
Appraising the Role of Astrocytes as Suppliers of Neuronal Glutathione Precursors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. General Facts about Glutathione
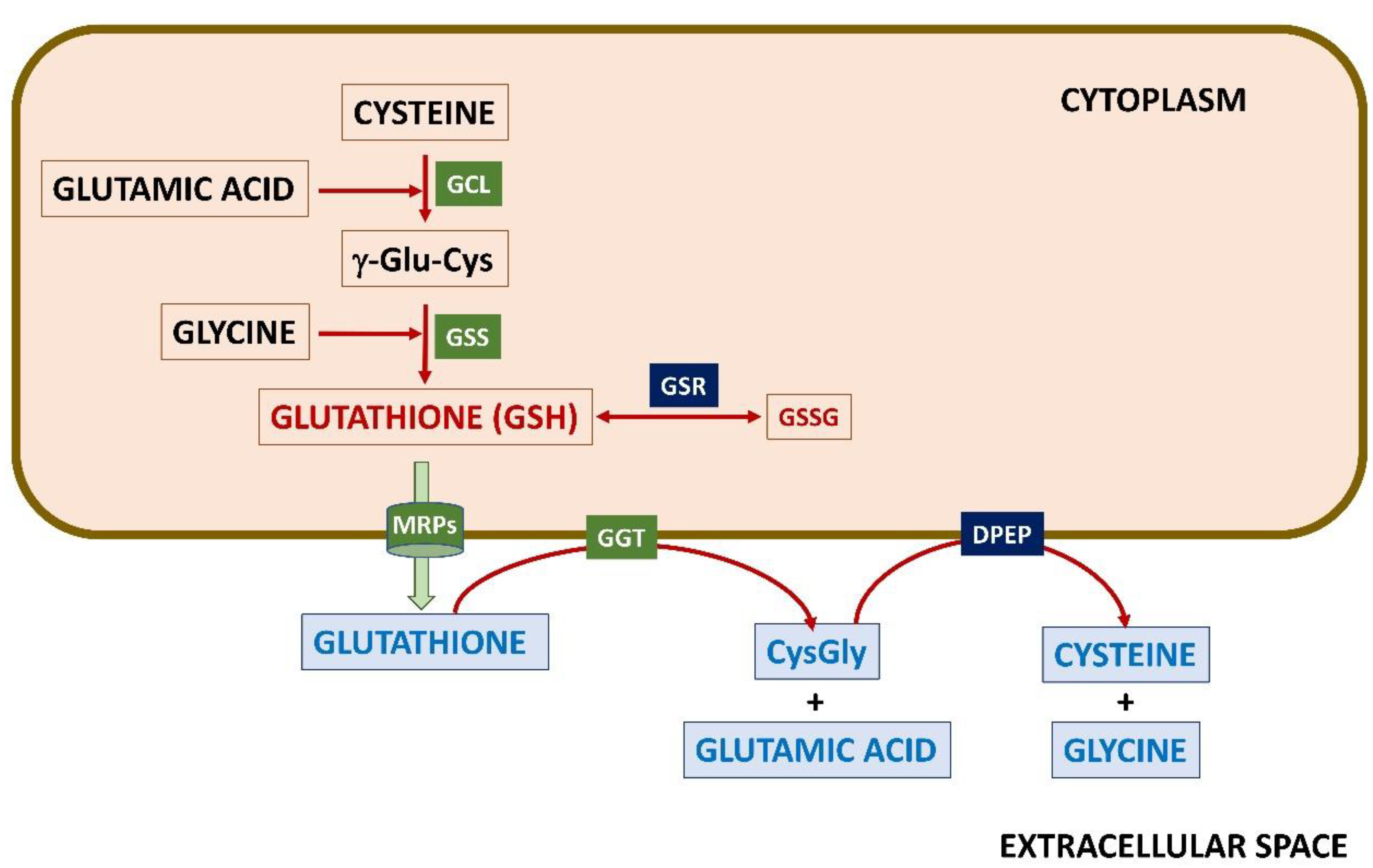
2.1. Glutathione Synthesis and Regulation
2.2. Availability and Uptake of Amino Acids for Glutathione Synthesis
2.2.1. Cysteine Uptake and Synthesis
2.2.2. Glutamate Uptake, Exchange, and Synthesis
2.2.3. Glycine Uptake and Synthesis
2.3. The γ-Glutamyl Cycle
3. General Facts about Astrocytes and Neurons and Their Energy Production
4. Glutathione Levels and Synthesis in Brain: Neurons and Astrocytes
4.1. Glutathione Levels and Distribution in Brain
4.2. Glutathione Metabolism in Brain Cells
4.2.1. Glutathione Levels in Brain Cells
4.2.2. Glutathione Synthesis and Recycling in Neurons and Astrocytes
4.2.3. Sources and Uptake of Reduced and Oxidized Cysteine for Glutathione Synthesis in Neurons and Astrocytes
4.2.4. Sources and Uptake of Glutamate for Glutathione Synthesis in Neurons and Astrocytes
4.2.5. Acquisition of Glycine for Glutathione Synthesis in Neurons and Astrocytes
4.2.6. Dipeptides as Precursors for Glutathione Synthesis
4.2.7. Glutathione Utilization in Detoxification
4.2.8. Export of Glutathione
5. Astrocytes as Suppliers of Glutathione Components: The Hypothesis
6. Putative Drawbacks of the Hypothesis and Additional Possibilities to Achieve Appropriate Neuronal Glutathione Levels
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Pellerin, L. Cellular mechanisms of brain energy metabolism. Relevance to functional brain imaging and to neurodegenerative disorders. Ann. N. Y. Acad. Sci. 1996, 777, 380–387. [Google Scholar] [CrossRef]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell. Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, S.J.; MacVicar, B.A. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 2004, 431, 195–199. [Google Scholar] [CrossRef]
- Takano, T.; Tian, G.F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2006, 9, 260–267. [Google Scholar] [CrossRef]
- Zonta, M.; Angulo, M.C.; Gobbo, S.; Rosengarten, B.; Hossmann, K.A.; Pozzan, T.; Carmignoto, G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 2003, 6, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.S.; Endo, S.; Kanai, N.; Schuster, V.L. Identification of lactate as a driving force for prostanoid transport by prostaglandin transporter PGT. Am. J. Physiol. Renal Physiol. 2002, 282, F1097–F1102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yamanishi, S.; Katsumura, K.; Kobayashi, T.; Puro, D.G. Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H925–H934. [Google Scholar] [CrossRef]
- Gordon, G.R.; Choi, H.B.; Rungta, R.L.; Ellis-Davies, G.C.; MacVicar, B.A. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 2008, 456, 745–749. [Google Scholar] [CrossRef]
- Ozugur, S.; Kunz, L.; Straka, H. Relationship between oxygen consumption and neuronal activity in a defined neural circuit. BMC Biol. 2020, 18, 76. [Google Scholar] [CrossRef]
- Kasischke, K.A.; Vishwasrao, H.D.; Fisher, P.J.; Zipfel, W.R.; Webb, W.W. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 2004, 305, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Pero, R.W.; Roush, G.C.; Markowitz, M.M.; Miller, D.G. Oxidative stress, DNA repair, and cancer susceptibility. Cancer Detect. Prev. 1990, 14, 555–561. [Google Scholar] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.; Rothschild, D.E.; Quinlan, C.L.; Scott, G.K.; Benz, C.C.; Brand, M.D. Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biol. 2014, 2, 901–909. [Google Scholar] [CrossRef]
- Banerjee, R. Redox outside the box: Linking extracellular redox remodeling with intracellular redox metabolism. J. Biol. Chem. 2012, 287, 4397–4402. [Google Scholar] [CrossRef]
- Kohen, R.; Vellaichamy, E.; Hrbac, J.; Gati, I.; Tirosh, O. Quantification of the overall reactive oxygen species scavenging capacity of biological fluids and tissues. Free Radic. Biol. Med. 2000, 28, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Rashed, M.N. The role of trace elements on hepatitis virus infections: A review. J. Trace Elem. Med. Biol. 2011, 25, 181–187. [Google Scholar] [CrossRef]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E.; Motley, A.K.; Winfrey, V.P.; Kurokawa, S.; Mitchell, S.L.; Zhang, W. Selenoprotein P and apolipoprotein E receptor-2 interact at the blood-brain barrier and also within the brain to maintain an essential selenium pool that protects against neurodegeneration. FASEB J. 2014, 28, 3579–3588. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Lassegue, B.; Ushio-Fukai, M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2175–2183. [Google Scholar] [CrossRef]
- Chance, B.; Schoener, B.; Oshino, R.; Itshak, F.; Nakase, Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J. Biol. Chem. 1979, 254, 4764–4771. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Raps, S.P.; Lai, J.C.; Hertz, L.; Cooper, A.J. Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res. 1989, 493, 398–401. [Google Scholar] [CrossRef]
- Makar, T.K.; Nedergaard, M.; Preuss, A.; Gelbard, A.S.; Perumal, A.S.; Cooper, A.J. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: Evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem. 1994, 62, 45–53. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of hepatic glutathione synthesis: Current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Gilbert, H.F. Biological disulfides: The third messenger? Modulation of phosphofructokinase activity by thiol/disulfide exchange. J. Biol. Chem. 1982, 257, 12086–12091. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, H.F. Molecular Perspectives and Clinical Implications. In Glutathione Centennial; Taniguchi, T., Higashi, T., Sakamoto, Y., Meister, A., Eds.; Academic Press: New York, NY, USA, 1989; pp. 73–87. [Google Scholar]
- Zhou, Y.; Harrison, D.E.; Love-Myers, K.; Chen, Y.; Grider, A.; Wickwire, K.; Burgess, J.R.; Stochelski, M.A.; Pazdro, R. Genetic analysis of tissue glutathione concentrations and redox balance. Free Radic. Biol. Med. 2014, 71, 157–164. [Google Scholar] [CrossRef]
- Meredith, M.J.; Reed, D.J. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J. Biol. Chem. 1982, 257, 3747–3753. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Kaplowitz, N.; Fernandez-Checa, J.C.; Kannan, R.; Garcia-Ruiz, C.; Ookhtens, M.; Yi, J.R. GSH transporters: Molecular characterization and role in GSH homeostasis. Biol. Chem. Hoppe Seyler 1996, 377, 267–273. [Google Scholar]
- Wang, J.Q.; Yang, Y.; Cai, C.Y.; Teng, Q.X.; Cui, Q.; Lin, J.; Assaraf, Y.G.; Chen, Z.S. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug. Resist. Updates 2021, 54, 100743. [Google Scholar] [CrossRef]
- Minich, T.; Riemer, J.; Schulz, J.B.; Wielinga, P.; Wijnholds, J.; Dringen, R. The multidrug resistance protein 1 (Mrp1), but not Mrp5, mediates export of glutathione and glutathione disulfide from brain astrocytes. J. Neurochem. 2006, 97, 373–384. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Kristal, B.S. Multiple roles of glutathione in the central nervous system. Biol. Chem. 1997, 378, 793–802. [Google Scholar] [PubMed]
- Singh, R.J.; Hogg, N.; Joseph, J.; Kalyanaraman, B. Mechanism of nitric oxide release from S-nitrosothiols. J. Biol. Chem. 1996, 271, 18596–18603. [Google Scholar] [CrossRef] [PubMed]
- Janaky, R.; Ogita, K.; Pasqualotto, B.A.; Bains, J.S.; Oja, S.S.; Yoneda, Y.; Shaw, C.A. Glutathione and signal transduction in the mammalian CNS. J. Neurochem. 1999, 73, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Iskusnykh, I.Y.; Zakharova, A.A.; Pathak, D. Glutathione in Brain Disorders and Aging. Molecules 2022, 27, 324. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; Mari, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 1997, 273, G7–G17. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione: Hepatocellular survival-death switch. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. S3), S3–S6. [Google Scholar] [CrossRef]
- Liu, H.; Wang, H.; Shenvi, S.; Hagen, T.M.; Liu, R.M. Glutathione metabolism during aging and in Alzheimer disease. Ann. N. Y Acad. Sci. 2004, 1019, 346–349. [Google Scholar] [CrossRef]
- Mantle, D.; Hargreaves, I.P. Mitochondrial Dysfunction and Neurodegenerative Disorders: Role of Nutritional Supplementation. Int. J. Mol. Sci. 2022, 23, 12603. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Dalton, T.P.; Chen, Y.; Schneider, S.N.; Nebert, D.W.; Shertzer, H.G. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic. Biol. Med. 2004, 37, 1511–1526. [Google Scholar] [CrossRef] [PubMed]
- Seelig, G.F.; Simondsen, R.P.; Meister, A. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J. Biol. Chem. 1984, 259, 9345–9347. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Anderson, M.E.; Meister, A. Amino acid sequence and function of the light subunit of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 1993, 268, 20578–20583. [Google Scholar] [CrossRef]
- Huang, C.S.; Chang, L.S.; Anderson, M.E.; Meister, A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 1993, 268, 19675–19680. [Google Scholar] [CrossRef]
- Dahl, E.L.; Mulcahy, R.T. Cell-type specific differences in glutamate cysteine ligase transcriptional regulation demonstrate independent subunit control. Toxicol. Sci. 2001, 61, 265–272. [Google Scholar] [CrossRef]
- Dasgupta, A.; Das, S.; Sarkar, P.K. Thyroid hormone promotes glutathione synthesis in astrocytes by up regulation of glutamate cysteine ligase through differential stimulation of its catalytic and modulator subunit mRNAs. Free Radic. Biol. Med. 2007, 42, 617–626. [Google Scholar] [CrossRef]
- Sun, W.M.; Huang, Z.Z.; Lu, S.C. Regulation of gamma-glutamylcysteine synthetase by protein phosphorylation. Biochem. J. 1996, 320 Pt 1, 321–328. [Google Scholar] [CrossRef]
- Koppal, T.; Drake, J.; Yatin, S.; Jordan, B.; Varadarajan, S.; Bettenhausen, L.; Butterfield, D.A. Peroxynitrite-induced alterations in synaptosomal membrane proteins: Insight into oxidative stress in Alzheimer’s disease. J. Neurochem. 1999, 72, 310–317. [Google Scholar] [CrossRef]
- Zarkovic, K. 4-hydroxynonenal and neurodegenerative diseases. Mol. Aspects Med. 2003, 24, 293–303. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.A.; Perez-Sala, D. Mammalian Sulfur Amino Acid Metabolism: A Nexus Between Redox Regulation, Nutrition, Epigenetics, and Detoxification. Antioxid. Redox Signal. 2018, 29, 408–452. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S.; Tateishi, N. Role of membrane transport in metabolism and function of glutathione in mammals. J. Membr. Biol. 1986, 89, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I. Sulfhydryl dependence in primary explant hematopoietic cells. Inhibition of growth in vitro with vitamin B12 compounds. Proc. Natl. Acad. Sci. USA 1975, 72, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Bannai, S. The synergistic action of the copper chelator bathocuproine sulphonate and cysteine in enhancing growth of L1210 cells in vitro. J. Cell. Physiol. 1985, 125, 151–155. [Google Scholar] [CrossRef]
- Wang, X.F.; Cynader, M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 2000, 74, 1434–1442. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef]
- Sagara, J.I.; Miura, K.; Bannai, S. Maintenance of neuronal glutathione by glial cells. J. Neurochem. 1993, 61, 1672–1676. [Google Scholar] [CrossRef]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef]
- Sagara, J.; Miura, K.; Bannai, S. Cystine uptake and glutathione level in fetal brain cells in primary culture and in suspension. J. Neurochem. 1993, 61, 1667–1671. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- Bannai, S. Induction of cystine and glutamate transport activity in human fibroblasts by diethyl maleate and other electrophilic agents. J. Biol. Chem. 1984, 259, 2435–2440. [Google Scholar] [CrossRef]
- Bannai, S.; Sato, H.; Ishii, T.; Sugita, Y. Induction of cystine transport activity in human fibroblasts by oxygen. J. Biol. Chem. 1989, 264, 18480–18484. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ishii, T.; Sugita, Y.; Bannai, S. Cystine uptake and glutathione level in endothelial cells exposed to oxidative stress. Am. J. Physiol. 1992, 262, C50–C58. [Google Scholar] [CrossRef]
- Liu, Z.; Dong, W.; Yang, B.; Peng, L.; Xia, X.; Pu, L.; Zhang, N.; Song, E.; Song, Y. Tetrachlorobenzoquinone-Induced Nrf2 Confers Neuron-like PC12 Cells Resistance to Endoplasmic Reticulum Stress via Regulating Glutathione Synthesis and Protein Thiol Homeostasis. Chem. Res. Toxicol. 2018, 31, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.P.; Larsson, H.P. Small-scale molecular motions accomplish glutamate uptake in human glutamate transporters. J. Neurosci. 2005, 25, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493 Pt 2, 419–423. [Google Scholar] [CrossRef]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998, 19, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 2008, 108, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef]
- Fournier, K.M.; Gonzalez, M.I.; Robinson, M.B. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J. Biol. Chem. 2004, 279, 34505–34513. [Google Scholar] [CrossRef]
- Gonzalez, M.I.; Kazanietz, M.G.; Robinson, M.B. Regulation of the neuronal glutamate transporter excitatory amino acid carrier-1 (EAAC1) by different protein kinase C subtypes. Mol. Pharmacol. 2002, 62, 901–910. [Google Scholar] [CrossRef]
- Davis, K.E.; Straff, D.J.; Weinstein, E.A.; Bannerman, P.G.; Correale, D.M.; Rothstein, J.D.; Robinson, M.B. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J. Neurosci. 1998, 18, 2475–2485. [Google Scholar] [CrossRef]
- Kalandadze, A.; Wu, Y.; Robinson, M.B. Protein kinase C activation decreases cell surface expression of the GLT-1 subtype of glutamate transporter. Requirement of a carboxyl-terminal domain and partial dependence on serine 486. J. Biol. Chem. 2002, 277, 45741–45750. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Shimizu, H.; Koike, T.; Furuya, S.; Watanabe, M. Neutral amino acid transporter ASCT1 is preferentially expressed in L-Ser-synthetic/storing glial cells in the mouse brain with transient expression in developing capillaries. J. Neurosci. 2003, 23, 550–560. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Pingitore, P.; Hedfalk, K.; Indiveri, C. Cysteine is not a substrate but a specific modulator of human ASCT2 (SLC1A5) transporter. FEBS Lett. 2015, 589, 3617–3623. [Google Scholar] [CrossRef]
- Banerjee, R. Catalytic promiscuity and heme-dependent redox regulation of H(2)S synthesis. Curr. Opin. Chem. Biol. 2017, 37, 115–121. [Google Scholar] [CrossRef]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Dayal, S.; Stabler, S.; Zhou, Y.; Wang, H.; Lentz, S.R.; Banerjee, R. Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R39–R46. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Thomas, M.; Ghorpade, A.; Gendelman, H.E.; Banerjee, R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J. Biol. Chem. 2006, 281, 35785–35793. [Google Scholar] [CrossRef]
- Stein, W.H.; Moore, S. The free amino acids of human blood plasma. J. Biol. Chem. 1954, 211, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef] [PubMed]
- Broer, S.; Gauthier-Coles, G. Amino Acid Homeostasis in Mammalian Cells with a Focus on Amino Acid Transport. J. Nutr. 2022, 152, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Paul, B.D.; Parker, G.M.; Hester, L.D.; Snowman, A.M.; Taniguchi, Y.; Kamiya, A.; Snyder, S.H.; Sawa, A. The glutathione cycle shapes synaptic glutamate activity. Proc. Natl. Acad. Sci. USA 2019, 116, 2701–2706. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Yelamanchi, S.D.; Jayaram, S.; Thomas, J.K.; Gundimeda, S.; Khan, A.A.; Singhal, A.; Prasad, T.S.K.; Pandey, A.; Somani, B.L.; Gowda, H. A pathway map of glutamate metabolism. J. Cell. Commun. Signal. 2016, 10, 69–75. [Google Scholar] [CrossRef]
- Plaitakis, A.; Metaxari, M.; Shashidharan, P. Nerve tissue-specific (GLUD2) and housekeeping (GLUD1) human glutamate dehydrogenases are regulated by distinct allosteric mechanisms: Implications for biologic function. J. Neurochem. 2000, 75, 1862–1869. [Google Scholar] [CrossRef]
- Lowry, M.; Hall, D.E.; Brosnan, J.T. Hydroxyproline metabolism by the rat kidney: Distribution of renal enzymes of hydroxyproline catabolism and renal conversion of hydroxyproline to glycine and serine. Metabolism 1985, 34, 955–961. [Google Scholar] [CrossRef]
- Brigham, M.P.; Stein, W.H.; Moore, S. The Concentrations of Cysteine and Cystine in Human Blood Plasma. J. Clin. Investig. 1960, 39, 1633–1638. [Google Scholar] [CrossRef]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Smith, M.; Henderson, M.J. Reference data for cerebrospinal fluid and the utility of amino acid measurement for the diagnosis of inborn errors of metabolism. Ann. Clin. Biochem. 2006, 43, 63–66. [Google Scholar] [CrossRef]
- Mason, S.; Reinecke, C.J.; Solomons, R. Cerebrospinal Fluid Amino Acid Profiling of Pediatric Cases with Tuberculous Meningitis. Front. Neurosci. 2017, 11, 534. [Google Scholar] [CrossRef] [PubMed]
- Conter, C.; Rolland, M.O.; Cheillan, D.; Bonnet, V.; Maire, I.; Froissart, R. Genetic heterogeneity of the GLDC gene in 28 unrelated patients with glycine encephalopathy. J. Inherit. Metab. Dis. 2006, 29, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, Z.; Dai, Z.; Yang, Y.; Wang, J.; Wu, G. Glycine metabolism in animals and humans: Implications for nutrition and health. Amino Acids 2013, 45, 463–477. [Google Scholar] [CrossRef]
- Darling, P.B.; Grunow, J.; Rafii, M.; Brookes, S.; Ball, R.O.; Pencharz, P.B. Threonine dehydrogenase is a minor degradative pathway of threonine catabolism in adult humans. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E877–E884. [Google Scholar] [CrossRef]
- Hanigan, M.H. Gamma-glutamyl transpeptidase: Redox regulation and drug resistance. Adv. Cancer Res. 2014, 122, 103–141. [Google Scholar]
- Zhang, H.; Liu, H.; Dickinson, D.A.; Liu, R.M.; Postlethwait, E.M.; Laperche, Y.; Forman, H.J. gamma-Glutamyl transpeptidase is induced by 4-hydroxynonenal via EpRE/Nrf2 signaling in rat epithelial type II cells. Free Radic. Biol. Med. 2006, 40, 1281–1292. [Google Scholar] [CrossRef]
- Chikhi, N.; Holic, N.; Guellaen, G.; Laperche, Y. Gamma-glutamyl transpeptidase gene organization and expression: A comparative analysis in rat, mouse, pig and human species. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 1999, 122, 367–380. [Google Scholar] [CrossRef]
- Tate, S.S.; Ross, L.L.; Meister, A. The -glutamyl cycle in the choroid plexus: Its possible function in amino acid transport. Proc. Natl. Acad. Sci. USA 1973, 70, 1447–1449. [Google Scholar] [CrossRef]
- Paolicchi, A.; Dominici, S.; Pieri, L.; Maellaro, E.; Pompella, A. Glutathione catabolism as a signaling mechanism. Biochem. Pharmacol. 2002, 64, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Paolicchi, A.; Sotiropuolou, M.; Perego, P.; Daubeuf, S.; Visvikis, A.; Lorenzini, E.; Franzini, M.; Romiti, N.; Chieli, E.; Leone, R.; et al. Gamma-Glutamyl transpeptidase catalyses the extracellular detoxification of cisplatin in a human cell line derived from the proximal convoluted tubule of the kidney. Eur. J. Cancer 2003, 39, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, S.; Leroy, P.; Paolicchi, A.; Pompella, A.; Wellman, M.; Galteau, M.M.; Visvikis, A. Enhanced resistance of HeLa cells to cisplatin by overexpression of gamma-glutamyltransferase. Biochem. Pharmacol. 2002, 64, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Buffo, A.; Gotz, M. The novel roles of glial cells revisited: The contribution of radial glia and astrocytes to neurogenesis. Curr. Top. Dev. Biol. 2005, 69, 67–99. [Google Scholar] [PubMed]
- Rakic, P. Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 2009, 10, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernandez, E.; Maestre, C.; Moncada, S.; Bolanos, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Mattson, M.P. PDGFs protect hippocampal neurons against energy deprivation and oxidative injury: Evidence for induction of antioxidant pathways. J. Neurosci. 1995, 15, 7095–7104. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Zilberter, Y. Critical state of energy metabolism in brain slices: The principal role of oxygen delivery and energy substrates in shaping neuronal activity. Front. Neuroenergetics 2011, 3, 9. [Google Scholar] [CrossRef]
- Hall, C.N.; Klein-Flugge, M.C.; Howarth, C.; Attwell, D. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Ho, M.S.; Parpura, V. Evolution of Neuroglia. Adv. Exp. Med. Biol. 2019, 1175, 15–44. [Google Scholar]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2015, 40, 2570–2582. [Google Scholar] [CrossRef]
- Mattson, M.P.; Shea, T.B. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Zorec, R. Gliotransmission: Exocytotic release from astrocytes. Brain Res. Rev. 2010, 63, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Heneka, M.T.; Montana, V.; Oliet, S.H.R.; Schousboe, A.; Haydon, P.G.; Stout, R.F., Jr.; Spray, D.C.; Reichenbach, A.; Pannicke, T.; et al. Glial cells in (patho)physiology. J. Neurochem. 2012, 121, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef]
- Danbolt, N.C.; Storm-Mathisen, J.; Kanner, B.I. An [Na+ + K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 1992, 51, 295–310. [Google Scholar] [CrossRef]
- Bergles, D.E.; Jahr, C.E. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 1997, 19, 1297–1308. [Google Scholar] [CrossRef]
- Kvamme, E. Synthesis of glutamate and its regulation. Prog. Brain Res. 1998, 116, 73–85. [Google Scholar]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake stimulates Na+,K+-ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J. Neurochem. 1997, 69, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, R.; Pierre, K.; Lengacher, S.; Magistretti, P.J.; Pellerin, L. Cell-specific expression pattern of monocarboxylate transporters in astrocytes and neurons observed in different mouse brain cortical cell cultures. J. Neurosci. Res. 2003, 73, 141–155. [Google Scholar] [CrossRef]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; Von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martín, A.; Romero-Gómez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell. Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Obel, L.F.; Walls, A.B.; Schousboe, A.; Faek, S.A.; Jajo, F.S.; Waagepetersen, H.S. Novel model of neuronal bioenergetics: Postsynaptic utilization of glucose but not lactate correlates positively with Ca2+ signalling in cultured mouse glutamatergic neurons. ASN Neuro 2012, 4, e00083. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Sonnewald, U.; Waagepetersen, H.S. Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J. Cereb. Blood Flow Metab. 2006, 26, 1285–1297. [Google Scholar] [CrossRef]
- Patel, A.B.; Lai, J.C.; Chowdhury, G.M.; Hyder, F.; Rothman, D.L.; Shulman, R.G.; Behar, K.L. Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc. Natl. Acad. Sci. USA 2014, 111, 5385–5390. [Google Scholar] [CrossRef]
- Lundgaard, I.; Li, B.; Xie, L.; Kang, H.; Sanggaard, S.; Haswell, J.D.R.; Sun, W.; Goldman, S.; Blekot, S.; Nielsen, M.; et al. Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun. 2015, 6, 6807. [Google Scholar] [CrossRef]
- Armbruster, M.; Naskar, S.; Garcia, J.P.; Sommer, M.; Kim, E.; Adam, Y.; Haydon, P.G.; Boyden, E.S.; Cohen, A.E.; Dulla, C.G. Neuronal activity drives pathway-specific depolarization of peripheral astrocyte processes. Nat. Neurosci. 2022, 25, 607–616. [Google Scholar] [CrossRef]
- Vicario, N.; Parenti, R. Connexins Signatures of the Neurovascular Unit and Their Physio-Pathological Functions. Int. J. Mol. Sci. 2022, 23, 9510. [Google Scholar] [CrossRef]
- Huang, S.-F.; Othman, A.; Koshkin, A.; Fischer, S.; Fischer, D.; Zamboni, N.; Ono, K.; Sawa, T.; Ogunshola, O.O. Astrocyte glutathione maintains endothelial barrier stability. Redox Biol. 2020, 34, 101576. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; McConnell, E.; Pare, J.-F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 2013, 339, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Hupfeld, K.E.; Hyatt, H.W.; Alvarez Jerez, P.; Mikkelsen, M.; Hass, C.J.; Edden, R.A.E.; Seidler, R.D.; Porges, E.C. In Vivo Brain Glutathione is Higher in Older Age and Correlates with Mobility. Cereb. Cortex 2021, 31, 4576–4594. [Google Scholar] [CrossRef]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006, 1090, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Slivka, A.; Spina, M.B.; Cohen, G. Reduced and oxidized glutathione in human and monkey brain. Neurosci. Lett. 1987, 74, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Krüger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef]
- Terpstra, M.; Henry, P.G.; Gruetter, R. Measurement of reduced glutathione (GSH) in human brain using LCModel analysis of difference-edited spectra. Magn. Reson. Med. 2003, 50, 19–23. [Google Scholar] [CrossRef]
- Akerboom, T.P.; Bilzer, M.; Sies, H. The relationship of biliary glutathione disulfide efflux and intracellular glutathione disulfide content in perfused rat liver. J. Biol. Chem. 1982, 257, 4248–4252. [Google Scholar] [CrossRef]
- Langeveld, C.H.; Schepens, E.; Jongenelen, C.A.; Stoof, J.C.; Hjelle, O.P.; Ottersen, O.P.; Drukarch, B. Presence of glutathione immunoreactivity in cultured neurones and astrocytes. Neuroreport 1996, 7, 1833–1836. [Google Scholar] [CrossRef]
- Slivka, A.; Mytilineou, C.; Cohen, G. Histochemical evaluation of glutathione in brain. Brain Res. 1987, 409, 275–284. [Google Scholar] [CrossRef]
- Philbert, M.A.; Beiswanger, C.M.; Waters, D.K.; Reuhl, K.R.; Lowndes, H.E. Cellular and regional distribution of reduced glutathione in the nervous system of the rat: Histochemical localization by mercury orange and o-phthaldialdehyde-induced histofluorescence. Toxicol. Appl. Pharmacol. 1991, 107, 215–227. [Google Scholar] [CrossRef]
- Orwar, O.; Li, X.; Andine, P.; Bergstrom, C.M.; Hagberg, H.; Folestad, S.; Sandberg, M. Increased intra- and extracellular concentrations of gamma-glutamylglutamate and related dipeptides in the ischemic rat striatum: Involvement of glutamyl transpeptidase. J. Neurochem. 1994, 63, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Chou, S.T.; Lin, N.N.; Liu, L.; Tsai, P.J.; Kuo, J.S.; Lai, J.S. Determination of extracellular glutathione in rat brain by microdialysis and high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B Biomed. Appl. 1994, 661, 231–235. [Google Scholar] [CrossRef]
- Zangerle, L.; Cuenod, M.; Winterhalter, K.H.; Do, K.Q. Screening of thiol compounds: Depolarization-induced release of glutathione and cysteine from rat brain slices. J. Neurochem. 1992, 59, 181–189. [Google Scholar] [CrossRef]
- Li, X.; Wallin, C.; Weber, S.G.; Sandberg, M. Net efflux of cysteine, glutathione and related metabolites from rat hippocampal slices during oxygen/glucose deprivation: Dependence on gamma-glutamyl transpeptidase. Brain Res. 1999, 815, 81–88. [Google Scholar] [CrossRef]
- Han, J.; Cheng, F.C.; Yang, Z.; Dryhurst, G. Inhibitors of mitochondrial respiration, iron (II), and hydroxyl radical evoke release and extracellular hydrolysis of glutathione in rat striatum and substantia nigra: Potential implications to Parkinson’s disease. J. Neurochem. 1999, 73, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Hamprecht, B. Glutathione restoration as indicator for cellular metabolism of astroglial cells. Dev. Neurosci. 1998, 20, 401–407. [Google Scholar] [CrossRef]
- Yudkoff, M.; Pleasure, D.; Cregar, L.; Lin, Z.P.; Nissim, I.; Stern, J.; Nissim, I. Glutathione turnover in cultured astrocytes: Studies with [15N]glutamate. J. Neurochem. 1990, 55, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Juurlink, B.H.; Schultke, E.; Hertz, L. Glutathione release and catabolism during energy substrate restriction in astrocytes. Brain Res. 1996, 710, 229–233. [Google Scholar] [CrossRef]
- Kranich, O.; Hamprecht, B.; Dringen, R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci. Lett. 1996, 219, 211–214. [Google Scholar] [CrossRef]
- Bolanos, J.P.; Heales, S.J.; Peuchen, S.; Barker, J.E.; Land, J.M.; Clark, J.B. Nitric oxide-mediated mitochondrial damage: A potential neuroprotective role for glutathione. Free Radic. Biol. Med. 1996, 21, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef]
- Voulgaris, D.; Nikolakopoulou, P.; Herland, A. Generation of Human iPSC-Derived Astrocytes with a mature star-shaped phenotype for CNS modeling. Stem Cell Rev. Rep. 2022, 18, 2494–2512. [Google Scholar] [CrossRef] [PubMed]
- Sagara, J.; Makino, N.; Bannai, S. Glutathione efflux from cultured astrocytes. J. Neurochem. 1996, 66, 1876–1881. [Google Scholar] [CrossRef]
- Rana, S.; Dringen, R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett. 2007, 415, 45–48. [Google Scholar] [CrossRef]
- Dringen, R.; Kranich, O.; Hamprecht, B. The gamma-glutamyl transpeptidase inhibitor acivicin preserves glutathione released by astroglial cells in culture. Neurochem. Res. 1997, 22, 727–733. [Google Scholar] [CrossRef]
- Kranich, O.; Dringen, R.; Sandberg, M.; Hamprecht, B. Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: Preference for cystine. Glia 1998, 22, 11–18. [Google Scholar] [CrossRef]
- McGann, J.C.; Mandel, G. Neuronal activity induces glutathione metabolism gene expression in astrocytes. Glia 2018, 66, 2024–2039. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.; Al-Mubarak, B.; Martel, M.-A.; McKay, S.; Wheelan, N.; Hasel, P.; Márkus, N.M.; Baxter, P.; Deighton, R.F.; Serio, A.; et al. Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat. Commun. 2015, 6, 7066. [Google Scholar] [CrossRef]
- Jimenez-Blasco, D.; Santofimia-Castaño, P.; Gonzalez, A.; Almeida, A.; Bolaños, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, S.; Almeida, A.; Bolanos, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Schnaar, R.L.; Coyle, J.T. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990, 4, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Hamprecht, B. N-acetylcysteine, but not methionine or 2-oxothiazolidine-4-carboxylate, serves as cysteine donor for the synthesis of glutathione in cultured neurons derived from embryonal rat brain. Neurosci. Lett. 1999, 259, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Han, J.; Lu, H.; Cui, C.; Yang, J.; Cui, Q.; Cai, J.; Zhou, Y.; Tang, C.; Xu, G.; et al. Cystathionine beta synthase-hydrogen sulfide system in paraventricular nucleus reduced high fatty diet induced obesity and insulin resistance by brain-adipose axis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3281–3291. [Google Scholar] [CrossRef]
- Robert, K.; Vialard, F.; Thiery, E.; Toyama, K.; Sinet, P.M.; Janel, N.; London, J. Expression of the cystathionine beta synthase (CBS) gene during mouse development and immunolocalization in adult brain. J. Histochem. Cytochem. 2003, 51, 363–371. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, E.; Devesa, A.; Garcia, C.; Puertes, I.R.; Pellin, A.; Vina, J.R. Biosynthesis and maintenance of GSH in primary astrocyte cultures: Role of L-cystine and ascorbate. Brain Res. 1995, 680, 157–163. [Google Scholar] [CrossRef]
- Enokido, Y.; Suzuki, E.; Iwasawa, K.; Namekata, K.; Okazawa, H.; Kimura, H. Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 2005, 19, 1854–1856. [Google Scholar] [CrossRef]
- Wan, X.; Ma, B.; Wang, X.; Guo, C.; Sun, J.; Cui, J.; Li, L. S-Adenosylmethionine Alleviates Amyloid-beta-Induced Neural Injury by Enhancing Trans-Sulfuration Pathway Activity in Astrocytes. J. Alzheimer’s Dis. 2020, 76, 981–995. [Google Scholar] [CrossRef]
- Ishii, I.; Akahoshi, N.; Yu, X.N.; Kobayashi, Y.; Namekata, K.; Komaki, G.; Kimura, H. Murine cystathionine gamma-lyase: Complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem. J. 2004, 381, 113–123. [Google Scholar] [CrossRef]
- Sharma, K.; Schmitt, S.; Bergner, C.G.; Tyanova, S.; Kannaiyan, N.; Manrique-Hoyos, N.; Kongi, K.; Cantuti, L.; Hanisch, U.-K.; Philips, M.A.; et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015, 18, 1819–1831. [Google Scholar] [CrossRef]
- Burdo, J.; Dargusch, R.; Schubert, D. Distribution of the cystine/glutamate antiporter system xc- in the brain, kidney, and duodenum. J. Histochem. Cytochem. 2006, 54, 549–557. [Google Scholar] [CrossRef]
- Seib, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the system x(C)- cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001, 902, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Himi, T.; Ikeda, M.; Yasuhara, T.; Nishida, M.; Morita, I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm. 2003, 110, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Curthoys, N.P.; Watford, M. Regulation of glutaminase activity and glutamine metabolism. Annu. Rev. Nutr. 1995, 15, 133–159. [Google Scholar] [CrossRef] [PubMed]
- Hogstad, S.; Svenneby, G.; Torgner, I.A.; Kvamme, E.; Hertz, L.; Schousboe, A. Glutaminase in neurons and astrocytes cultured from mouse brain: Kinetic properties and effects of phosphate, glutamate, and ammonia. Neurochem. Res. 1988, 13, 383–388. [Google Scholar] [CrossRef]
- Zafra, F.; Gomeza, J.; Olivares, L.; Aragon, C.; Gimenez, C. Regional distribution and developmental variation of the glycine transporters GLYT1 and GLYT2 in the rat CNS. Eur. J. Neurosci. 1995, 7, 1342–1352. [Google Scholar] [CrossRef]
- Dringen, R.; Kranich, O.; Loschmann, P.A.; Hamprecht, B. Use of dipeptides for the synthesis of glutathione by astroglia-rich primary cultures. J. Neurochem. 1997, 69, 868–874. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Cabrera, R.; Fernandez-Fernandez, S.; Bobo-Jimenez, V.; Escobar, J.; Sastre, J.; Almeida, A.; Bolanos, J.P. gamma-Glutamylcysteine detoxifies reactive oxygen species by acting as glutathione peroxidase-1 cofactor. Nat. Commun. 2012, 3, 718. [Google Scholar] [CrossRef]
- Lai, L.; Tan, T.M. Role of glutathione in the multidrug resistance protein 4 (MRP4/ABCC4)-mediated efflux of cAMP and resistance to purine analogues. Biochem. J. 2002, 361, 497–503. [Google Scholar] [CrossRef]
- Okamura, T.; Okada, M.; Kikuchi, T.; Wakizaka, H.; Zhang, M.R. Mechanisms of glutathione-conjugate efflux from the brain into blood: Involvement of multiple transporters in the course. J. Cereb. Blood Flow Metab. 2020, 40, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Tulpule, K.; Hohnholt, M.C.; Dringen, R. Formaldehyde metabolism and formaldehyde-induced stimulation of lactate production and glutathione export in cultured neurons. J. Neurochem. 2013, 125, 260–272. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Schulz, J.B.; Dringen, R. Glutathione release from cultured brain cells: Multidrug resistance protein 1 mediates the release of GSH from rat astroglial cells. J. Neurosci. Res. 2002, 69, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Messing, A. Alexander disease. Handb. Clin. Neurol. 2018, 148, 693–700. [Google Scholar] [PubMed]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.-M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef]
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell. Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef]
- Viedma-Poyatos, A.; de Pablo, Y.; Pekny, M.; Perez-Sala, D. The cysteine residue of glial fibrillary acidic protein is a critical target for lipoxidation and required for efficient network organization. Free Radic. Biol. Med. 2018, 120, 380–394. [Google Scholar] [CrossRef]
- Viedma-Poyatos, A.; Gonzalez-Jimenez, P.; Pajares, M.A.; Perez-Sala, D. Alexander disease GFAP R239C mutant shows increased susceptibility to lipoxidation and elicits mitochondrial dysfunction and oxidative stress. Redox Biol. 2022, 55, 102415. [Google Scholar] [CrossRef] [PubMed]
- Garfinkel, D. A simulation study of the metabolism and compartmentation in brain of glutamate, aspartate, the Krebs cycle, and related metabolites. J. Biol. Chem. 1966, 241, 3918–3929. [Google Scholar] [CrossRef]
- Desagher, S.; Glowinski, J.; Premont, J. Astrocytes protect neurons from hydrogen peroxide toxicity. J. Neurosci. 1996, 16, 2553–2562. [Google Scholar] [CrossRef]
- Stone, R.; Stewart, V.C.; Hurst, R.D.; Clark, J.B.; Heales, S.J. Astrocyte nitric oxide causes neuronal mitochondrial damage, but antioxidant release limits neuronal cell death. Ann. N. Y Acad. Sci. 1999, 893, 400–403. [Google Scholar] [CrossRef]
- Gutbier, S.; Spreng, A.S.; Delp, J.; Schildknecht, S.; Karreman, C.; Suciu, I.; Brunner, T.; Groettrup, M.; Leist, M. Prevention of neuronal apoptosis by astrocytes through thiol-mediated stress response modulation and accelerated recovery from proteotoxic stress. Cell Death Differ. 2018, 25, 2101–2117. [Google Scholar] [CrossRef] [PubMed]
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kölle, P.; Tschoep, K.; Issels, R.D.; Daniel, P.T.; et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility vs. resistance to cell death. Oncogene 2008, 27, 1618–1628. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Elsherbiny, N.M.; Zaidi, Y.; Rajpurohit, P. Homocysteine and Age-Related Central Nervous System Diseases: Role of Inflammation. Int. J. Mol. Sci. 2021, 22, 6259. [Google Scholar] [CrossRef]
- Borowczyk, K.; Shih, D.M.; Jakubowski, H. Metabolism and neurotoxicity of homocysteine thiolactone in mice: Evidence for a protective role of paraoxonase 1. J. Alzheimer’s Dis. 2012, 30, 225–231. [Google Scholar] [CrossRef]
- Hogg, N. The effect of cyst(e)ine on the auto-oxidation of homocysteine. Free Radic. Biol. Med. 1999, 27, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Gharbi, T.; Zhang, Z.; Yang, G.Y. The Function of Astrocyte Mediated Extracellular Vesicles in Central Nervous System Diseases. Front. Cell Dev. Biol. 2020, 8, 568889. [Google Scholar] [CrossRef] [PubMed]
- Schnatz, A.; Muller, C.; Brahmer, A.; Kramer-Albers, E.M. Extracellular Vesicles in neural cell interaction and CNS homeostasis. FASEB Bioadv. 2021, 3, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, S.; Gevi, F.; Pietrucci, D.; Cavinato, L.; Luly, F.R.; Pascucci, L.; Petrini, S.; Ascenzioni, F.; Zolla, L.; Chillemi, G.; et al. Anti-Inflammatory Potential of Cow, Donkey and Goat Milk Extracellular Vesicles as Revealed by Metabolomic Profile. Nutrients 2020, 12, 2908. [Google Scholar] [CrossRef] [PubMed]
- Harmati, M.; Bukva, M.; Boroczky, T.; Buzas, K.; Gyukity-Sebestyen, E. The role of the metabolite cargo of extracellular vesicles in tumor progression. Cancer Metastasis Rev. 2021, 40, 1203–1221. [Google Scholar] [CrossRef]
- Caserta, S.; Ghezzi, P. Release of redox enzymes and micro-RNAs in extracellular vesicles, during infection and inflammation. Free Radic. Biol. Med. 2021, 169, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, S.; Bonetto, V.; Fratelli, M.; Gianazza, E.; Eberini, I.; Massignan, T.; Salmona, M.; Chang, G.; Holmgren, A.; Ghezzi, P. Glutathionylation of human thioredoxin: A possible crosstalk between the glutathione and thioredoxin systems. Proc. Natl. Acad. Sci. USA 2002, 99, 9745–9749. [Google Scholar] [CrossRef]
- Froes, M.M.; Correia, A.H.; Garcia-Abreu, J.; Spray, D.C.; Campos de Carvalho, A.C.; Neto, M.V. Gap-junctional coupling between neurons and astrocytes in primary central nervous system cultures. Proc. Natl. Acad. Sci. USA 1999, 96, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Maubecin, V.; Garcia-Hernandez, F.; Williams, J.T.; Van Bockstaele, E.J. Functional coupling between neurons and glia. J. Neurosci. 2000, 20, 4091–4098. [Google Scholar] [CrossRef]
- Rozental, R.; Andrade-Rozental, A.F.; Zheng, X.; Urban, M.; Spray, D.C.; Chiu, F.C. Gap junction-mediated bidirectional signaling between human fetal hippocampal neurons and astrocytes. Dev. Neurosci. 2001, 23, 420–431. [Google Scholar] [CrossRef]
- Nadarajah, B.; Thomaidou, D.; Evans, W.H.; Parnavelas, J.G. Gap junctions in the adult cerebral cortex: Regional differences in their distribution and cellular expression of connexins. J. Comp. Neurol. 1996, 376, 326–342. [Google Scholar] [CrossRef]
- Dobrenis, K.; Chang, H.Y.; Pina-Benabou, M.H.; Woodroffe, A.; Lee, S.C.; Rozental, R.; Spray, D.C.; Scemes, E. Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. J. Neurosci. Res. 2005, 82, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes—From electrical signals to organelle transfer. J. Cell Sci. 2012, 125, 1089–1098. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Sala, D.; Pajares, M.A. Appraising the Role of Astrocytes as Suppliers of Neuronal Glutathione Precursors. Int. J. Mol. Sci. 2023, 24, 8059. https://doi.org/10.3390/ijms24098059
Pérez-Sala D, Pajares MA. Appraising the Role of Astrocytes as Suppliers of Neuronal Glutathione Precursors. International Journal of Molecular Sciences. 2023; 24(9):8059. https://doi.org/10.3390/ijms24098059
Chicago/Turabian StylePérez-Sala, Dolores, and María A. Pajares. 2023. "Appraising the Role of Astrocytes as Suppliers of Neuronal Glutathione Precursors" International Journal of Molecular Sciences 24, no. 9: 8059. https://doi.org/10.3390/ijms24098059
APA StylePérez-Sala, D., & Pajares, M. A. (2023). Appraising the Role of Astrocytes as Suppliers of Neuronal Glutathione Precursors. International Journal of Molecular Sciences, 24(9), 8059. https://doi.org/10.3390/ijms24098059