood-brain barrier. Pharm. Res. 2005, 22, 1259–1268. [Google Scholar] [CrossRef]
- Chauvin, B.; Drouot, S.; Barrail-Tran, A.; Taburet, A.M. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin. Pharmacokinet. 2013, 52, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Leowattana, W. Angiotensin-converting enzyme 2 receptors, chronic liver diseases, common medications, and clinical outcomes in coronavirus disease 2019 patients. World J. Virol. 2021, 10, 86–96. [Google Scholar] [CrossRef]
- Grando, M.; Balbi, M.; Zeppieri, M. COVID-19-induced liver injury in adult patients: A brief overview. World J. Virol. 2022, 11, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Zhou, J.; Zhu, H.; Chen, Y.; Lu, Y.; Zhang, T.; Yu, H.; Wang, L.; Xu, H.; Wang, Z.; et al. The feasibility, safety, and efficacy of Paxlovid treatment in SARS-CoV-2-infected children aged 6-14 years: A cohort study. Ann. Transl. Med. 2022, 10, 619. [Google Scholar] [CrossRef]
- Marzolini, C.; Kuritzkes, D.R.; Marra, F.; Boyle, A.; Gibbons, S.; Flexner, C.; Pozniak, A.; Boffito, M.; Waters, L.; Burger, D.; et al. Recommendations for the Management of Drug-Drug Interactions Between the COVID-19 Antiviral Nirmatrelvir/Ritonavir (Paxlovid) and Comedications. Clin. Pharmacol. Ther. 2022, 112, 1191–1200. [Google Scholar] [CrossRef]
- Abrams, D.I.; Hilton, J.F.; Leiser, R.J.; Shade, S.B.; Elbeik, T.A.; Aweeka, F.T.; Benowitz, N.L.; Bredt, B.M.; Kosel, B.; Aberg, J.A.; et al. Short-term effects of cannabinoids in patients with HIV-1 infection: A randomized, placebo-controlled clinical trial. Ann. Intern. Med. 2003, 139, 258–266. [Google Scholar] [CrossRef]
- Kharasch, E.D.; Bedynek, P.S.; Walker, A.; Whittington, D.; Hoffer, C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics: II. Ritonavir effects on CYP3A and P-glycoprotein activities. Clin. Pharmacol. Ther. 2008, 84, 506–512. [Google Scholar] [CrossRef]
- Foster, A.J.; Prime, L.H.; Gustafsson, F.; Temesi, D.G.; Isin, E.M.; Midlöv, J.; Castagnoli, N., Jr.; Kenna, J.G. Bioactivation of the cannabinoid receptor antagonist rimonabant to a cytotoxic iminium ion metabolite. Chem. Res. Toxicol. 2013, 26, 124–135. [Google Scholar] [CrossRef]
- Campbell, S.D.; Gadel, S.; Friedel, C.; Crafford, A.; Regina, K.J.; Kharasch, E.D. Influence of HIV antiretrovirals on methadone N-demethylation and transport. Biochem. Pharmacol. 2015, 95, 115–125. [Google Scholar] [CrossRef]
- Svedmyr, A.; Hack, H.; Anderson, B.J. Interactions of the protease inhibitor, ritonavir, with common anesthesia drugs. Paediatr. Anaesth. 2022, 32, 1091–1099. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2023, 77, 619–639. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N.; Lau, M.Y.; Kao, E.; Petrovic, L.M.; Lee, A.S. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology 2011, 54, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Ji, C. Advances and New Concepts in Alcohol-Induced Organelle Stress, Unfolded Protein Responses and Organ Damage. Biomolecules 2015, 5, 1099–1121. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef]
- Sasaki, K.; Yoshida, H. Golgi stress response and organelle zones. FEBS Lett. 2019, 593, 2330–2340. [Google Scholar] [CrossRef]
- Flint, O.P.; Noor, M.A.; Hruz, P.W.; Hylemon, P.B.; Yarasheski, K.; Kotler, D.P.; Parker, R.A.; Bellamine, A. The role of protease inhibitors in the pathogenesis of HIV-associated lipodystrophy: Cellular mechanisms and clinical implications. Toxicol. Pathol. 2009, 37, 65–77. [Google Scholar] [CrossRef]
- Kao, E.; Shinohara, M.; Feng, M.; Lau, M.Y.; Ji, C. Human immunodeficiency virus protease inhibitors modulate Ca2+ homeostasis and potentiate alcoholic stress and injury in mice and primary mouse and human hepatocytes. Hepatology 2012, 56, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Wu, X.; Gurley, E.C.; Kennedy, E.; Hylemon, P.B.; Pandak, W.M.; Sanyal, A.J.; Zhou, H. The role of CCAAT enhancer-binding protein homologous protein in human immunodeficiency virus protease-inhibitor-induced hepatic lipotoxicity in mice. Hepatology 2013, 57, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Zha, B.S.; Wan, X.; Zhang, X.; Zha, W.; Zhou, J.; Wabitsch, M.; Wang, G.; Lyall, V.; Hylemon, P.B.; Zhou, H. HIV protease inhibitors disrupt lipid metabolism by activating endoplasmic reticulum stress and inhibiting autophagy activity in adipocytes. PLoS ONE 2013, 8, e59514. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Peng, K.; Zhou, H. HIV protease inhibitors in gut barrier dysfunction and liver injury. Curr. Opin. Pharmacol. 2014, 19, 61–66. [Google Scholar] [CrossRef]
- Han, H.; He, Y.; Hu, J.; Lau, R.; Lee, H.; Ji, C. Disrupted ER-to-Golgi Trafficking Underlies Anti-HIV Drugs and Alcohol-Induced Cellular Stress and Hepatic Injury. Hepatol. Commun. 2017, 1, 122–139. [Google Scholar] [CrossRef]
- Han, H.; He, Y.; Johnson, H.; Mishra, P.; Lee, H.; Ji, C. Protective Effects of Facilitated Removal of Blood Alcohol and Acetaldehyde Against Liver Injury in Animal Models Fed Alcohol and Anti-HIV Drugs. Alcohol. Clin. Exp. Res. 2019, 43, 1091–1102. [Google Scholar] [CrossRef]
- Hu, J.; Han, H.; Lau, M.Y.; Lee, H.; MacVeigh-Aloni, M.; Ji, C. Effects of combined alcohol and anti-HIV drugs on cellular stress responses in primary hepatocytes and hepatic stellate and kupffer cells. Alcohol. Clin. Exp. Res. 2015, 39, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Alegre, F.; Funes, H.A.; Victor, V.M.; Barrachina, M.D.; Blas-Garcia, A.; Esplugues, J.V. ER stress in human hepatic cells treated with Efavirenz: Mitochondria again. J. Hepatol. 2013, 59, 780–789. [Google Scholar] [CrossRef]
- El Hoss, S.; Bahr, G.M.; Echtay, K.S. Lopimune-induced mitochondrial toxicity is attenuated by increased uncou-pling protein-2 level in treated mouse hepatocytes. Biochem. J. 2015, 468, 401–407. [Google Scholar] [CrossRef]
- De Gassart, A.; Bujisic, B.; Zaffalon, L.; Decosterd, L.A.; Di Micco, A.; Frera, G.; Tallant, R.; Martinon, F. An inhibitor of HIV-1 protease modulates constitutive eIF2α dephosphorylation to trigger a specific integrated stress response. Proc. Natl. Acad. Sci. USA 2016, 113, E117–E126. [Google Scholar] [CrossRef]
- Khalatbari, A.; Mishra, P.; Han, H.; He, Y.; MacVeigh-Aloni, M.; Ji, C. Ritonavir and Lopinavir Suppress RCE1 and CAAX Rab Proteins Sensitizing the Liver to Organelle Stress and Injury. Hepatol. Commun. 2020, 4, 932–944. [Google Scholar] [CrossRef]
- Khalatbari, A.; Aghazadeh, Z.; Ji, C. Adverse Effects of Anti-Covid-19 Drug Candidates and Alcohol on Cellular Stress Responses of Hepatocytes. Hepatol. Commun. 2022, 6, 1262–1277. [Google Scholar] [CrossRef]
- Duden, R. ER-to-Golgi transport: COP I and COP II function (Review). Mol. Membr. Biol. 2003, 20, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef]
- Peotter, J.; Kasberg, W.; Pustova, I.; Audhya, A. COPII-mediated trafficking at the ER/ERGIC interface. Traffic 2019, 20, 491–503. [Google Scholar] [CrossRef]
- Ji, C. Dissecting the Role of Disturbed ER-Golgi Trafficking in Antivirals and Alcohol Abuse-Induced Pathogenesis of Liver Disorders. J. Drug Abus. 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Mesmin, B.; Kovacs, D.; D’Angelo, G. Lipid exchange and signaling at ER-Golgi contact sites. Curr. Opin. Cell Biol. 2019, 57, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.; Auclair, M.; Sterlingot, H.; Kornprobst, M.; Capeau, J. Some HIV protease inhibitors alter lamin A/C maturation and stability, SREBP-1 nuclear localization and adipocyte differentiation. Aids 2003, 17, 2437–2444. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Hudon, S.E.; Farber, E.A.; Chang, S.Y.; Hrycyna, C.A.; Young, S.G.; Fong, L.G. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc. Natl. Acad. Sci. USA 2007, 104, 13432–13437. [Google Scholar] [CrossRef]
- Clark, K.M.; Jenkins, J.L.; Fedoriw, N.; Dumont, M.E. Human CaaX protease ZMPSTE24 expressed in yeast: Structure and inhibition by HIV protease inhibitors. Protein Sci. 2017, 26, 242–257. [Google Scholar] [CrossRef]
- Coffinier, C.; Hudon, S.E.; Lee, R.; Farber, E.A.; Nobumori, C.; Miner, J.H.; Andres, D.A.; Spielmann, H.P.; Hrycyna, C.A.; Fong, L.G.; et al. A potent HIV protease inhibitor, darunavir, does not inhibit ZMPSTE24 or lead to an accumulation of farnesyl-prelamin A in cells. J. Biol. Chem. 2008, 283, 9797–9804. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, B.W.; Mehrtash, A.B.; Broshar, C.L.; Runnebohm, A.M.; Snow, B.J.; Scanameo, L.N.; Hochstrasser, M.; Rubenstein, E.M. Endoplasmic reticulum stress differentially inhibits endoplasmic reticulum and inner nuclear membrane protein quality control degradation pathways. J. Biol. Chem. 2019, 294, 19814–19830. [Google Scholar] [CrossRef]
- Runnebohm, A.M.; Richards, K.A.; Irelan, C.B.; Turk, S.M.; Vitali, H.E.; Indovina, C.J.; Rubenstein, E.M. Overlapping function of Hrd1 and Ste24 in translocon quality control provides robust channel surveillance. J. Biol. Chem. 2020, 295, 16113–16120. [Google Scholar] [CrossRef]
- Barrowman, J.; Michaelis, S. ZMPSTE24, an integral membrane zinc metalloprotease with a connection to progeroid disorders. Biol. Chem. 2009, 390, 761–773. [Google Scholar] [CrossRef]
- Afonso, P.; Auclair, M.; Boccara, F.; Vantyghem, M.C.; Katlama, C.; Capeau, J.; Vigouroux, C.; Caron-Debarle, M. LMNA mutations resulting in lipodystrophy and HIV protease inhibitors trigger vascular smooth muscle cell senescence and calcification: Role of ZMPSTE24 downregulation. Atherosclerosis 2016, 245, 200–211. [Google Scholar] [CrossRef]
- Hampton, S.E.; Dore, T.M.; Schmidt, W.K. Rce1: Mechanism and inhibition. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 157–174. [Google Scholar] [CrossRef]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Goud, B.; Liu, S.; Storrie, B. Rab proteins as major determinants of the Golgi complex structure. Small GTPases 2018, 9, 66–75. [Google Scholar] [CrossRef]
- Pulido, M.R.; Diaz-Ruiz, A.; Jiménez-Gómez, Y.; Garcia-Navarro, S.; Gracia-Navarro, F.; Tinahones, F.; López-Miranda, J.; Frühbeck, G.; Vázquez-Martínez, R.; Malagón, M.M. Rab18 dynamics in adipocytes in relation to lipogenesis, lipolysis and obesity. PLoS ONE 2011, 6, e22931. [Google Scholar] [CrossRef] [PubMed]
- Rasineni, K.; McVicker, B.L.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Rab GTPases associate with isolated lipid droplets (LDs) and show altered content after ethanol administration: Potential role in alcohol-impaired LD metabolism. Alcohol. Clin. Exp. Res. 2014, 38, 327–335. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.S. Rab proteins as regulators of lipid droplet formation and lipolysis. Cell Biol. Int. 2016, 40, 1026–1032. [Google Scholar] [CrossRef]
- Chandra, S.; Murthy, S.N.; Mondal, D.; Agrawal, K.C. Therapeutic effects of Nigella sativa on chronic HAART-induced hyperinsulinemia in rats. Can. J. Physiol. Pharmacol. 2009, 87, 300–309. [Google Scholar] [CrossRef]
- Khazdair, M.R.; Ghafari, S.; Sadeghi, M. Possible therapeutic effects of Nigella sativa and its thymoquinone on COVID-19. Pharm. Biol. 2021, 59, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Tania, M.; Asad, A.; Li, T.; Islam, M.S.; Islam, S.B.; Hossen, M.M.; Bhuiyan, M.R.; Khan, M.A. Thymoquinone against infectious diseases: Perspectives in recent pandemics and future therapeutics. Iran J. Basic Med. Sci. 2021, 24, 1014–1022. [Google Scholar]
- Badary, O.A.; Hamza, M.S.; Tikamdas, R. Thymoquinone: A Promising Natural Compound with Potential Benefits for COVID-19 Prevention and Cure. Drug Des. Dev. Ther. 2021, 15, 1819–1833. [Google Scholar] [CrossRef]
- Nzuza, S.; Zondi, S.; Owira, P.M.O. Naringin prevents HIV-1 protease inhibitors-induced metabolic complications in vivo. PLoS ONE 2017, 12, e0183355. [Google Scholar] [CrossRef]
- Berginc, K.; Trontelj, J.; Kristl, A. The influence of aged garlic extract on the uptake of saquinavir and darunavir into HepG2 cells and rat liver slices. Drug Metab. Pharmacokinet. 2010, 25, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Oluwafeyisetan, A.; Olubunmi, A.; Peter, O. Naringin Ameliorates HIV-1 Nucleoside Reverse Transcriptase Inhibitors-Induced Mitochondrial Toxicity. Curr. HIV Res. 2016, 14, 506–516. [Google Scholar] [CrossRef]
- Su, Q.J.; Lu, Z.Z.; Deng, Q.Y.; Wei, B.M. Alcoholic Extract of Lotus Leaves Improves Lipid Profile in Rats with HIV Protease Inhibitor-induced Dyslipidaemia. West Indian Med. J. 2015, 64, 195–200. [Google Scholar]
- Ma, Z.; Zhang, D.; Sun, J.; Zhang, Q.; Qiao, Y.; Zhu, Y.; Niu, J.; Ren, Q.; Zhou, L.; Wen, A.; et al. Formononetin Inhibits Hepatic I/R-Induced Injury through Regulating PHB2/PINK1/Parkin Pathway. Oxid Med. Cell Longev. 2022, 2022, 6481192. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.R.; Karimi-Sales, E. Molecular mechanisms of protective roles of isoflavones against chemicals-induced liver injuries. Chem. Biol. Interact. 2020, 329, 109213. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.K. Flavonoid compounds of buah merah (Pandanus conoideus Lamk) as a potent SARS-CoV-2 main protease inhibitor: In silico approach. Future J. Pharm. Sci. 2021, 7, 158. [Google Scholar] [CrossRef]
- Hu, T.; Wang, J.; Li, W.; Liu, M.; Han, N.; Yuan, M.; Du, L.; Tang, H. Endoplasmic Reticulum Stress in Hepatitis B Virus and Hepatitis C Virus Infection. Viruses 2022, 14, 2630. [Google Scholar] [CrossRef] [PubMed]
- Aoe, T. Pathological Aspects of COVID-19 as a Conformational Disease and the Use of Pharmacological Chaperones as a Potential Therapeutic Strategy. Front. Pharmacol. 2020, 11, 1095. [Google Scholar] [CrossRef]
- Mahameed, M.; Boukeileh, S.; Obiedat, A.; Darawshi, O.; Dipta, P.; Rimon, A.; McLennan, G.; Fassler, R.; Reichmann, D.; Karni, R.; et al. Pharmacological induction of selective endoplasmic reticulum retention as a strategy for cancer therapy. Nat. Commun. 2020, 11, 1304. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Nissar, A.U.; Sharma, L.; Mudasir, M.A.; Nazir, L.A.; Umar, S.A.; Sharma, P.R.; Vishwakarma, R.A.; Tasduq, S.A. Chemical chaperone 4-phenyl butyric acid (4-PBA) reduces hepatocellular lipid accumulation and lipotoxicity through induction of autophagy. J. Lipid Res. 2017, 58, 1855–1868. [Google Scholar] [CrossRef]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef]
- Kusaczuk, M. Tauroursodeoxycholate-Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Komoike, Y. Experimental Evidence Shows Salubrinal, an eIF2α Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage. Int. J. Mol. Sci. 2015, 16, 16275–16287. [Google Scholar] [CrossRef] [PubMed]
- Vierling, P.; Greiner, J. Prodrugs of HIV protease inhibitors. Curr. Pharm. Des. 2003, 9, 1755–1770. [Google Scholar] [CrossRef]
- Agarwal, S.; Boddu, S.H.; Jain, R.; Samanta, S.; Pal, D.; Mitra, A.K. Peptide prodrugs: Improved oral absorption of lopinavir, a HIV protease inhibitor. Int. J. Pharm. 2008, 359, 7–14. [Google Scholar] [CrossRef]
- Subbaiah, M.A.M.; Mandlekar, S.; Desikan, S.; Ramar, T.; Subramani, L.; Annadurai, M.; Desai, S.D.; Sinha, S.; Jenkins, S.M.; Krystal, M.R.; et al. Design, Synthesis, and Pharmacokinetic Evaluation of Phosphate and Amino Acid Ester Prodrugs for Improving the Oral Bioavailability of the HIV-1 Protease Inhibitor Atazanavir. J. Med. Chem. 2019, 62, 3553–3574. [Google Scholar] [CrossRef]
- Subbaiah, M.A.M.; Ramar, T.; Subramani, L.; Desai, S.D.; Sinha, S.; Mandlekar, S.; Jenkins, S.M.; Krystal, M.R.; Subramanian, M.; Sridhar, S.; et al. (Carbonyl)oxyalkyl linker-based amino acid prodrugs of the HIV-1 protease inhibitor atazanavir that enhance oral bioavailability and plasma trough concentration. Eur. J. Med. Chem. 2020, 207, 112749. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Sharma, J.M.; Jain, A.K.; Mahajan, R.R. Surface-stabilized lopinavir nanoparticles enhance oral bioavailability without coadministration of ritonavir. Nanomedicine 2013, 8, 1639–1655. [Google Scholar] [CrossRef]
- Pham, K.; Li, D.; Guo, S.; Penzak, S.; Dong, X. Development and in vivo evaluation of child-friendly lopinavir/ritonavir pediatric granules utilizing novel in situ self-assembly nanoparticles. J. Control. Release 2016, 226, 88–97. [Google Scholar] [CrossRef]
- Wong, H.L.; Chattopadhyay, N.; Wu, X.Y.; Bendayan, R. Nanotechnology applications for improved delivery of antiretroviral drugs to the brain. Adv. Drug Deliv. Rev. 2010, 62, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.D.; Roy, I.; Xu, G.; Yong, K.T.; Ding, H.; Aalinkeel, R.; Reynolds, J.; Sykes, D.; Nair, B.B.; Lin, E.Y.; et al. Enhancing the delivery of anti retroviral drug “Saquinavir” across the blood brain barrier using nanoparticles. Curr. HIV Res. 2010, 8, 396–404. [Google Scholar] [CrossRef]
- Dou, H.; Destache, C.J.; Morehead, J.R.; Mosley, R.L.; Boska, M.D.; Kingsley, J.; Gorantla, S.; Poluektova, L.; Nelson, J.A.; Chaubal, M.; et al. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood 2006, 108, 2827–2835. [Google Scholar] [CrossRef]
- Nowacek, A.S.; McMillan, J.; Miller, R.; Anderson, A.; Rabinow, B.; Gendelman, H.E. Nanoformulated antiretroviral drug combinations extend drug release and antiretroviral responses in HIV-1-infected macrophages: Implications for neuroAIDS therapeutics. J. Neuroimmune Pharmacol. 2010, 5, 592–601. [Google Scholar] [CrossRef]
- Duan, J.; Freeling, J.P.; Koehn, J.; Shu, C.; Ho, R.J. Evaluation of atazanavir and darunavir interactions with lipids for developing pH-responsive anti-HIV drug combination nanoparticles. J. Pharm. Sci. 2014, 103, 2520–2529. [Google Scholar] [CrossRef] [PubMed]
- Kurd, M.; Sadegh Malvajerd, S.; Rezaee, S.; Hamidi, M.; Derakhshandeh, K. Oral delivery of indinavir using mPEG-PCL nanoparticles: Preparation, optimization, cellular uptake, transport and pharmacokinetic evaluation. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2123–2133. [Google Scholar] [CrossRef]
- Singh, G.; Pai, R.S. Atazanavir-loaded Eudragit RL 100 nanoparticles to improve oral bioavailability: Optimization and in vitro/in vivo appraisal. Drug Deliv. 2016, 23, 532–539. [Google Scholar] [CrossRef]
- Augustine, R.; Ashkenazi, D.L.; Arzi, R.S.; Zlobin, V.; Shofti, R.; Sosnik, A. Nanoparticle-in-microparticle oral drug delivery system of a clinically relevant darunavir/ritonavir antiretroviral combination. Acta Biomater. 2018, 74, 344–359. [Google Scholar] [CrossRef]
- Schmidt, W.K.; Tam, A.; Fujimura-Kamada, K.; Michaelis, S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 11175–11180. [Google Scholar] [CrossRef] [PubMed]
- Pryor, E.E., Jr.; Horanyi, P.S.; Clark, K.M.; Fedoriw, N.; Connelly, S.M.; Koszelak-Rosenblum, M.; Zhu, G.; Malkowski, M.G.; Wiener, M.C.; Dumont, M.E. Structure of the integral membrane protein CAAX protease Ste24p. Science 2013, 339, 1600–1604. [Google Scholar] [CrossRef]
- Mehmood, S.; Marcoux, J.; Gault, J.; Quigley, A.; Michaelis, S.; Young, S.; Carpenter, L.; Robinson, C. Mass spectrometry captures off-target drug binding and provides mechanistic insights into the human metalloprotease ZMPSTE24. Nat. Chem. 2016, 8, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Goblirsch, B.R.; Wiener, M.C. Ste24: An Integral Membrane Protein Zinc Metalloprotease with Provocative Structure and Emergent Biology. J. Mol. Biol. 2020, 432, 5079–5090. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Chapsal, B.D.; Weber, I.T.; Mitsuya, H. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res 2008, 41, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Weber, I.T.; Mitsuya, H. Beyond darunavir: Recent development of next generation HIV-1 protease inhibitors to combat drug resistance. Chem. Commun. 2022, 58, 11762–11782. [Google Scholar] [CrossRef]
- La Monica, G.; Lauria, A.; Bono, A.; Martorana, A. Off-Target-Based Design of Selective HIV-1 PROTEASE Inhibitors. Int. J. Mol. Sci. 2021, 22, 6070. [Google Scholar] [CrossRef] [PubMed]
- Yoosefian, M.; Moghani, M.Z.; Juan, A. In silico evaluation of atazanavir as a potential HIV main protease inhibitor and its comparison with new designed analogs. Comput. Biol. Med. 2022, 145, 105523. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Drug Names | Molecular Factors and Pathways | Pathological Consequences |
|---|---|---|
| IDV, LPV, NFV, RTV, SQV | GLUT4, IRS1&2, GLUT2, Insulin Signaling | |
| AKT/PKA, PKCε, JNK1 ApoB, C/EBPα, PPARγ, SREBP | Insulin Resistance, Dyslipidemia | |
| ATV, NFV, RTV | ROS, UCP2, CYP450, Nrf2, HO1, GST, SREBP | |
| IDV, NFV, RTV, SQV RTV, SQV ATV, IDV | OCT1 MRP2/ABCC2, OATP1B3 UDPGT1A1, UDPGT1A3, UDPGT1A7 | Dysfunction of Transporters, Hyperbilirubinemia |
| RTV, LPV | FXR, PXR, SREBP, HNF4α, CYP3A4, GLUT2 G0/G1 Arrest, NFκB/Akt Signaling, BAX, BCL2 Caspase 3&8, TNFα, IL-1β, IL-6, IL-10, eNOS | Lipid Accumulation, Cell Death, Immune Dysfunction |
| RTV, Tipranavir IDV, NFV, Cannabinoids, Alcohol, ATV, DRV, LPV, Azoles SQV, RTV, Rifampicin ETV, RTV, Cobicistat, Statins, Telaprevir RTV, DRV, LPV, MPV, NMV, Amoxicillin, Interferon, Ribavirin | CYP3A | |
| CYP3A4 | ||
| CYP45014DM | ||
| CYP3A4 OATP1B1, P-gp, CYP3A4, CYP3A5 | Elevation of ALT and AST, Biliary and Hepatic Injuries | |
| GST, ACE2, CYP450 | ||
| APV, LPV, RTV, DRV, DEX, RDV, EFV, DTG, NFV, Alcohol | UPR, IRE1, ATF6, PERK, CHOP, ER Stress C/EBPβ, CREBP3, TFE3, Rab Proteins Golgi Stress, SREBP, ACC, FAS, SCD1 | Cell Death, Inflammation, Fatty Liver, Liver Fibrosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, C. Molecular Factors and Pathways of Hepatotoxicity Associated with HIV/SARS-CoV-2 Protease Inhibitors. Int. J. Mol. Sci. 2023, 24, 7938. https://doi.org/10.3390/ijms24097938
Ji C. Molecular Factors and Pathways of Hepatotoxicity Associated with HIV/SARS-CoV-2 Protease Inhibitors. International Journal of Molecular Sciences. 2023; 24(9):7938. https://doi.org/10.3390/ijms24097938
Chicago/Turabian StyleJi, Cheng. 2023. "Molecular Factors and Pathways of Hepatotoxicity Associated with HIV/SARS-CoV-2 Protease Inhibitors" International Journal of Molecular Sciences 24, no. 9: 7938. https://doi.org/10.3390/ijms24097938