
The Potential Biotherapeutic Targets of Contrast-Induced Acute Kidney Injury
Abstract
:1. Introduction
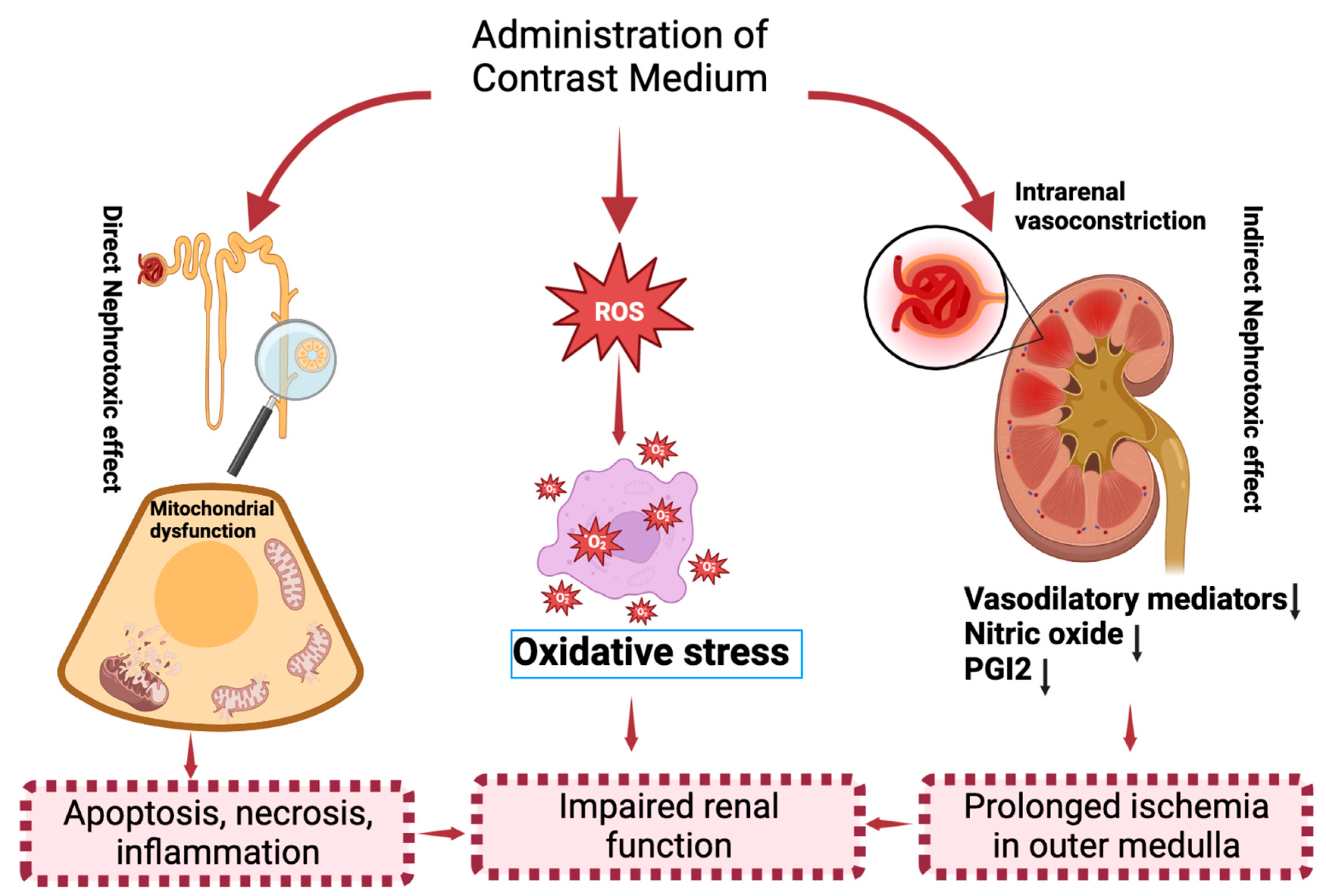
2. Molecular Mechanisms of CI−AKI
3. The Potential Biomarkers of CI−AKI
4. The Biological Targets of CI−AKI
4.1. Apoptotic Targets
4.2. Inflammatory Targets
4.3. Oxidative Stress Targets
4.4. Epigenetic Targets
5. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.-J. Acute kidney injury. Nat. Rev. Dis. Prim. 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Dumas, S.J.; Meta, E.; Borri, M.; Luo, Y.; Li, X.; Rabelink, T.J.; Carmeliet, P. Phenotypic diversity and metabolic specialization of renal endothelial cells. Nat. Rev. Nephrol. 2021, 17, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerdá, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Naseer, A.; Sumida, K.; Molnar, M.Z.; Potukuchi, P.K.; Thomas, F.; Streja, E.; Heung, M.; Abbott, K.C.; Saran, R. Abrupt decline in kidney function precipitating initiation of chronic renal replacement therapy. Kidney Int. Rep. 2018, 3, 602–609. [Google Scholar] [CrossRef]
- Ostermann, M.; Joannidis, M. Acute kidney injury 2016: Diagnosis and diagnostic workup. Crit. Care 2016, 20, 299. [Google Scholar] [CrossRef]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303. [Google Scholar]
- Weiner, I.D.; Verlander, J.W. Renal ammonia metabolism and transport. Compr. Physiol. 2013, 3, 201. [Google Scholar]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef]
- Abdulkhaleq, L.; Assi, M.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.; Hezmee, M. The crucial roles of inflammatory mediators in inflammation: A review. Vet. World 2018, 11, 627. [Google Scholar] [CrossRef]
- Chevalier, R.L. The proximal tubule is the primary target of injury and progression of kidney disease: Role of the glomerulotubular junction. Am. J. Physiol.-Ren. Physiol. 2016, 311, F145–F161. [Google Scholar] [CrossRef]
- Ludes, P.-O.; De Roquetaillade, C.; Chousterman, B.G.; Pottecher, J.; Mebazaa, A. Role of damage-associated molecular patterns in septic acute kidney injury, from injury to recovery. Front. Immunol. 2021, 12, 606622. [Google Scholar] [CrossRef]
- Maaniitty, T.; Stenström, I.; Uusitalo, V.; Ukkonen, H.; Kajander, S.; Bax, J.J.; Saraste, A.; Knuuti, J. Incidence of persistent renal dysfunction after contrast enhanced coronary CT angiography in patients with suspected coronary artery disease. Int. J. Cardiovasc. Imaging 2016, 32, 1567–1575. [Google Scholar] [CrossRef]
- Shams, E.; Mayrovitz, H.N. Contrast-induced nephropathy: A review of mechanisms and risks. Cureus 2021, 13, e14842. [Google Scholar] [CrossRef]
- Evola, S.; Lunetta, M.; Macaione, F.; Fonte, G.; Milana, G.; Corrado, E.; Bonura, F.; Novo, G.; Hoffmann, E.; Novo, S. Risk factors for contrast induced nephropathy: A study among Italian patients. Indian Heart J. 2012, 64, 484–491. [Google Scholar] [CrossRef]
- Jung, S.C.; Kim, S.H.; Cho, J.Y. A comparison of the use of contrast media with different iodine concentrations for multidetector CT of the kidney. Korean J. Radiol. 2011, 12, 714–721. [Google Scholar] [CrossRef]
- Sendeski, M.M. Pathophysiology of renal tissue damage by iodinated contrast media. Clin. Exp. Pharmacol. Physiol. 2011, 38, 292–299. [Google Scholar] [CrossRef]
- Andreucci, M.; Faga, T.; Pisani, A.; Sabbatini, M.; Michael, A. Acute kidney injury by radiographic contrast media: Pathogenesis and prevention. BioMed Res. Int. 2014, 2014, 362725. [Google Scholar] [CrossRef]
- Mamoulakis, C.; Tsarouhas, K.; Fragkiadoulaki, I.; Heretis, I.; Wilks, M.F.; Spandidos, D.A.; Tsitsimpikou, C.; Tsatsakis, A. Contrast-induced nephropathy: Basic concepts, pathophysiological implications and prevention strategies. Pharmacol. Ther. 2017, 180, 99–112. [Google Scholar] [CrossRef]
- Cho, E.; Ko, G.-J. The Pathophysiology and the Management of Radiocontrast-Induced Nephropathy. Diagnostics 2022, 12, 180. [Google Scholar] [CrossRef]
- Palli, E.; Makris, D.; Papanikolaou, J.; Garoufalis, G.; Zakynthinos, E. Contrast-induced nephropathy in aged critically ill patients. Oxidative Med. Cell. Longev. 2014, 2014, 756469. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Suo, M.; Liu, L.; Qi, Y.; Zhang, C.; Xie, L.; Zheng, X.; Ma, C.; Li, J.; Yang, J. Melatonin alleviates contrast-induced acute kidney injury by activation of Sirt3. Oxidative Med. Cell. Longev. 2021, 2021, 6668887. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.-J.; Kim, J.H.; Chang, Y.K.; Park, C.W.; Kim, S.Y.; Hong, Y.A. Inhibition of xanthine oxidoreductase protects against contrast-induced renal tubular injury by activating adenosine monophosphate-activated protein kinase. Free Radic. Biol. Med. 2019, 145, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Regner, K.R.; Roman, R.J. Role of medullary blood flow in the pathogenesis of renal ischemia-reperfusion injury. Curr. Opin. Nephrol. Hypertens. 2012, 21, 33. [Google Scholar] [CrossRef]
- Mehran, R.; Nikolsky, E. Contrast-induced nephropathy: Definition, epidemiology, and patients at risk. Kidney Int. 2006, 69, S11–S15. [Google Scholar] [CrossRef]
- Seeliger, E.; Sendeski, M.; Rihal, C.S.; Persson, P.B. Contrast-induced kidney injury: Mechanisms, risk factors, and prevention. Eur. Heart J. 2012, 33, 2007–2015. [Google Scholar] [CrossRef]
- Solomon, R.; Dauerman, H.L. Contrast-induced acute kidney injury. Circulation 2010, 122, 2451–2455. [Google Scholar] [CrossRef]
- Hashemi, M.; Karami-Tehrani, F.; Ghavami, S. Cytotoxicity effect of Cladribine on the MCF-7 human breast cancer cell line. Iran. Biomed. J. 2004, 8, 7–12. [Google Scholar]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.; Hanson, S.Y. Radiographic contrast media–induced tubular injury: Evaluation of oxidant stress and plasma membrane integrity. Kidney Int. 2003, 64, 128–139. [Google Scholar] [CrossRef]
- Quintavalle, C.; Brenca, M.; De Micco, F.; Fiore, D.; Romano, S.; Romano, M.; Apone, F.; Bianco, A.; Zabatta, M.; Troncone, G. In vivo and in vitro assessment of pathways involved in contrast media-induced renal cells apoptosis. Cell Death Dis. 2011, 2, e155. [Google Scholar] [CrossRef]
- Nelson, J.B.; Udan, M.S.; Guruli, G.; Pflug, B.R. Endothelin-1 inhibits apoptosis in prostate cancer. Neoplasia 2005, 7, 631–637. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, N.; Shi, G.; Wang, H. Geniposide ameliorated sepsis-induced acute kidney injury by activating PPARγ. Aging 2020, 12, 22744. [Google Scholar] [CrossRef]
- Ashraf, M.I.; Ebner, M.; Wallner, C.; Haller, M.; Khalid, S.; Schwelberger, H.; Koziel, K.; Enthammer, M.; Hermann, M.; Sickinger, S. A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury. Cell Commun. Signal. 2014, 12, 6. [Google Scholar] [CrossRef]
- Chang, K.-C.; Hsu, C.-C.; Liu, S.-H.; Su, C.-C.; Yen, C.-C.; Lee, M.-J.; Chen, K.-L.; Ho, T.-J.; Hung, D.-Z.; Wu, C.-C. Cadmium induces apoptosis in pancreatic β-cells through a mitochondria-dependent pathway: The role of oxidative stress-mediated c-Jun N-terminal kinase activation. PLoS ONE 2013, 8, e54374. [Google Scholar] [CrossRef]
- Cobley, J.N.; Husi, H. Immunological techniques to assess protein thiol redox state: Opportunities, challenges and solutions. Antioxidants 2020, 9, 315. [Google Scholar] [CrossRef]
- Ranneh, Y.; Ali, F.; Akim, A.M.; Hamid, H.A.; Khazaai, H.; Fadel, A. Crosstalk between reactive oxygen species and pro-inflammatory markers in developing various chronic diseases: A review. Appl. Biol. Chem. 2017, 60, 327–338. [Google Scholar] [CrossRef]
- Hansen, P.R. Chronic inflammatory diseases and atherosclerotic cardiovascular disease: Innocent bystanders or partners in crime? Curr. Pharm. Des. 2018, 24, 281–290. [Google Scholar] [CrossRef]
- Li, Y.; Ren, K. The mechanism of contrast-induced acute kidney injury and its association with diabetes mellitus. Contrast Media Mol. Imaging 2020, 2020, 3295176. [Google Scholar] [CrossRef] [PubMed]
- Lorenzatti, A.J.; Servato, M.L. New evidence on the role of inflammation in CVD risk. Curr. Opin. Cardiol. 2019, 34, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Sendeski, M.M.; Persson, A.B.; Liu, Z.Z.; Busch, J.F.; Weikert, S.; Persson, P.B.; Hippenstiel, S.; Patzak, A. Iodinated contrast media cause endothelial damage leading to vasoconstriction of human and rat vasa recta. Am. J. Physiol.-Ren. Physiol. 2012, 303, F1592–F1598. [Google Scholar] [CrossRef] [PubMed]
- Tsarouhas, K.; Tsitsimpikou, C.; Papantoni, X.; Lazaridou, D.; Koutouzis, M.; Mazzaris, S.; Rezaee, R.; Mamoulakis, C.; Georgoulias, P.; Nepka, C. Oxidative stress and kidney injury in trans-radial catheterization. Biomed. Rep. 2018, 8, 417–425. [Google Scholar] [CrossRef]
- Cervantes-Gracia, K.; Raja, K.; Llanas-Cornejo, D.; Cobley, J.N.; Megson, I.L.; Chahwan, R.; Husi, H. Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy. Vessel Plus 2020, 4, 27. [Google Scholar] [CrossRef]
- Van Biesen, W.; Vanholder, R.; Lameire, N. Defining acute renal failure: RIFLE and beyond. Clin. J. Am. Soc. Nephrol. 2006, 1, 1314–1319. [Google Scholar] [CrossRef]
- Parikh, C.R.; Abraham, E.; Ancukiewicz, M.; Edelstein, C.L. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J. Am. Soc. Nephrol. 2005, 16, 3046–3052. [Google Scholar] [CrossRef]
- Peng, Z.-Y. The biomarkers for acute kidney injury: A clear road ahead? J. Transl. Intern. Med. 2016, 4, 95–98. [Google Scholar] [CrossRef]
- Vaidya, V.S.; Ferguson, M.A.; Bonventre, J.V. Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 463. [Google Scholar] [CrossRef]
- Koyner, J.L.; Vaidya, V.S.; Bennett, M.R.; Ma, Q.; Worcester, E.; Akhter, S.A.; Raman, J.; Jeevanandam, V.; O’Connor, M.F.; Devarajan, P. Urinary biomarkers in the clinical prognosis and early detection of acute kidney injury. Clin. J. Am. Soc. Nephrol. 2010, 5, 2154–2165. [Google Scholar] [CrossRef]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.R.; Portilla, D.; Okusa, M.D. A basic science view of acute kidney injury biomarkers. Nephrol. Dial. Transplant. 2014, 29, 1301–1311. [Google Scholar] [CrossRef]
- Mamoulakis, C.; Fragkiadoulaki, I.; Karkala, P.; Georgiadis, G.; Zisis, I.-E.; Stivaktakis, P.; Kalogeraki, A.; Tsiaoussis, I.; Burykina, T.; Lazopoulos, G. Contrast-induced nephropathy in an animal model: Evaluation of novel biomarkers in blood and tissue samples. Toxicol. Rep. 2019, 6, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Tufro, A.; Teichman, J.; Woda, C.; Villegas, G. Semaphorin3a inhibits ureteric bud branching morphogenesis. Mech. Dev. 2008, 125, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Her, G.M.; Chiang, C.C.; Chen, W.Y.; Wu, J.L. In vivo studies of liver-type fatty acid binding protein (L-FABP) gene expression in liver of transgenic zebrafish (Danio rerio). FEBS Lett. 2003, 538, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, B.; Chen, S.; Li, H.; Liu, J.; Mai, Z.; Chen, E.; Zhou, C.; Sun, G.; Guo, Z. Novel biomarkers for post-contrast acute kidney injury identified from long non-coding RNA expression profiles. Int. J. Biol. Sci. 2021, 17, 882. [Google Scholar] [CrossRef]
- Malyszko, J.; Bachorzewska-Gajewska, H.; Koc-Zorawska, E.; Malyszko, J.S.; Kobus, G.; Dobrzycki, S. Midkine: A novel and early biomarker of contrast-induced acute kidney injury in patients undergoing percutaneous coronary interventions. BioMed Res. Int. 2015, 2015, 879509. [Google Scholar] [CrossRef]
- Kato, K.; Sato, N.; Yamamoto, T.; Iwasaki, Y.K.; Tanaka, K.; Mizuno, K. Valuable markers for contrast-induced nephropathy in patients undergoing cardiac catheterization. Circ. J. 2008, 72, 1499–1505. [Google Scholar] [CrossRef]
- Ren, L.; Ji, J.; Fang, Y.; Jiang, S.; Lin, Y.; Bo, J.; Qian, J.; Xu, X.; Ding, X. Assessment of urinary N-acetyl-β-glucosaminidase as an early marker of contrast-induced nephropathy. J. Int. Med Res. 2011, 39, 647–653. [Google Scholar] [CrossRef]
- Vijayasimha, M.; Padma, V.V.; Mujumdar, S.K.D.; Satyanarayana, P.; Yadav, A. Kidney injury molecule-1: A urinary biomarker for contrast-induced acute kidney injury. Med. J. Dr. DY Patil Univ. 2014, 7, 321. [Google Scholar] [CrossRef]
- Hirsch, R.; Dent, C.; Pfriem, H.; Allen, J.; Beekman, R.H.; Ma, Q.; Dastrala, S.; Bennett, M.; Mitsnefes, M.; Devarajan, P. NGAL is an early predictive biomarker of contrast-induced nephropathy in children. Pediatr. Nephrol. 2007, 22, 2089–2095. [Google Scholar] [CrossRef]
- Ling, W.; Zhaohui, N.; Ben, H.; Leyi, G.; Jian, L.; Huili, D.; Jiaqi, Q. Urinary IL-18 and NGAL as early predictive biomarkers in contrast-induced nephropathy after coronary angiography. Nephron Clin. Pract. 2008, 108, c176–c181. [Google Scholar] [CrossRef]
- Pahade, J.K.; LeBedis, C.A.; Raptopoulos, V.D.; Avigan, D.E.; Yam, C.S.; Kruskal, J.B.; Pedrosa, I. Incidence of Contrast-Induced Nephropathy in Patients With Multiple Myeloma Undergoing Contrast-Enhanced CT. Am. J. Roentgenol. 2011, 196, 1094–1101. [Google Scholar] [CrossRef]
- Sadat, U.; Walsh, S.R.; Norden, A.G.; Gillard, J.H.; Boyle, J.R. Does oral N-acetylcysteine reduce contrast-induced renal injury in patients with peripheral arterial disease undergoing peripheral angiography? A randomized-controlled study. Angiology 2011, 62, 225–230. [Google Scholar] [CrossRef]
- Aspelin, P.; Aubry, P.; Fransson, S.-G.; Strasser, R.; Willenbrock, R.; Berg, K.J. Nephrotoxic effects in high-risk patients undergoing angiography. N. Engl. J. Med. 2003, 348, 491–499. [Google Scholar] [CrossRef]
- Chao, A.; Major, K.; Kumar, S.R.; Patel, K.; Trujillo, I.; Hood, D.B.; Rowe, V.L.; Weaver, F.A. Carbon dioxide digital subtraction angiography–assisted endovascular aortic aneurysm repair in the azotemic patient. J. Vasc. Surg. 2007, 45, 451–460. [Google Scholar] [CrossRef]
- Mehran, R.; Dangas, G.D.; Weisbord, S.D. Contrast-associated acute kidney injury. N. Engl. J. Med. 2019, 380, 2146–2155. [Google Scholar] [CrossRef]
- Leoncini, M.; Toso, A.; Maioli, M.; Tropeano, F.; Villani, S.; Bellandi, F. Early high-dose rosuvastatin for contrast-induced nephropathy prevention in acute coronary syndrome: Results from the PRATO-ACS Study (Protective Effect of Rosuvastatin and Antiplatelet Therapy on contrast-induced acute kidney injury and myocardial damage in patients with Acute Coronary Syndrome). J. Am. Coll. Cardiol. 2014, 63, 71–79. [Google Scholar]
- Quintavalle, C.; Fiore, D.; De Micco, F.; Visconti, G.; Focaccio, A.; Golia, B.; Ricciardelli, B.; Donnarumma, E.; Bianco, A.; Zabatta, M.A. Impact of a high loading dose of atorvastatin on contrast-induced acute kidney injury. Circulation 2012, 126, 3008–3016. [Google Scholar] [CrossRef]
- Er, F.; Nia, A.M.; Dopp, H.; Hellmich, M.; Dahlem, K.M.; Caglayan, E.; Kubacki, T.; Benzing, T.; Erdmann, E.; Burst, V. Ischemic preconditioning for prevention of contrast medium–induced nephropathy: Randomized pilot RenPro Trial (Renal Protection Trial). Circulation 2012, 126, 296–303. [Google Scholar] [CrossRef]
- Er, F.; Nia, A.M.; Dopp, H.; Dahlem, K.M.; Caglayan, E.; Erdmann, E.; Gassanov, N.; Hellmich, M.; Burst, V.; Kubacki, T. Response to Letter Regarding Article,“Ischemic Preconditioning for Prevention of Contrast Medium–Induced Nephropathy: Randomized Pilot RenPro-Trial (Renal Protection Trial)”. Circulation 2013, 127, e536. [Google Scholar] [CrossRef] [PubMed]
- Persson, P.B.; Hansell, P.; Liss, P. Pathophysiology of contrast medium–induced nephropathy. Kidney Int. 2005, 68, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-H.; Lee, T.-S.; Lin, S.-J.; Yeh, Y.-C.; Lu, T.-M.; Hsu, C.-P. DDAH-2 alleviates contrast medium iopromide-induced acute kidney injury through nitric oxide synthase. Clin. Sci. 2019, 133, 2361–2378. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, C.; Donnarumma, E.; Fiore, D.; Briguori, C.; Condorelli, G. Therapeutic strategies to prevent contrast-induced acute kidney injury. Curr. Opin. Cardiol. 2013, 28, 676–682. [Google Scholar] [CrossRef]
- Hizoh, I.; Sträter, J.; Schick, C.; Kübler, W.; Haller, C. Radiocontrast-induced DNA fragmentation of renal tubular cells in vitro: Role of hypertonicity. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 1998, 13, 911–918. [Google Scholar] [CrossRef]
- Zhang, F.; Lu, Z.; Wang, F. Advances in the pathogenesis and prevention of contrast-induced nephropathy. Life Sci. 2020, 259, 118379. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- McCullough, P.A.; Choi, J.P.; Feghali, G.A.; Schussler, J.M.; Stoler, R.M.; Vallabahn, R.C.; Mehta, A. Contrast-induced acute kidney injury. J. Am. Coll. Cardiol. 2016, 68, 1465–1473. [Google Scholar] [CrossRef]
- Zhang, J.; Duarte, C.G.; Ellis, S. Contrast medium-and mannitol-induced apoptosis in heart and kidney of SHR rats. Toxicol. Pathol. 1999, 27, 427–435. [Google Scholar] [CrossRef]
- Romano, G.; Briguori, C.; Quintavalle, C.; Zanca, C.; Rivera, N.V.; Colombo, A.; Condorelli, G. Contrast agents and renal cell apoptosis. Eur. Heart J. 2008, 29, 2569–2576. [Google Scholar] [CrossRef]
- Martinou, J.-C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Saikumar, P.; Dong, Z.; Patel, Y.; Hall, K.; Hopfer, U.; Weinberg, J.M.; Venkatachalam, M. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene 1998, 17, 3401–3415. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Wang, Z.; Gall, J.M.; Bonegio, R.; Havasi, A.; Illanes, K.; Schwartz, J.H.; Borkan, S.C. Nucleophosmin, a critical Bax cofactor in ischemia-induced cell death. Mol. Cell. Biol. 2013, 33, 1916–1924. [Google Scholar] [CrossRef]
- Vaara, S.T.; Lakkisto, P.; Immonen, K.; Tikkanen, I.; Ala-Kokko, T.; Pettilä, V.; Group, F.S. Urinary biomarkers indicative of apoptosis and acute kidney injury in the critically ill. PLoS ONE 2016, 11, e0149956. [Google Scholar] [CrossRef]
- Roth, G.A.; Lebherz-Eichinger, D.; Ankersmit, H.J.; Hacker, S.; Hetz, H.; Vukovich, T.; Perne, A.; Reiter, T.; Farr, A.; Hörl, W.H. Increased total cytokeratin-18 serum and urine levels in chronic kidney disease. Clin. Chim. Acta 2011, 412, 713–717. [Google Scholar] [CrossRef]
- Kim, J.Y.; Barua, S.; Huang, M.Y.; Park, J.; Yenari, M.A.; Lee, J.E. Heat shock protein 70 (HSP70) induction: Chaperonotherapy for neuroprotection after brain injury. Cells 2020, 9, 2020. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Pérez-Villalva, R.; Cortés-González, C.; Ojeda-Cervantes, M.; Gamba, G.; Morales-Buenrostro, L.E.; Bobadilla, N.A. Hsp72 is an early and sensitive biomarker to detect acute kidney injury. EMBO Mol. Med. 2011, 3, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) as new tools for cancer therapy: First steps from bench to bedside. Target. Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Q.; Sun, L.; Chen, L.; Li, Y.; Huang, B.; Liu, Y.; Jiang, C. miR-30e-5p regulates autophagy and apoptosis by targeting Beclin1 involved in contrast-induced acute kidney injury. Curr. Med. Chem. 2021, 28, 7974–7984. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chung, A.C.; Yu, X.; Lan, H.Y. MicroRNAs in diabetic kidney disease. Int. J. Endocrinol. 2014, 2014, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Karbiener, M.; Neuhold, C.; Opriessnig, P.; Prokesch, A.; Bogner-Strauss, J.G.; Scheideler, M. MicroRNA-30c promotes human adipocyte differentiation and co-represses PAI-1 and ALK2. RNA Biol. 2011, 8, 850–860. [Google Scholar] [CrossRef]
- Wang, K.; Bei, W.J.; Liu, Y.H.; Li, H.L.; Chen, S.Q.; Lin, K.Y.; Zhou, Z.L.; Chen, J.Y.; Liu, Y.; Tan, N. miR-21 attenuates contrast-induced renal cell apoptosis by targeting PDCD4. Mol. Med. Rep. 2017, 16, 6757–6763. [Google Scholar] [CrossRef]
- Liu, C.; Tong, Z.; Tan, J.; Xin, Z.; Wang, Z.; Tian, L. MicroRNA-21-5p targeting PDCD4 suppresses apoptosis via regulating the PI3K/AKT/FOXO1 signaling pathway in tongue squamous cell carcinoma. Exp. Ther. Med. 2019, 18, 3543–3551. [Google Scholar] [CrossRef]
- Askari, H.; Raeis-Abdollahi, E.; Abazari, M.; Akrami, H.; Vakili, S.; Savardashtaki, A.; Tajbakhsh, A.; Sanadgol, N.; Azarnezhad, A.; Rahmati, L.; et al. Recent findings on the role of microRNAs in genetic kidney diseases. Mol. Biol. Rep. 2022, 49, 7039–7056. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, Y.; Harris, D.C. Pathogenic and protective role of macrophages in kidney disease. Am. J. Physiol.-Ren. Physiol. 2013, 305, F3–F11. [Google Scholar] [CrossRef]
- Rogers, N.M.; Ferenbach, D.A.; Isenberg, J.S.; Thomson, A.W.; Hughes, J. Dendritic cells and macrophages in the kidney: A spectrum of good and evil. Nat. Rev. Nephrol. 2014, 10, 625–643. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Kawakami, T.; Lichtnekert, J.; Thompson, L.J.; Karna, P.; Bouabe, H.; Hohl, T.M.; Heinecke, J.W.; Ziegler, S.F.; Nelson, P.J.; Duffield, J.S. Resident renal mononuclear phagocytes comprise five discrete populations with distinct phenotypes and functions. J. Immunol. 2013, 191, 3358–3372. [Google Scholar] [CrossRef]
- Chung, H.; Vilaysane, A.; Lau, A.; Stahl, M.; Morampudi, V.; Bondzi-Simpson, A.; Platnich, J.; Bracey, N.; French, M.; Beck, P. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ. 2016, 23, 1331–1346. [Google Scholar] [CrossRef]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar] [CrossRef]
- Kim, Y.G.; Kim, S.-M.; Kim, K.-P.; Lee, S.-H.; Moon, J.-Y. The role of inflammasome-dependent and inflammasome-independent NLRP3 in the kidney. Cells 2019, 8, 1389. [Google Scholar] [CrossRef]
- Lau, A.; Chung, H.; Komada, T.; Platnich, J.M.; Sandall, C.F.; Choudhury, S.R.; Chun, J.; Naumenko, V.; Surewaard, B.G.; Nelson, M.C. Renal immune surveillance and dipeptidase-1 contribute to contrast-induced acute kidney injury. J. Clin. Investig. 2018, 128, 2894–2913. [Google Scholar] [CrossRef]
- Jha, A.K.; Gairola, S.; Kundu, S.; Doye, P.; Syed, A.M.; Ram, C.; Murty, U.S.; Naidu, V.; Sahu, B.D. Toll-like receptor 4: An attractive therapeutic target for acute kidney injury. Life Sci. 2021, 271, 119155. [Google Scholar] [CrossRef]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Fan, J.; Zhang, X.; Luan, J.; Bian, Q.; Ding, T.; Wang, Y.; Wang, Z.; Song, P. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017, 8, e2937. [Google Scholar] [CrossRef]
- Yang, J.-S.; Peng, Y.-R.; Tsai, S.-C.; Tyan, Y.-S.; Lu, C.-C.; Chiu, H.-Y.; Chiu, Y.-J.; Kuo, S.-C.; Tsai, Y.-F.; Lin, P.-C. The molecular mechanism of contrast-induced nephropathy (CIN) and its link to in vitro studies on iodinated contrast media (CM). Biomedicine 2018, 8, 1. [Google Scholar] [CrossRef]
- Shayan, M.; Elyasi, S. Cilastatin as a protective agent against drug-induced nephrotoxicity: A literature review. Expert Opin. Drug Saf. 2020, 19, 999–1010. [Google Scholar] [CrossRef]
- Hori, Y.; Aoki, N.; Kuwahara, S.; Hosojima, M.; Kaseda, R.; Goto, S.; Iida, T.; De, S.; Kabasawa, H.; Kaneko, R. Megalin blockade with cilastatin suppresses drug-induced nephrotoxicity. J. Am. Soc. Nephrol. 2017, 28, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, V.; Venkatadri, R.; Dogan, M.; Sharma, R. The Yin and Yang of alarmins in regulation of acute kidney injury. Front. Med. 2020, 7, 441. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.R.; Ebenezer, P.J.; Saini, Y.; Francis, J. Angiotensin II-induced hypertensive renal inflammation is mediated through HMGB1-TLR4 signaling in rat tubulo-epithelial cells. Exp. Cell Res. 2015, 335, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, C.J.; Go, Y.S.; Lee, H.Y.; Kim, M.-G.; Oh, S.W.; Cho, W.Y.; Im, S.-H.; Jo, S.K. Intestinal microbiota control acute kidney injury severity by immune modulation. Kidney Int. 2020, 98, 932–946. [Google Scholar] [CrossRef]
- Yue, R.; Zuo, C.; Zeng, J.; Su, B.; Tao, Y.; Huang, S.; Zeng, R. Atorvastatin attenuates experimental contrast-induced acute kidney injury: A role for TLR4/MyD88 signaling pathway. Ren. Fail. 2017, 39, 643–651. [Google Scholar] [CrossRef]
- Ehrchen, J.M.; Sunderkötter, C.; Foell, D.; Vogl, T.; Roth, J. The endogenous Toll–like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukoc. Biol. 2009, 86, 557–566. [Google Scholar] [CrossRef]
- Tan, X.; Zheng, X.; Huang, Z.; Lin, J.; Xie, C.; Lin, Y. Involvement of S100A8/A9-TLR4-NLRP3 inflammasome pathway in contrast-induced acute kidney injury. Cell. Physiol. Biochem. 2017, 43, 209–222. [Google Scholar] [CrossRef]
- Meyers, J.H.; Sabatos, C.A.; Chakravarti, S.; Kuchroo, V.K. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol. Med. 2005, 11, 362–369. [Google Scholar] [CrossRef]
- Vilà, M.R.; Kaplan, G.G.; Feigelstock, D.; Nadal, M.; Morote, J.; Porta, R.; Bellmunt, J.; Meseguer, A. Hepatitis A virus receptor blocks cell differentiation and is overexpressed in clear cell renal cell carcinoma. Kidney Int. 2004, 65, 1761–1773. [Google Scholar] [CrossRef]
- Kim, H.Y.; Chang, Y.-J.; Chuang, Y.-T.; Lee, H.-H.; Kasahara, D.I.; Martin, T.; Hsu, J.T.; Savage, P.B.; Shore, S.A.; Freeman, G.J. T-cell immunoglobulin and mucin domain 1 deficiency eliminates airway hyperreactivity triggered by the recognition of airway cell death. J. Allergy Clin. Immunol. 2013, 132, 414–425.e416. [Google Scholar] [CrossRef]
- Song, J.; Yu, J.; Prayogo, G.W.; Cao, W.; Wu, Y.; Jia, Z.; Zhang, A. Understanding kidney injury molecule 1: A novel immune factor in kidney pathophysiology. Am. J. Transl. Res. 2019, 11, 1219. [Google Scholar]
- Ajay, A.K.; Kim, T.-M.; Ramirez-Gonzalez, V.; Park, P.J.; Frank, D.A.; Vaidya, V.S. A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J. Am. Soc. Nephrol. 2014, 25, 105–118. [Google Scholar] [CrossRef]
- Collier, J.B.; Schnellmann, R.G. Extracellular signal–regulated kinase 1/2 regulates mouse kidney injury molecule-1 expression physiologically and following ischemic and septic renal injury. J. Pharmacol. Exp. Ther. 2017, 363, 419–427. [Google Scholar] [CrossRef]
- Brooks, C.R.; Yeung, M.Y.; Brooks, Y.S.; Chen, H.; Ichimura, T.; Henderson, J.M.; Bonventre, J.V. KIM-1-/TIM-1-mediated phagocytosis links ATG 5-/ULK 1-dependent clearance of apoptotic cells to antigen presentation. EMBO J. 2015, 34, 2441–2464. [Google Scholar] [CrossRef]
- Zhang, Z.; Cai, C.X. Kidney injury molecule-1 (KIM-1) mediates renal epithelial cell repair via ERK MAPK signaling pathway. Mol. Cell. Biochem. 2016, 416, 109–116. [Google Scholar] [CrossRef]
- Luo, M.; Liu, Z.; Hu, Z.; He, Q. Quercetin improves contrast-induced acute kidney injury through the HIF-1α/lncRNA NEAT1/HMGB1 pathway. Pharm. Biol. 2022, 60, 889–898. [Google Scholar] [CrossRef]
- Zhang, L.; Ni, Y.-H.; Cao, G.-Y.; Zhang, J.-Y.; Yi, B.-J.; Pang, Z.-H.; Ma, H.-J.; Yin, K.-M. Chronic Intermittent Hypobaric Hypoxia Prevents Contrast-Induced Acute Kidney Injury By Modulating The HIF-1α Signaling Pathway. bioRxiv 2022. [Google Scholar] [CrossRef]
- Roshanzamir, F.; Yazdanparast, R. Quercetin attenuates cell apoptosis of oxidant-stressed SK-N-MC cells while suppressing up-regulation of the defensive element, HIF-1α. Neuroscience 2014, 277, 780–793. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Y.; Tang, Z.; Hu, D.; Yao, C.; Yang, L. LncRNA NEAT1 regulated inflammation and apoptosis in a rat model of sepsis-induced acute kidney injury via MiR-27a-3p/TAB3 axis. Biosci. Biotechnol. Biochem. 2020, 84, 2215–2227. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, X.; Zhang, B. Depression of lncRNA NEAT1 antagonizes LPS-evoked acute injury and inflammatory response in alveolar epithelial cells via HMGB1-RAGE signaling. Mediat. Inflamm. 2020, 2020, 8019467. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, J.; Wang, P.; Fan, H.; Hou, S.; Gong, Y. Major signaling pathways and key mediators of macrophages in acute kidney injury. Mol. Med. Rep. 2021, 23, 455. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, B.; Tang, K.; Dong, X.; Xue, L.; Su, G.; Jin, Y. Aquaporin 1 alleviates acute kidney injury via PI3K-mediated macrophage M2 polarization. Inflamm. Res. 2020, 69, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Saritemur, M.; Un, H.; Cadirci, E.; Karakus, E.; Akpinar, E.; Halici, Z.; Ugan, R.A.; Karaman, A.; Atmaca, H.T. Tnf-inhibition by infliximab as a new target for the prevention of glycerol-contrast-induced nephropathy. Environ. Toxicol. Pharmacol. 2015, 39, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Rajawat, Y.; Hilioti, Z.; Bossis, I. Autophagy: A target for retinoic acids. Autophagy 2010, 6, 1224–1226. [Google Scholar] [CrossRef]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschläger, N.; Schlee, M.; Rothenfusser, S.; Barchet, W. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1β production. Nat. Immunol. 2010, 11, 63–69. [Google Scholar] [CrossRef]
- Chiba, T.; Skrypnyk, N.I.; Skvarca, L.B.; Penchev, R.; Zhang, K.X.; Rochon, E.R.; Fall, J.L.; Paueksakon, P.; Yang, H.; Alford, C.E. Retinoic acid signaling coordinates macrophage-dependent injury and repair after AKI. J. Am. Soc. Nephrol. 2016, 27, 495–508. [Google Scholar] [CrossRef]
- Dugbartey, G.J.; Redington, A.N. Prevention of contrast-induced nephropathy by limb ischemic preconditioning: Underlying mechanisms and clinical effects. Am. J. Physiol.-Ren. Physiol. 2018, 314, F319–F328. [Google Scholar] [CrossRef]
- Ozkok, S.; Ozkok, A. Contrast-induced acute kidney injury: A review of practical points. World J. Nephrol. 2017, 6, 86. [Google Scholar] [CrossRef]
- Ricciardi, C.A.; Gnudi, L. The endoplasmic reticulum stress and the unfolded protein response in kidney disease: Implications for vascular growth factors. J. Cell. Mol. Med. 2020, 24, 12910–12919. [Google Scholar] [CrossRef]
- Scharnweber, T.; Alhilali, L.; Fakhran, S. Contrast-induced acute kidney injury: Pathophysiology, manifestations, prevention, and management. Magn. Reson. Imaging Clin. 2017, 25, 743–753. [Google Scholar] [CrossRef]
- Yang, X.; Yan, X.; Yang, D.; Zhou, J.; Song, J.; Yang, D. Rapamycin attenuates mitochondrial injury and renal tubular cell apoptosis in experimental contrast-induced acute kidney injury in rats. Biosci. Rep. 2018, 38, BSR20180876. [Google Scholar] [CrossRef]
- Freeman, B.D.; Machado, F.S.; Tanowitz, H.B.; Desruisseaux, M.S. Endothelin-1 and its role in the pathogenesis of infectious diseases. Life Sci. 2014, 118, 110–119. [Google Scholar] [CrossRef]
- Hong, Y.A.; Park, C.W. Catalytic Antioxidants in the Kidney. Antioxidants 2021, 10, 130. [Google Scholar] [CrossRef]
- Pisani, A.; Sabbatini, M.; Riccio, E.; Rossano, R.; Andreucci, M.; Capasso, C.; De Luca, V.; Carginale, V.; Bizzarri, M.; Borrelli, A. Effect of a recombinant manganese superoxide dismutase on prevention of contrast-induced acute kidney injury. Clin. Exp. Nephrol. 2014, 18, 424–431. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Jeong, B.Y.; Lee, H.Y.; Park, C.G.; Kang, J.; Yu, S.-L.; Choi, D.-r.; Han, S.-Y.; Park, M.H.; Cho, S.; Lee, S.Y. Oxidative stress caused by activation of NADPH oxidase 4 promotes contrast-induced acute kidney injury. PLoS ONE 2018, 13, e0191034. [Google Scholar] [CrossRef]
- Bohacek, J.; Mansuy, I.M. Epigenetic inheritance of disease and disease risk. Neuropsychopharmacology 2013, 38, 220–236. [Google Scholar] [CrossRef]
- Tanemoto, F.; Mimura, I. Therapies targeting epigenetic alterations in acute kidney injury-to-chronic kidney disease transition. Pharmaceuticals 2022, 15, 123. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Tang, J.; Zhuang, S. Epigenetics in acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2015, 24, 351. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Romo, R.; Berman, N.; Gómez, A.; Bobadilla, N.A. Epigenetic regulation in the acute kidney injury to chronic kidney disease transition. Nephrology 2015, 20, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Adiyanti, S.S.; Loho, T. Acute kidney injury (AKI) biomarker. Acta Med. Indones. 2012, 44, 246–255. [Google Scholar] [PubMed]
- Cong, B.; Zhang, Q.; Cao, X. The function and regulation of TET2 in innate immunity and inflammation. Protein Cell 2021, 12, 165–173. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 2017, 170, 1079–1095.e1020. [Google Scholar] [CrossRef]
- Bomsztyk, K.; Denisenko, O. Epigenetic alterations in acute kidney injury. In Seminars in Nephrology; Elsevier: Amsterdam, The Netherlands, 2013; pp. 327–340. [Google Scholar]
- Yuan, H.; Marmorstein, R. Histone acetyltransferases: Rising ancient counterparts to protein kinases. Biopolymers 2013, 99, 98–111. [Google Scholar] [CrossRef]
- Havasi, A.; Haegele, J.A.; Gall, J.M.; Blackmon, S.; Ichimura, T.; Bonegio, R.G.; Panchenko, M.V. Histone acetyl transferase (HAT) HBO1 and JADE1 in epithelial cell regeneration. Am. J. Pathol. 2013, 182, 152–162. [Google Scholar] [CrossRef]
- Liu, F.; Zong, M.; Wen, X.; Li, X.; Wang, J.; Wang, Y.; Jiang, W.; Li, X.; Guo, Z.; Qi, H. Silencing of histone deacetylase 9 expression in podocytes attenuates kidney injury in diabetic nephropathy. Sci. Rep. 2016, 6, 33676. [Google Scholar] [CrossRef]
- Lu, H.-C.; Dai, W.-N.; Liu, Z.-W.; Liu, H.; He, L.-Y. HDAC9 promotes the susceptibility of diabetes to Contrast-induced acute kidney injury by regulating TXNIP/Trx1 pathway. Res. Sq. 2022. preprint. [Google Scholar] [CrossRef]
- Karnewar, S.; Neeli, P.K.; Panuganti, D.; Kotagiri, S.; Mallappa, S.; Jain, N.; Jerald, M.K.; Kotamraju, S. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: Relevance in age-associated vascular dysfunction. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 1115–1128. [Google Scholar] [CrossRef]
- Sack, M.N.; Finkel, T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb. Perspect. Biol. 2012, 4, a013102. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, X.; Shao, X.; Wang, H.; Liu, X.; Ke, X.; Xiong, C.; Wei, L.; Zou, H. tert-Butylhydroquinone treatment alleviates contrast-induced nephropathy in rats by activating the Nrf2/Sirt3/SOD2 signaling pathway. Oxidative Med. Cell. Longev. 2019, 2019, 4657651. [Google Scholar] [CrossRef]
- Metzger, E.; Imhof, A.; Patel, D.; Kahl, P.; Hoffmeyer, K.; Friedrichs, N.; Müller, J.M.; Greschik, H.; Kirfel, J.; Ji, S. Phosphorylation of histone H3T6 by PKCβI controls demethylation at histone H3K4. Nature 2010, 464, 792–796. [Google Scholar] [CrossRef]
- Guo, C.; Wei, Q.; Su, Y.; Dong, Z. SUMOylation occurs in acute kidney injury and plays a cytoprotective role. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 482–489. [Google Scholar] [CrossRef]
- Rossetto, D.; Avvakumov, N.; Côté, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef]
- Tang, J.; Liu, N.; Tolbert, E.; Ponnusamy, M.; Ma, L.; Gong, R.; Bayliss, G.; Yan, H.; Zhuang, S. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am. J. Pathol. 2013, 183, 160–172. [Google Scholar] [CrossRef]
- Ribitsch, W.; Horina, J.H.; Quehenberger, F.; Rosenkranz, A.R.; Schilcher, G. Contrast induced acute kidney injury and its impact on mid-term kidney function, cardiovascular events and mortality. Sci. Rep. 2019, 9, 16896. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Xin, S.; Liu, J.; Sun, G.; Chen, S.; Cen, X.; Dai, X.; He, Y.; Song, F. Risk factors for contrast-induced acute kidney injury (CI−AKI): Protocol for systematic review and meta-analysis. BMJ Open 2019, 9, e030048. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Position in Kidney | Function in Kidney | Reference |
|---|---|---|---|
| Semaphorin 3A | Developing glomerulus, adult podocytes and collecting tubules | Inhibitory effect on ureteric bud branching | [54] |
| Liver-type fatty acid-binding protein (L-FABP) | Cytoplasm of human renal proximal tubules | Regulator of fatty acid metabolism | [55] |
| lnc-HILPDA | Collecting tubules | Regulation of oxidative stress, vascular endothelial cell damage | [56] |
| lnc-PRND | Collecting tubules | Regulation of oxidative stress, vascular endothelial cell damage | [57] |
| Midkine (MK) | Epithelium of the proximal tubules | Regulation of oxidative stress | [58] |
| Cystatin C (Cys-C) | Nucleated cells, filtered by glomerulus, and reabsorbed by proximal tubule cells | Indicator of reduced kidney function | [59] |
| N-Acetyl-β-glucosaminidase (NAG) | Proximal tubule lysosomal enzyme | Marker of occult renal dysfunction | [60] |
| Kidney injury molecule-1 (KIM-1] | Upregulated in dedifferentiated proximal tubule cells | Cell phagocytosis, repair processes, and anti-inflammation, and is shed into urine | [61] |
| Neutrophil gelatinase-associated lipocalin (NGAL) | Expression upregulated in proximal tubule cells after renal injury | Renal tubular epithelial recovery, bacterial defense and inflammation | [62] |
| Interleukin-18 (IL-18) | Expressed in distal tubule cells; expression may be induced in proximal tubules | Regulator of T cell function in the kidneys during hypertension | [63] |
| β2-Microglobulin (β2M) | Filtered by the glomerulus and reabsorbed by the proximal tubule cells | Marker of chronic kidney dysfunction. | [64] |
| Retinol-binding protein (RBP) | Filtered by the glomerulus and reabsorbed by the proximal tubule cells | Marker in assessing prophylactic treatments | [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, A.S.; Li, X. The Potential Biotherapeutic Targets of Contrast-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2023, 24, 8254. https://doi.org/10.3390/ijms24098254
Cheng AS, Li X. The Potential Biotherapeutic Targets of Contrast-Induced Acute Kidney Injury. International Journal of Molecular Sciences. 2023; 24(9):8254. https://doi.org/10.3390/ijms24098254
Chicago/Turabian StyleCheng, Alice Shasha, and Xiaogang Li. 2023. "The Potential Biotherapeutic Targets of Contrast-Induced Acute Kidney Injury" International Journal of Molecular Sciences 24, no. 9: 8254. https://doi.org/10.3390/ijms24098254