
Immune Dysfunction in Medication-Related Osteonecrosis of the Jaw
 ,
,  , ,
, ,  ,
, 

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. An Immunosuppressed Milieu Favors ONJ Induced by ARDs
3. Bacterial Infections as Both Cause and Consequence of Immune Dysfunction in MRONJ
4. Antiresorptive Treatments Exert Different Effects on Immune System Subsets
4.1. γδ T Cells
4.2. T Regulatory Cells and T Helper-17 Cells
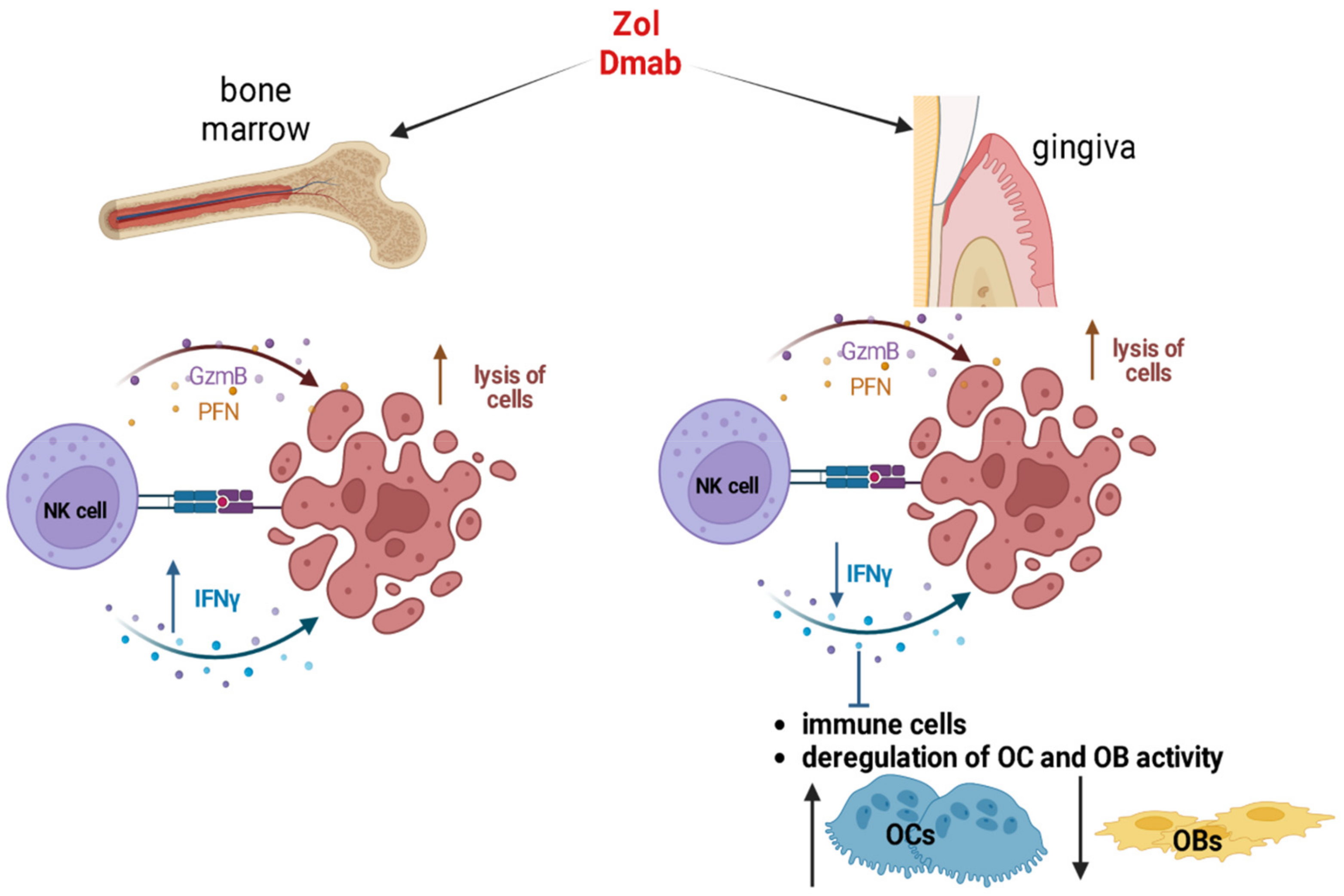
4.3. Natural Killer Cells
4.4. Macrophages
4.5. Dendritic Cells
4.6. Neutrophils
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marx, R.E. Pamidronate (Aredia) and zoledronate (Zometa) induced avascular necrosis of the jaws: A growing epidemic. J. Oral Maxillofac. Surg. 2003, 61, 1115–1117. [Google Scholar] [CrossRef]
- Fusco, V.; Santini, D.; Armento, G.; Tonini, G.; Campisi, G. Osteonecrosis of jaw beyond antiresorptive (bone-targeted) agents: New horizons in oncology. Expert Opin. Drug. Saf. 2016, 15, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Nicolatou-Galitis, O.; Schiodt, M.; Mendes, R.A.; Ripamonti, C.; Hope, S.; Drudge-Coates, L.; Niepel, D.; Van den Wyngaert, T. Medication-related osteonecrosis of the jaw: Definition and best practice for prevention, diagnosis, and treatment. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 127, 117–135. [Google Scholar] [CrossRef] [Green Version]
- Eguia, A.; Bagan-Debon, L.; Cardona, F. Review and update on drugs related to the development of osteonecrosis of the jaw. Med. Oral Patol. Oral Cir. Bucal 2020, 25, e71–e83. [Google Scholar] [CrossRef]
- Yarom, N.; Shapiro, C.L.; Peterson, D.E.; Van Poznak, C.H.; Bohlke, K.; Ruggiero, S.L.; Migliorati, C.A.; Khan, A.; Morrison, A.; Anderson, H.; et al. Medication-Related Osteonecrosis of the Jaw: MASCC/ISOO/ASCO Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 2270–2290. [Google Scholar] [CrossRef]
- Ruggiero, S.L.; Dodson, T.B.; Fantasia, J.; Goodday, R.; Aghaloo, T.; Mehrotra, B.; O’Ryan, F.; American Association of Oral and Maxillofacial Surgeons. American Association of Oral and Maxillofacial Surgeons position paper on medication-related osteonecrosis of the jaw—2014 update. J. Oral Maxillofac. Surg. 2014, 72, 1938–1956. [Google Scholar] [CrossRef]
- Bedogni, A.; Fusco, V.; Agrillo, A.; Campisi, G. Learning from experience. Proposal of a refined definition and staging system for bisphosphonate-related osteonecrosis of the jaw (BRONJ). Oral Dis. 2012, 18, 621–623. [Google Scholar] [CrossRef] [Green Version]
- Fusco, V.; Santini, D.; Campisi, G.; Bertoldo, F.; Lanzetta, G.; Ibrahim, T.; Bertetto, O.; Numico, G.; Addeo, A.; Berruti, A.; et al. Comment on Medication-Related Osteonecrosis of the Jaw: MASCC/ISOO/ASCO Clinical Practice Guideline Summary. JCO Oncol. Pract. 2020, 16, 142–145. [Google Scholar] [CrossRef]
- Otto, S.; Pautke, C.; Van den Wyngaert, T.; Niepel, D.; Schiodt, M. Medication-related osteonecrosis of the jaw: Prevention, diagnosis and management in patients with cancer and bone metastases. Cancer Treat. Rev. 2018, 69, 177–187. [Google Scholar] [CrossRef]
- Campisi, G.; Mauceri, R.; Bedogni, A.; Fusco, V. Re: AAOMS Position Paper on Medication-Related Osteonecrosis of the Jaw-2022 Update. J. Oral Maxillofac. Surg. 2022, 80, 1723–1724. [Google Scholar] [CrossRef]
- Reid, I.R. Osteonecrosis of the jaw: Who gets it, and why? Bone 2009, 44, 4–10. [Google Scholar] [CrossRef]
- Huja, S.S.; Fernandez, S.A.; Hill, K.J.; Li, Y. Remodeling dynamics in the alveolar process in skeletally mature dogs. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2006, 288, 1243–1249. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.R.; Burr, D.B. The pathogenesis of bisphosphonate-related osteonecrosis of the jaw: So many hypotheses, so few data. J. Oral Maxillofac. Surg. 2009, 67, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Hanley, D.A.; Adachi, J.D.; Bell, A.; Brown, V. Denosumab: Mechanism of action and clinical outcomes. Int. J. Clin. Pract. 2012, 66, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Bensi, C.; Giovacchini, F.; Lomurno, G.; Eramo, S.; Barraco, G.; Tullio, A. Quality of life in patients affected by medication-related osteonecrosis of the jaws: A systematic review. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2021, 132, 182–189. [Google Scholar] [CrossRef]
- Peng, J.; Wang, H.; Liu, Z.; Xu, Z.L.; Wang, M.X.; Chen, Q.M.; Wu, M.L.; Ren, X.L.; Liang, Q.H.; Liu, F.P.; et al. Real-world study of antiresorptive-related osteonecrosis of jaw based on the US food and drug administration adverse event reporting system database. Front. Pharmacol. 2022, 13, 1017391. [Google Scholar] [CrossRef]
- Marx, R.E.; Sawatari, Y.; Fortin, M.; Broumand, V. Bisphosphonate-induced exposed bone (osteonecrosis/osteopetrosis) of the jaws: Risk factors, recognition, prevention, and treatment. J. Oral Maxillofac. Surg. 2005, 63, 1567–1575. [Google Scholar] [CrossRef]
- Walter, C.; Pabst, A.; Ziebart, T.; Klein, M.; Al-Nawas, B. Bisphosphonates affect migration ability and cell viability of HUVEC, fibroblasts and osteoblasts in vitro. Oral Dis. 2011, 17, 194–199. [Google Scholar] [CrossRef]
- Otto, S.; Pautke, C.; Martin Jurado, O.; Nehrbass, D.; Stoddart, M.J.; Ehrenfeld, M.; Zeiter, S. Further development of the MRONJ minipig large animal model. J. Craniomaxillofac. Surg. 2017, 45, 1503–1514. [Google Scholar] [CrossRef]
- Francisconi, C.F.; Vieira, A.E.; Azevedo, M.C.S.; Tabanez, A.P.; Fonseca, A.C.; Trombone, A.P.F.; Letra, A.; Silva, R.M.; Sfeir, C.S.; Little, S.R.; et al. RANKL Triggers Treg-Mediated Immunoregulation in Inflammatory Osteolysis. J. Dent. Res. 2018, 97, 917–927. [Google Scholar] [CrossRef]
- Park, S.; Kanayama, K.; Kaur, K.; Tseng, H.C.; Banankhah, S.; Quje, D.T.; Sayre, J.W.; Jewett, A.; Nishimura, I. Osteonecrosis of the Jaw Developed in Mice: Disease Variants Regulated by gammadelta T Cells in Oral Mucosal Barrier Immunity. J. Biol. Chem. 2015, 290, 17349–17366. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Xu, R.; Du, J.; Fu, Y.; Li, S.; Zhang, P.; Liu, L.; Jiang, H. Zoledronic acid promotes TLR-4-mediated M1 macrophage polarization in bisphosphonate-related osteonecrosis of the jaw. FASEB J. 2019, 33, 5208–5219. [Google Scholar] [CrossRef]
- Kalyan, S. It May Seem Inflammatory, but Some T Cells Are Innately Healing to the Bone. J. Bone Miner. Res. 2016, 31, 1997–2000. [Google Scholar] [CrossRef] [Green Version]
- Kalyan, S.; Quabius, E.S.; Wiltfang, J.; Monig, H.; Kabelitz, D. Can peripheral blood gammadelta T cells predict osteonecrosis of the jaw? An immunological perspective on the adverse drug effects of aminobisphosphonate therapy. J. Bone Miner. Res. 2013, 28, 728–735. [Google Scholar] [CrossRef]
- Salesi, N.; Pistilli, R.; Marcelli, V.; Govoni, F.A.; Bozza, F.; Bossone, G.; Venturelli, V.; Di Cocco, B.; Pacetti, U.; Ciorra, A.; et al. Bisphosphonates and oral cavity avascular bone necrosis: A review of twelve cases. Anticancer Res. 2006, 26, 3111–3115. [Google Scholar] [CrossRef]
- Muratsu, D.; Yoshiga, D.; Taketomi, T.; Onimura, T.; Seki, Y.; Matsumoto, A.; Nakamura, S. Zoledronic acid enhances lipopolysaccharide-stimulated proinflammatory reactions through controlled expression of SOCS1 in macrophages. PLoS ONE 2013, 8, e67906. [Google Scholar] [CrossRef] [Green Version]
- Norton, J.T.; Hayashi, T.; Crain, B.; Cho, J.S.; Miller, L.S.; Corr, M.; Carson, D.A. Cutting edge: Nitrogen bisphosphonate-induced inflammation is dependent upon mast cells and IL-1. J. Immunol. 2012, 188, 2977–2980. [Google Scholar] [CrossRef] [Green Version]
- Rossini, M.; Adami, S.; Viapiana, O.; Fracassi, E.; Ortolani, R.; Vella, A.; Zanotti, R.; Tripi, G.; Idolazzi, L.; Gatti, D. Long-term effects of amino-bisphosphonates on circulating gammadelta T cells. Calcif. Tissue Int. 2012, 91, 395–399. [Google Scholar] [CrossRef]
- Welton, J.L.; Morgan, M.P.; Marti, S.; Stone, M.D.; Moser, B.; Sewell, A.K.; Turton, J.; Eberl, M. Monocytes and gammadelta T cells control the acute-phase response to intravenous zoledronate: Insights from a phase IV safety trial. J. Bone Miner. Res. 2013, 28, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Uluckan, O.; Jimenez, M.; Karbach, S.; Jeschke, A.; Grana, O.; Keller, J.; Busse, B.; Croxford, A.L.; Finzel, S.; Koenders, M.; et al. Chronic skin inflammation leads to bone loss by IL-17-mediated inhibition of Wnt signaling in osteoblasts. Sci. Transl. Med. 2016, 8, 330ra337. [Google Scholar] [CrossRef]
- Pollinger, B.; Junt, T.; Metzler, B.; Walker, U.A.; Tyndall, A.; Allard, C.; Bay, S.; Keller, R.; Raulf, F.; Di Padova, F.; et al. Th17 cells, not IL-17+ gammadelta T cells, drive arthritic bone destruction in mice and humans. J. Immunol. 2011, 186, 2602–2612. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.; Okamoto, K.; Nakashima, T.; Nitta, T.; Hori, S.; Iwakura, Y.; Takayanagi, H. IL-17-producing gammadelta T cells enhance bone regeneration. Nat. Commun. 2016, 7, 10928. [Google Scholar] [CrossRef] [Green Version]
- Soma, T.; Iwasaki, R.; Sato, Y.; Kobayashi, T.; Nakamura, S.; Kaneko, Y.; Ito, E.; Okada, H.; Watanabe, H.; Miyamoto, K.; et al. Tooth extraction in mice administered zoledronate increases inflammatory cytokine levels and promotes osteonecrosis of the jaw. J. Bone Miner. Metab. 2021, 39, 372–384. [Google Scholar] [CrossRef]
- Soma, T.; Iwasaki, R.; Sato, Y.; Kobayashi, T.; Ito, E.; Matsumoto, T.; Kimura, A.; Miyamoto, K.; Matsumoto, M.; Nakamura, M.; et al. Osteonecrosis development by tooth extraction in zoledronate treated mice is inhibited by active vitamin D analogues, anti-inflammatory agents or antibiotics. Sci. Rep. 2022, 12, 19. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Kim, T.; Kim, S.; Williams, D.W.; Esmaeili, M.; Hong, C.; Shin, K.H.; Kang, M.K.; Park, N.H.; et al. Indigenous microbiota protects development of medication-related osteonecrosis induced by periapical disease in mice. Int. J. Oral Sci. 2022, 14, 16. [Google Scholar] [CrossRef]
- Shibahara, T. Antiresorptive Agent-Related Osteonecrosis of the Jaw (ARONJ): A Twist of Fate in the Bone. Tohoku J. Exp. Med. 2019, 247, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Russmueller, G.; Seemann, R.; Weiss, K.; Stadler, V.; Speiss, M.; Perisanidis, C.; Fuereder, T.; Willinger, B.; Sulzbacher, I.; Steininger, C. The association of medication-related osteonecrosis of the jaw with Actinomyces spp. infection. Sci. Rep. 2016, 6, 31604. [Google Scholar] [CrossRef]
- Nair, S.P.; Meghji, S.; Wilson, M.; Reddi, K.; White, P.; Henderson, B. Bacterially induced bone destruction: Mechanisms and misconceptions. Infect. Immun. 1996, 64, 2371–2380. [Google Scholar] [CrossRef] [Green Version]
- Meghji, S.; Crean, S.J.; Hill, P.A.; Sheikh, M.; Nair, S.P.; Heron, K.; Henderson, B.; Mawer, E.B.; Harris, M. Surface-associated protein from Staphylococcus aureus stimulates osteoclastogenesis: Possible role in S. aureus-induced bone pathology. Br. J. Rheumatol. 1998, 37, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- De Ceulaer, J.; Tacconelli, E.; Vandecasteele, S.J. Actinomyces osteomyelitis in bisphosphonate-related osteonecrosis of the jaw (BRONJ): The missing link? Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1873–1880. [Google Scholar] [CrossRef]
- Schipmann, S.; Metzler, P.; Rossle, M.; Zemann, W.; von Jackowski, J.; Obwegeser, J.A.; Gratz, K.W.; Jacobsen, C. Osteopathology associated with bone resorption inhibitors—Which role does Actinomyces play? A presentation of 51 cases with systematic review of the literature. J. Oral Pathol. Med. 2013, 42, 587–593. [Google Scholar] [CrossRef]
- Cerrato, A.; Zanette, G.; Boccuto, M.; Angelini, A.; Valente, M.; Bacci, C. Actinomyces and MRONJ: A retrospective study and a literature review. J. Stomatol. Oral Maxillofac. Surg. 2021, 122, 499–504. [Google Scholar] [CrossRef]
- Sedghizadeh, P.P.; Stanley, K.; Caligiuri, M.; Hofkes, S.; Lowry, B.; Shuler, C.F. Oral bisphosphonate use and the prevalence of osteonecrosis of the jaw: An institutional inquiry. J. Am. Dent. Assoc. 2009, 140, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Lesclous, P.; Abi Najm, S.; Carrel, J.P.; Baroukh, B.; Lombardi, T.; Willi, J.P.; Rizzoli, R.; Saffar, J.L.; Samson, J. Bisphosphonate-associated osteonecrosis of the jaw: A key role of inflammation? Bone 2009, 45, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Gursoy, U.K.; Kononen, E. Understanding the roles of gingival beta-defensins. J. Oral Microbiol. 2012, 4, 15127. [Google Scholar] [CrossRef]
- Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [CrossRef] [Green Version]
- Abiko, Y.; Saitoh, M.; Nishimura, M.; Yamazaki, M.; Sawamura, D.; Kaku, T. Role of beta-defensins in oral epithelial health and disease. Med. Mol. Morphol. 2007, 40, 179–184. [Google Scholar] [CrossRef]
- Morita, M.; Iwasaki, R.; Sato, Y.; Kobayashi, T.; Watanabe, R.; Oike, T.; Nakamura, S.; Keneko, Y.; Miyamoto, K.; Ishihara, K.; et al. Elevation of pro-inflammatory cytokine levels following anti-resorptive drug treatment is required for osteonecrosis development in infectious osteomyelitis. Sci. Rep. 2017, 7, 46322. [Google Scholar] [CrossRef]
- Shuster, A.; Reiser, V.; Trejo, L.; Ianculovici, C.; Kleinman, S.; Kaplan, I. Comparison of the histopathological characteristics of osteomyelitis, medication-related osteonecrosis of the jaw, and osteoradionecrosis. Int. J. Oral Maxillofac. Surg. 2019, 48, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Stockmann, P.; Wehrhan, F.; Schwarz-Furlan, S.; Stelzle, F.; Trabert, S.; Neukam, F.W.; Nkenke, E. Increased human defensine levels hint at an inflammatory etiology of bisphosphonate-associated osteonecrosis of the jaw: An immunohistological study. J. Transl. Med. 2011, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Warnke, P.H.; Springer, I.N.; Russo, P.A.; Wiltfang, J.; Essig, H.; Kosmahl, M.; Sherry, E.; Acil, Y. Innate immunity in human bone. Bone 2006, 38, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Deschner, J.; Jager, A.; Wenghoefer, M.; Bayer, S.; Jepsen, S.; Allam, J.P.; Novak, N.; Meyer, R.; Winter, J. Human beta-defensins differently affect proliferation, differentiation, and mineralization of osteoblast-like MG63 cells. J. Cell. Physiol. 2012, 227, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Varoga, D.; Tohidnezhad, M.; Paulsen, F.; Wruck, C.J.; Brandenburg, L.; Mentlein, R.; Lippross, S.; Hassenpflug, J.; Besch, L.; Muller, M.; et al. The role of human beta-defensin-2 in bone. J. Anat. 2008, 213, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Thiel, Y.; Ghayor, C.; Lindhorst, D.; Essig, H.; Weber, F.; Rucker, M.; Schumann, P. Antimicrobial peptide gene expression in medication-related osteonecrosis of the jaw. Pathol. Res. Pract. 2020, 216, 153245. [Google Scholar] [CrossRef]
- Zhang, Q.; Atsuta, I.; Liu, S.; Chen, C.; Shi, S.; Shi, S.; Le, A.D. IL-17-mediated M1/M2 macrophage alteration contributes to pathogenesis of bisphosphonate-related osteonecrosis of the jaws. Clin. Cancer Res. 2013, 19, 3176–3188. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Sun, Y.; Kanayama, K.; Morinaga, K.; Hokugo, A.; Nishimura, I.; Jewett, A. Augmentation of IFN-gamma by bone marrow derived immune cells in the presence of severe suppression of IFN-gamma in gingivae induced by zoledronic acid and denosumab in Hu-BLT mice model of ONJ. Front. Endocrinol. 2023, 14, 1111627. [Google Scholar] [CrossRef]
- Elsayed, R.; Kurago, Z.; Cutler, C.W.; Arce, R.M.; Gerber, J.; Celis, E.; Sultan, H.; Elashiry, M.; Meghil, M.; Sun, C.; et al. Role of dendritic cell-mediated immune response in oral homeostasis: A new mechanism of osteonecrosis of the jaw. FASEB J. 2020, 34, 2595–2608. [Google Scholar] [CrossRef]
- Hagelauer, N.; Pabst, A.M.; Ziebart, T.; Ulbrich, H.; Walter, C. In vitro effects of bisphosphonates on chemotaxis, phagocytosis, and oxidative burst of neutrophil granulocytes. Clin. Oral Investig. 2015, 19, 139–148. [Google Scholar] [CrossRef]
- Gkouveris, I.; Soundia, A.; Gouveris, P.; Zouki, D.; Hadaya, D.; Tetradis, S. Macrophage Involvement in Medication-Related Osteonecrosis of the Jaw (MRONJ): A Comprehensive, Short Review. Cancers 2022, 14, 330. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Hayday, A.C. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity 2009, 31, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Morita, C.T.; Tanaka, Y.; Nieves, E.; Brenner, M.B.; Bloom, B.R. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature 1995, 375, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, S.; Kabelitz, D. Defining the nature of human gammadelta T cells: A biographical sketch of the highly empathetic. Cell. Mol. Immunol. 2013, 10, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jameson, J.; Havran, W.L. Skin gammadelta T-cell functions in homeostasis and wound healing. Immunol. Rev. 2007, 215, 114–122. [Google Scholar] [CrossRef]
- Kabelitz, D.; Kalyan, S.; Oberg, H.H.; Wesch, D. Human Vdelta2 versus non-Vdelta2 gammadelta T cells in antitumor immunity. Oncoimmunology 2013, 2, e23304. [Google Scholar] [CrossRef]
- Chen, Z.W. Immune biology of Ag-specific gammadelta T cells in infections. Cell. Mol. Life Sci. 2011, 68, 2409–2417. [Google Scholar] [CrossRef] [Green Version]
- Kalyan, S.; Wang, J.; Quabius, E.S.; Huck, J.; Wiltfang, J.; Baines, J.F.; Kabelitz, D. Systemic immunity shapes the oral microbiome and susceptibility to bisphosphonate-associated osteonecrosis of the jaw. J. Transl. Med. 2015, 13, 212. [Google Scholar] [CrossRef] [Green Version]
- Lo Presti, E.; Toia, F.; Oieni, S.; Buccheri, S.; Turdo, A.; Mangiapane, L.R.; Campisi, G.; Caputo, V.; Todaro, M.; Stassi, G.; et al. Squamous Cell Tumors Recruit gammadelta T Cells Producing either IL17 or IFNgamma Depending on the Tumor Stage. Cancer Immunol. Res. 2017, 5, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef]
- Kunzmann, V.; Bauer, E.; Wilhelm, M. Gamma/delta T-cell stimulation by pamidronate. N. Engl. J. Med. 1999, 340, 737–738. [Google Scholar] [CrossRef]
- Croes, M.; Oner, F.C.; van Neerven, D.; Sabir, E.; Kruyt, M.C.; Blokhuis, T.J.; Dhert, W.J.A.; Alblas, J. Proinflammatory T cells and IL-17 stimulate osteoblast differentiation. Bone 2016, 84, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; D’Amelio, P.; Robinson, J.; Walker, L.D.; Vaccaro, C.; Luo, T.; Tyagi, A.M.; Yu, M.; Reott, M.; Sassi, F.; et al. IL-17A Is Increased in Humans with Primary Hyperparathyroidism and Mediates PTH-Induced Bone Loss in Mice. Cell. Metab. 2015, 22, 799–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movila, A.; Mawardi, H.; Nishimura, K.; Kiyama, T.; Egashira, K.; Kim, J.Y.; Villa, A.; Sasaki, H.; Woo, S.B.; Kawai, T. Possible pathogenic engagement of soluble Semaphorin 4D produced by gammadeltaT cells in medication-related osteonecrosis of the jaw (MRONJ). Biochem. Biophys. Res. Commun. 2016, 480, 42–47. [Google Scholar] [CrossRef]
- Schledzewski, K.; Falkowski, M.; Moldenhauer, G.; Metharom, P.; Kzhyshkowska, J.; Ganss, R.; Demory, A.; Falkowska-Hansen, B.; Kurzen, H.; Ugurel, S.; et al. Lymphatic endothelium-specific hyaluronan receptor LYVE-1 is expressed by stabilin-1+, F4/80+, CD11b+ macrophages in malignant tumours and wound healing tissue in vivo and in bone marrow cultures in vitro: Implications for the assessment of lymphangiogenesis. J. Pathol. 2006, 209, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Gober, H.J.; Kistowska, M.; Angman, L.; Jeno, P.; Mori, L.; De Libero, G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idrees, A.S.; Sugie, T.; Inoue, C.; Murata-Hirai, K.; Okamura, H.; Morita, C.T.; Minato, N.; Toi, M.; Tanaka, Y. Comparison of gammadelta T cell responses and farnesyl diphosphate synthase inhibition in tumor cells pretreated with zoledronic acid. Cancer Sci. 2013, 104, 536–542. [Google Scholar] [CrossRef] [Green Version]
- Roelofs, A.J.; Jauhiainen, M.; Monkkonen, H.; Rogers, M.J.; Monkkonen, J.; Thompson, K. Peripheral blood monocytes are responsible for gammadelta T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br. J. Haematol. 2009, 144, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Gan, Y.H.; Wallace, M.; Malkovsky, M. Fas-dependent, activation-induced cell death of gammadelta cells. J. Biol. Regul. Homeost. Agents 2001, 15, 277–285. [Google Scholar]
- Green, D.R.; Droin, N.; Pinkoski, M. Activation-induced cell death in T cells. Immunol. Rev. 2003, 193, 70–81. [Google Scholar] [CrossRef]
- Roberts, N.A.; White, A.J.; Jenkinson, W.E.; Turchinovich, G.; Nakamura, K.; Withers, D.R.; McConnell, F.M.; Desanti, G.E.; Benezech, C.; Parnell, S.M.; et al. Rank signaling links the development of invariant gammadelta T cell progenitors and Aire(+) medullary epithelium. Immunity 2012, 36, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Loser, K.; Mehling, A.; Loeser, S.; Apelt, J.; Kuhn, A.; Grabbe, S.; Schwarz, T.; Penninger, J.M.; Beissert, S. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat. Med. 2006, 12, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, A.; Kallies, A. The Regulatory T Cell: Jack-Of-All-Trades. Trends Immunol. 2015, 36, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuiri, T.; Kim, I.; Yamaza, T.; Akiyama, K.; Zhang, Q.; Li, Y.; Chen, C.; Chen, W.; Wang, S.; Le, A.D.; et al. Cell-based immunotherapy with mesenchymal stem cells cures bisphosphonate-related osteonecrosis of the jaw-like disease in mice. J. Bone Miner. Res. 2010, 25, 1668–1679. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, N.I.; Cowan, J.E.; Nakamura, K.; Bacon, A.; Baik, S.; White, A.J.; Parnell, S.M.; Jenkinson, E.J.; Jenkinson, W.E.; Anderson, G. Osteoprotegerin-Mediated Homeostasis of Rank+ Thymic Epithelial Cells Does Not Limit Foxp3+ Regulatory T Cell Development. J. Immunol. 2015, 195, 2675–2682. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, M.M.; Takayanagi, H. The immune system, bone and RANKL. Arch. Biochem. Biophys. 2014, 561, 118–123. [Google Scholar] [CrossRef]
- Kang, B.; Cheong, S.; Chaichanasakul, T.; Bezouglaia, O.; Atti, E.; Dry, S.M.; Pirih, F.Q.; Aghaloo, T.L.; Tetradis, S. Periapical disease and bisphosphonates induce osteonecrosis of the jaws in mice. J. Bone Miner. Res. 2013, 28, 1631–1640. [Google Scholar] [CrossRef] [Green Version]
- Aghaloo, T.L.; Cheong, S.; Bezouglaia, O.; Kostenuik, P.; Atti, E.; Dry, S.M.; Pirih, F.Q.; Tetradis, S. RANKL inhibitors induce osteonecrosis of the jaw in mice with periapical disease. J. Bone Miner. Res. 2014, 29, 843–854. [Google Scholar] [CrossRef]
- Demoulin, S.A.; Somja, J.; Duray, A.; Guenin, S.; Roncarati, P.; Delvenne, P.O.; Herfs, M.F.; Hubert, P.M. Cervical (pre)neoplastic microenvironment promotes the emergence of tolerogenic dendritic cells via RANKL secretion. Oncoimmunology 2015, 4, e1008334. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Yang, L.; Silva, H.M.; Trzeciak, A.; Choi, Y.; Schwab, S.R.; Dustin, M.L.; Lafaille, J.J. Increased generation of Foxp3(+) regulatory T cells by manipulating antigen presentation in the thymus. Nat. Commun. 2016, 7, 10562. [Google Scholar] [CrossRef] [Green Version]
- Farag, S.S.; Caligiuri, M.A. Human natural killer cell development and biology. Blood Rev. 2006, 20, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Kanayama, K.; Wu, Q.Q.; Gumrukcu, S.; Nishimura, I.; Jewett, A. Zoledronic acid mediated differential activation of NK cells in different organs of WT and Rag2(−/−) mice; stark differences between the bone marrow and gingivae. Cell. Immunol. 2022, 375, 104526. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Grassi, F.; Ryan, M.R.; Terauchi, M.; Page, K.; Yang, X.; Weitzmann, M.N.; Pacifici, R. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J. Clin. Invest. 2007, 117, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.C.; Kanayama, K.; Kaur, K.; Park, S.H.; Park, S.; Kozlowska, A.; Sun, S.; McKenna, C.E.; Nishimura, I.; Jewett, A. Bisphosphonate-induced differential modulation of immune cell function in gingiva and bone marrow in vivo: Role in osteoclast-mediated NK cell activation. Oncotarget 2015, 6, 20002–20025. [Google Scholar] [CrossRef] [PubMed]
- Soderstrom, K.; Stein, E.; Colmenero, P.; Purath, U.; Muller-Ladner, U.; de Matos, C.T.; Tarner, I.H.; Robinson, W.H.; Engleman, E.G. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc. Natl. Acad. Sci. USA 2010, 107, 13028–13033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowska, A.K.; Topchyan, P.; Kaur, K.; Tseng, H.C.; Teruel, A.; Hiraga, T.; Jewett, A. Differentiation by NK cells is a prerequisite for effective targeting of cancer stem cells/poorly differentiated tumors by chemopreventive and chemotherapeutic drugs. J. Cancer 2017, 8, 537–554. [Google Scholar] [CrossRef] [PubMed]
- Roato, I.; Vitale, M. The Uncovered Role of Immune Cells and NK Cells in the Regulation of Bone Metastasis. Front. Endocrinol. 2019, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Patntirapong, S.; Poolgesorn, M. Alteration of macrophage viability, differentiation, and function by bisphosphonates. Oral Dis. 2018, 24, 1294–1302. [Google Scholar] [CrossRef]
- Paschalidi, P.; Gkouveris, I.; Soundia, A.; Kalfarentzos, E.; Vardas, E.; Georgaki, M.; Kostakis, G.; Erovic, B.M.; Tetradis, S.; Perisanidis, C.; et al. The role of M1 and M2 macrophage polarization in progression of medication-related osteonecrosis of the jaw. Clin. Oral Investig. 2021, 25, 2845–2857. [Google Scholar] [CrossRef]
- Yang, X.; Xu, X.; Chen, J.; Wang, Q.; Wang, G.; Ai, X.; Wang, X.; Pan, J. Zoledronic acid regulates the synthesis and secretion of IL-1beta through Histone methylation in macrophages. Cell. Death Discov. 2020, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Hoefert, S.; Hoefert, C.S.; Albert, M.; Munz, A.; Grimm, M.; Northoff, H.; Reinert, S.; Alexander, D. Zoledronate but not denosumab suppresses macrophagic differentiation of THP-1 cells. An aetiologic model of bisphosphonate-related osteonecrosis of the jaw (BRONJ). Clin. Oral Investig. 2015, 19, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, S.; Kuroshima, S.; Hayano, H.; Nakajima, K.; Kakehashi, H.; Ishisaki, A.; Sawase, T. Dynamic polarization shifting from M1 to M2 macrophages in reduced osteonecrosis of the jaw-like lesions by cessation of anti-RANKL antibody in mice. Bone 2020, 141, 115560. [Google Scholar] [CrossRef] [PubMed]
- Hayano, H.; Kuroshima, S.; Sasaki, M.; Tamaki, S.; Inoue, M.; Ishisaki, A.; Sawase, T. Distinct immunopathology in the early stages between different antiresorptives-related osteonecrosis of the jaw-like lesions in mice. Bone 2020, 135, 115308. [Google Scholar] [CrossRef] [PubMed]
- Akita, Y.; Kuroshima, S.; Nakajima, K.; Hayano, H.; Kanai, R.; Sasaki, M.; Sawase, T. Effect of anti-angiogenesis induced by chemotherapeutic monotherapy, chemotherapeutic/bisphosphonate combination therapy and anti-VEGFA mAb therapy on tooth extraction socket healing in mice. J. Bone Miner. Metab. 2018, 36, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Kapsenberg, M.L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 2003, 3, 984–993. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chao, K.S.; Yang, Y.C.; Hsu, M.L.; Lin, C.P.; Chen, Y.Y. Zoledronic acid, an aminobisphosphonate, modulates differentiation and maturation of human dendritic cells. Immunopharmacol. Immunotoxicol. 2009, 31, 499–508. [Google Scholar] [CrossRef]
- Wolf, A.M.; Rumpold, H.; Tilg, H.; Gastl, G.; Gunsilius, E.; Wolf, D. The effect of zoledronic acid on the function and differentiation of myeloid cells. Haematologica 2006, 91, 1165–1171. [Google Scholar]
- Bando, K.; Kuroishi, T.; Tada, H.; Oizumi, T.; Tanaka, Y.; Takahashi, T.; Mizoguchi, I.; Sugawara, S.; Endo, Y. Nitrogen-containing bisphosphonates and lipopolysaccharide mutually augment inflammation via adenosine triphosphate (ATP)-mediated and interleukin 1beta (IL-1beta)-mediated production of neutrophil extracellular traps (NETs). J. Bone Miner. Res. 2021, 36, 1866–1878. [Google Scholar] [CrossRef]
- Nomura, I.; Goleva, E.; Howell, M.D.; Hamid, Q.A.; Ong, P.Y.; Hall, C.F.; Darst, M.A.; Gao, B.; Boguniewicz, M.; Travers, J.B.; et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J. Immunol. 2003, 171, 3262–3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, J.W.; Forster, C.; Sun, C.; Peel, S.; Glogauer, M. Zoledronate and pamidronate depress neutrophil functions and survival in mice. Br. J. Pharmacol. 2012, 165, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Pretto, E.; Pacheco, M.; Moreno, A.; Messa, O.; Gnecco, J. Bisphosphonate-induced osteonecrosis of the jaws: Clinical, imaging, and histopathology findings. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 118, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Hokugo, A.; Christensen, R.; Chung, E.M.; Sung, E.C.; Felsenfeld, A.L.; Sayre, J.W.; Garrett, N.; Adams, J.S.; Nishimura, I. Increased prevalence of bisphosphonate-related osteonecrosis of the jaw with vitamin D deficiency in rats. J. Bone Miner. Res. 2010, 25, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- de Barros Silva, P.G.; de Oliveira, C.C.; Brizeno, L.; Wong, D.; Lima Junior, R.; Goncalves, R.P.; Sousa, F.B.; Mota, M.; de Albuquerque Ribeiro, R.; Alves, A. Immune cellular profile of bisphosphonate-related osteonecrosis of the jaw. Oral Dis. 2016, 22, 649–657. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roato, I.; Mauceri, R.; Notaro, V.; Genova, T.; Fusco, V.; Mussano, F. Immune Dysfunction in Medication-Related Osteonecrosis of the Jaw. Int. J. Mol. Sci. 2023, 24, 7948. https://doi.org/10.3390/ijms24097948
Roato I, Mauceri R, Notaro V, Genova T, Fusco V, Mussano F. Immune Dysfunction in Medication-Related Osteonecrosis of the Jaw. International Journal of Molecular Sciences. 2023; 24(9):7948. https://doi.org/10.3390/ijms24097948
Chicago/Turabian StyleRoato, Ilaria, Rodolfo Mauceri, Vincenzo Notaro, Tullio Genova, Vittorio Fusco, and Federico Mussano. 2023. "Immune Dysfunction in Medication-Related Osteonecrosis of the Jaw" International Journal of Molecular Sciences 24, no. 9: 7948. https://doi.org/10.3390/ijms24097948