

Genetic Profiling of Sodium Channels in Diabetic Painful and Painless and Idiopathic Painful and Painless Neuropathies

 , , , , , , , , , , ,
, , , , , , , , , , ,
Abstract
:1. Introduction
2. Results
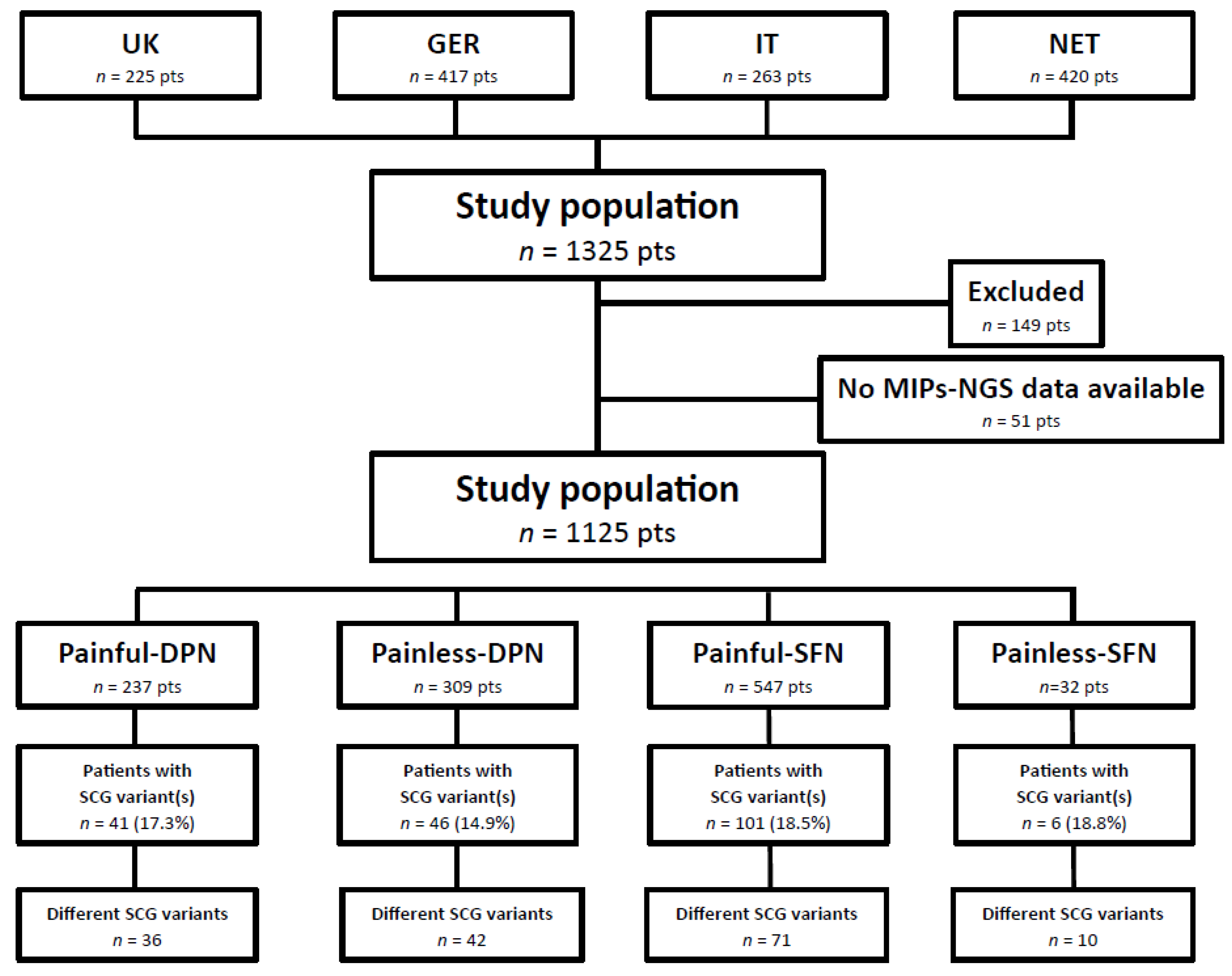
2.1. Patient Characteristics
2.2. Performance of SCG-smMIPs-NGS
2.3. SCG Variants in Painful-DPN Patients
2.4. SCG Variants in Painless-DPN Patients
2.5. SCG Variants in Painful-SFN Patients
2.6. SCG Variants in Painless-SFN Patients
2.7. Shared SCG Variants in Painful and Painless DPN and SFN
2.8. Mutation Frequencies and Distribition of SCG Variants in Patients with Painful and Painless DPN and Painful and Painless SFN
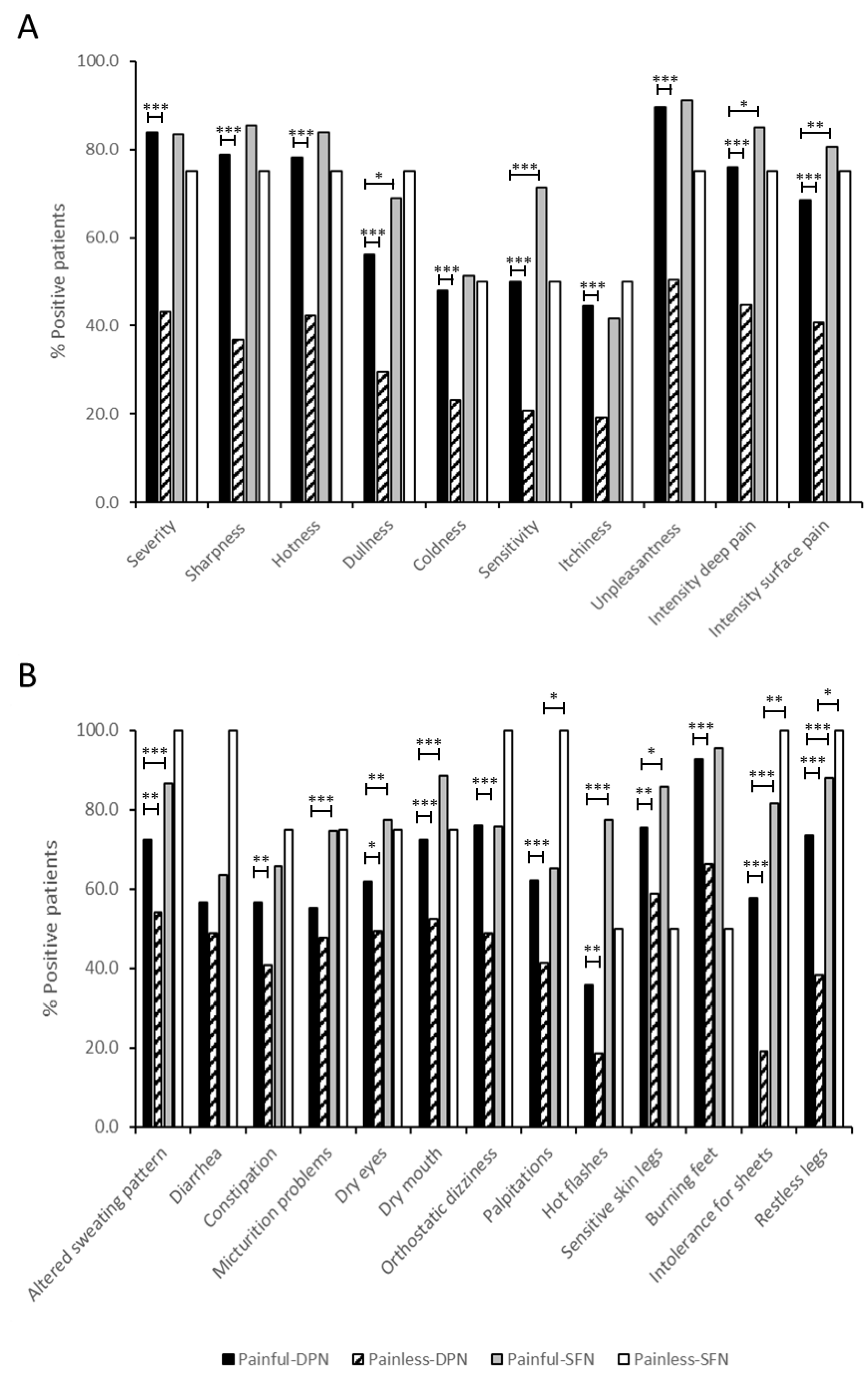
2.9. Clinical Features of Painful-DPN, Painless-DPN, Painful-SFN, and Painless-SFN Patients with and without SCG Variants
3. Discussion
4. Methods
4.1. Study Population
4.2. Inclusion/Exclusion Criteria and Definitions of Painful/Non-Painful Neuropathy
4.3. Clinical Assessments
4.4. SCG Mutation Analysis by smMIPs-NGS
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jensen, T.S.; Baron, R.; Haanpää, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.C.; Treede, R.-D. A new definition of neuropathic pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, B.A.; Merkies, I.S.J.; Gerrits, M.M.; Waxman, S.G.; Hoeijmakers, J.G.J.; Faber, C.G. Painful neuropathies: The emerging role of sodium channelopathies. J. Peripher. Nerv. Syst. 2014, 19, 53–65. [Google Scholar] [CrossRef]
- Hoeijmakers, J.G.; Faber, C.G.; Lauria, G.; Merkies, I.S.; Waxman, S.G. Small-fibre neuropathies—Advances in diagnosis, pathophysiology and management. Nat. Rev. Neurol. 2012, 8, 369–379. [Google Scholar] [CrossRef] [PubMed]
- de Greef, B.T.A.; Hoeijmakers, J.G.J.; Gorissen-Brouwers, C.M.L.; Geerts, M.; Faber, C.G.; Merkies, I.S.J. Associated conditions in small fiber neuropathy—A large cohort study and review of the literature. Eur. J. Neurol. 2018, 25, 348–355. [Google Scholar] [CrossRef]
- Bril, V.; Breiner, A.; Perkins, B.A.; Zochodne, D. Neuropathy. Can. J. Diabetes 2018, 42, S217–S221. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, S.; Boulton, A.J.M.; Dyck, P.J.; Freeman, R.; Horowitz, M.; Kempler, P.; Lauria, G.; Malik, R.A.; Spallone, V.; Vinik, A.; et al. Diabetic neuropathies: Update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 2010, 33, 2285–2293. [Google Scholar] [CrossRef]
- Davies, M.; Brophy, S.; Williams, R.; Taylor, A. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care 2006, 29, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.; Brandenburg, N.A.; Dukes, E.; Hoffman, D.L.; Tai, K.-S.; Stacey, B. Pain severity in diabetic peripheral neuropathy is associated with patient functioning, symptom levels of anxiety and depression, and sleep. J. Pain Symptom Manag. 2005, 30, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Lauria, G.; Merkies, I.S.J.; Cheng, X.; Han, C.; Ahn, H.-S.; Persson, A.-K.; Hoeijmakers, J.G.J.; Gerrits, M.M.; Pierro, T.; et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef]
- Faber, C.G.; Hoeijmakers, J.G.J.; Ahn, H.-S.; Cheng, X.; Han, C.; Choi, J.-S.; Estacion, M.; Lauria, G.; Vanhoutte, E.K.; Gerrits, M.M.; et al. Gain of function Na V1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 2012, 71, 26–39. [Google Scholar] [CrossRef]
- Eijkenboom, I.; Sopacua, M.; Hoeijmakers, J.G.J.; De Greef, B.T.A.; Lindsey, P.; Almomani, R.; Marchi, M.; Vanoevelen, J.; Smeets, H.J.M.; Waxman, S.G.; et al. Yield of peripheral sodium channels gene screening in pure small fibre neuropathy. J. Neurol. Neurosurg. Psychiatry 2019, 90, 342–352. [Google Scholar] [CrossRef]
- Alsaloum, M.; Estacion, M.; Almomani, R.; Gerrits, M.M.; Bönhof, G.J.; Ziegler, D.; Malik, R.; Ferdousi, M.; Lauria, G.; Merkies, I.S.; et al. A gain-of-function sodium channel β2-subunit mutation in painful diabetic neuropathy. Mol. Pain 2019, 15, 1744806919849802. [Google Scholar] [CrossRef]
- Blesneac, I.; Themistocleous, A.C.; Fratter, C.; Conrad, L.J.; Ramirez, J.D.; Cox, J.J.; Tesfaye, S.; Shillo, P.R.; Rice, A.S.; Tucker, S.J.; et al. Rare NaV1.7 variants associated with painful diabetic peripheral neuropathy. Pain 2018, 159, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ou, S.-W.; Wang, Y.-J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534–554. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Cregg, R.; Laguda, B.; Werdehausen, R.; Cox, J.J.; Linley, J.E.; Ramirez, J.D.; Bodi, I.; Markiewicz, M.; Howell, K.J.; Chen, Y.-C.; et al. Novel mutations mapping to the fourth sodium channel domain of nav1.7 result in variable clinical manifestations of primary erythromelalgia. NeuroMolecular Med. 2013, 15, 265–278. [Google Scholar] [CrossRef]
- Huang, J.; Han, C.; Estacion, M.; Vasylyev, D.; Hoeijmakers, J.G.J.; Gerrits, M.M.; Tyrrell, L.; Lauria, G.; Faber, C.G.; Dib-Hajj, S.D.; et al. Gain-of-function mutations in sodium channel NaV1.9 in painful neuropathy. Brain 2014, 137, 1627–1642. [Google Scholar] [CrossRef]
- Singh, N.A.; Pappas, C.; Dahle, E.J.; Claes, L.R.F.; Pruess, T.H.; De Jonghe, P.; Thompson, J.; Dixon, M.; Gurnett, C.A.; Peiffer, A.; et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009, 5, e1000649. [Google Scholar] [CrossRef]
- Fernández-Marmiesse, A.; Roca, I.; Díaz-Flores, F.; Cantarín, V.; Pérez-Poyato, M.S.; Fontalba, A.; Laranjeira, F.; Quintans, S.; Moldovan, O.; Felgueroso, B.; et al. Rare Variants in 48 Genes Account for 42% of Cases of Epilepsy With or Without Neurodevelopmental Delay in 246 Pediatric Patients. Front. Neurosci. 2019, 13, 1135. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Patowary, A.; Stanaway, I.B.; McCord, E.; Nesbitt, R.R.; Archer, M.; Scheuer, T.; Nickerson, D.; Raskind, W.H.; Wijsman, E.M.; et al. Association of rare missense variants in the second intracellular loop of NaV1.7 sodium channels with familial autism. Mol. Psychiatry 2018, 23, 231–239. [Google Scholar] [CrossRef]
- Savio-Galimberti, E.; Weeke, P.; Muhammad, R.; Blair, M.; Ansari, S.; Short, L.; Atack, T.C.; Kor, K.; Vanoye, C.G.; Olesen, M.S.; et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc. Res. 2014, 104, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Harkin, L.A.; Grinton, B.E.; Dibbens, L.M.; Turner, S.J.; Zielinski, M.A.; Xu, R.; Jackson, G.; Adams, J.; Connellan, M.; et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 2007, 130, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef]
- Han, C.; Vasylyev, D.; Macala, L.J.; Gerrits, M.M.; Hoeijmakers, J.G.J.; Bekelaar, K.J.; Dib-Hajj, S.D.; Faber, C.G.; Merkies, I.S.J.; Waxman, S.G. The G1662S NaV1.8 mutation in small fibre neuropathy: Impaired inactivation underlying DRG neuron hyperexcitability. J. Neurol. Neurosurg. Psychiatry 2014, 85, 499–505. [Google Scholar] [CrossRef]
- Abou Ziki, M.D.; Seidelmann, S.B.; Smith, E.; Atteya, G.; Jiang, Y.; Fernandes, R.G.; Marieb, M.A.; Akar, J.G.; Mani, A. Deleterious protein-altering mutations in the SCN10A voltage-gated sodium channel gene are associated with prolonged QT. Clin. Genet. 2018, 93, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Darbar, D.; Kaiser, D.W.; Jiramongkolchai, K.; Chopra, S.; Donahue, B.S.; Kannankeril, P.J.; Roden, D.M. Mutations in Sodium Channel β1- and β2-Subunits Associated With Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2009, 2, 268–275. [Google Scholar] [CrossRef]
- van Lint, F.H.M.; Mook, O.R.F.; Alders, M.; Bikker, H.; Lekanne dit Deprez, R.H.; Christiaans, I. Large next-generation sequencing gene panels in genetic heart disease: Yield of pathogenic variants and variants of unknown significance. Neth. Heart J. 2019, 27, 304–309. [Google Scholar] [CrossRef]
- Meglič, A.; Perkovič-Benedik, M.; Trebušak Podkrajšek, K.; Bertok, S. Painful micturition in a small child: An unusual clinical picture of paroxysmal extreme pain disorder. Pediatr. Nephrol. 2014, 29, 1643–1646. [Google Scholar] [CrossRef]
- Di Stefano, G.; Yuan, J.-H.; Cruccu, G.; Waxman, S.G.; Dib-Hajj, S.D.; Truini, A. Familial trigeminal neuralgia—A systematic clinical study with a genomic screen of the neuronal electrogenisome. Cephalalgia 2020, 40, 767–777. [Google Scholar] [CrossRef]
- Labau, J.I.R.; Estacion, M.; Tanaka, B.S.; de Greef, B.T.A.; Hoeijmakers, J.G.J.; Geerts, M.; Gerrits, M.M.; Smeets, H.J.M.; Faber, C.G.; Merkies, I.S.J.; et al. Differential effect of lacosamide on Nav1.7 variants from responsive and non-responsive patients with small fibre neuropathy. Brain 2020, 143, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Kambouris, M.; Thevenon, J.; Soldatos, A.; Cox, A.; Stephen, J.; Ben-Omran, T.; Al-Sarraj, Y.; Boulos, H.; Bone, W.; Mullikin, J.C.; et al. Biallelic SCN10A mutations in neuromuscular disease and epileptic encephalopathy. Ann. Clin. Transl. Neurol. 2017, 4, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Mulley, J.C.; Hodgson, B.; McMahon, J.M.; Iona, X.; Bellows, S.; Mullen, S.A.; Farrell, K.; Mackay, M.; Sadleir, L.; Bleasel, A.; et al. Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia 2013, 54, e122–e126. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, J.; Lecca, M.R.; Russo, G.; Bartsch, C.; Medeiros-Domingo, A.; Berger, W.; Haas, C. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur. J. Hum. Genet. 2017, 25, 404–409. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Geha, P.; Waxman, S.G. Sodium channels in pain disorders: Pathophysiology and prospects for treatment. Pain 2017, 158, S97–S107. [Google Scholar] [CrossRef]
- Wadhawan, S.; Pant, S.; Golhar, R.; Kirov, S.; Thompson, J.; Jacobsen, L.; Qureshi, I.; Ajroud-Driss, S.; Freeman, R.; Simpson, D.M.; et al. NaV channel variants in patients with painful and nonpainful peripheral neuropathy. Neurol. Genet. 2017, 3, e207. [Google Scholar] [CrossRef]
- Lauria, G.; Ziegler, D.; Malik, R.; Merkies, I.S.J.; Waxman, S.G.; Faber, C.G.; On Behalf of the PROPANE Study Group. The Role of Sodium Channels in Painful Diabetic and Idiopathic Neuropathy. Curr. Diabetes Rep. 2014, 14, 538. [Google Scholar] [CrossRef]
- Estacion, M.; Harty, T.P.; Choi, J.-S.; Tyrrell, L.; Dib-Hajj, S.D.; Waxman, S.G. A sodium channel gene SCN9A polymorphism that increases nociceptor excitability. Ann. Neurol. 2009, 66, 862–866. [Google Scholar] [CrossRef]
- Bennett, D.L.H.; Woods, C.G. Painful and painless channelopathies. Lancet Neurol. 2014, 13, 587–599. [Google Scholar] [CrossRef]
- Young, E.E.; Lariviere, W.R.; Belfer, I. Genetic basis of pain variability: Recent advances. J. Med. Genet. 2012, 49, 1–9. [Google Scholar] [CrossRef]
- Yuan, J.-H.; Estacion, M.; Mis, M.A.; Tanaka, B.S.; Schulman, B.R.; Chen, L.; Liu, S.; Dib-Hajj, F.B.; Dib-Hajj, S.D.; Waxman, S.G. KCNQ variants and pain modulation: A missense variant in Kv7.3 contributes to pain resilience. Brain Commun. 2021, 3, fcab212. [Google Scholar] [CrossRef]
- Mis, M.A.; Yang, Y.; Tanaka, B.S.; Gomis-Perez, C.; Liu, S.; Dib-Hajj, F.; Adi, T.; Garcia-Milian, R.; Schulman, B.R.; Dib-Hajj, S.D.; et al. Resilience to pain: A peripheral component identified using induced pluripotent stem cells and dynamic clamp. J. Neurosci. 2019, 39, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Estacion, M.; Vohra, B.P.S.; Liu, S.; Hoeijmakers, J.; Faber, C.G.; Merkies, I.S.J.; Lauria, G.; Black, J.A.; Waxman, S.G. Ca2+ toxicity due to reverse Na+/Ca2+ exchange contributes to degeneration of neurites of DRG neurons induced by a neuropathy-associated Nav1.7 mutation. J. Neurophysiol. 2015, 114, 1554–1564. [Google Scholar] [CrossRef]
- Lee, S.-I.; Hoeijmakers, J.G.J.; Faber, C.G.; Merkies, I.S.J.; Lauria, G.; Waxman, S.G. The small fiber neuropathy NaV1.7 I228M mutation: Impaired neurite integrity via bioenergetic and mitotoxic mechanisms, and protection by dexpramipexole. J. Neurophysiol. 2020, 123, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G.; Merkies, I.S.J.; Gerrits, M.M.; Dib-Hajj, S.D.; Lauria, G.; Cox, J.J.; Wood, J.N.; Woods, C.G.; Drenth, J.P.H.; Faber, C.G. Sodium channel genes in pain-related disorders: Phenotype–genotype associations and recommendations for clinical use. Lancet Neurol. 2014, 13, 1152–1160. [Google Scholar] [CrossRef]
- Huang, J.; Vanoye, C.G.; Cutts, A.; Goldberg, Y.P.; Dib-Hajj, S.D.; Cohen, C.J.; Waxman, S.G.; George, A.L. Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J. Clin. Investig. 2017, 127, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yang, Y.; de Greef, B.T.A.; Hoeijmakers, J.G.J.; Gerrits, M.M.; Verhamme, C.; Qu, J.; Lauria, G.; Merkies, I.S.J.; Faber, C.G.; et al. The Domain II S4-S5 Linker in Nav1.9: A Missense Mutation Enhances Activation, Impairs Fast Inactivation, and Produces Human Painful Neuropathy. NeuroMol. Med. 2015, 17, 158–169. [Google Scholar] [CrossRef]
- Treede, R.-D.; Jensen, T.S.; Campbell, J.N.; Cruccu, G.; Dostrovsky, J.O.; Griffin, J.W.; Hansson, P.; Hughes, R.; Nurmikko, T.; Serra, J. Neuropathic pain: Redefinition and a grading system for clinical and research purposes. Neurology 2008, 70, 1630–1635. [Google Scholar] [CrossRef]
- Mücke, M.; Cuhls, H.; Radbruch, L.; Baron, R.; Maier, C.; Tölle, T.; Treede, R.-D.; Rolke, R. Quantitative sensory testing (QST). English version. Schmerz 2021, 35, 153–160. [Google Scholar] [CrossRef]
- Farrar, J.T.; Young, J.P., Jr.; LaMoreaux, L.; Werth, J.L.; Poole, R.M. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain 2001, 94, 149–158. [Google Scholar] [CrossRef]
- Galer, B.S.; Jensen, M.P. Development and preliminary validation of a pain measure specific to neuropathic pain: The Neuropathic Pain Scale. Neurology 1997, 48, 332–338. [Google Scholar] [CrossRef]
- Bakkers, M.; Merkies, I.S.J.; Lauria, G.; Devigili, G.; Penza, P.; Lombardi, R.; Hermans, M.C.; van Nes, S.I.; De Baets, M.; Faber, C.G. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology 2009, 73, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Almomani, R.; Marchi, M.; Sopacua, M.; Lindsey, P.; Salvi, E.; de Koning, B.; Santoro, S.; Magri, S.; Smeets, H.J.M.; Boneschi, F.M.; et al. Evaluation of molecular inversion probe versus TruSeq® custom methods for targeted next-generation sequencing. PLoS ONE 2020, 15, e0248250. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Painful-DPN Patients (n = 237) | Painless-DPN Patients (n = 309) | Painful-SFN Patients (n = 547) | Painless-SFN Patients (n = 32) | ||
|---|---|---|---|---|---|
| Male (n) | 150/237 (63.3%) | 236/309 (76.4%) | 220/547 (40.2%) | 15/32 (46.9%) | |
| Mean age at recruitment (in years ± SD) | 63.70 ± 10.42 no data missing from patients | 64.92 ± 11.88 no data missing from patients | 53.56 ± 14.09 no data missing from patients | 54.69 ± 17.11 no data missing from patients | |
| Mean age of onset neuropathy (in years ± SD) | 58.43 ± 11.26 data missing from 69 patients | 62.29 ± 11.82 data missing from 128 patients | 53.56 ± 14.09 no data missing from patients | 45.79 ± 14.16 data missing from 13 patients | |
| Mean duration of neuropathy (in years ± SD) | 6.70 ± 5.86 data missing from 70 patients | 5.62 ± 6.60 data missing from 140 patients | 7.91 ± 9.65 data missing from 218 patients | 6.42 ± 6.30 data missing from 13 patients | |
| Familial cases of neuropathy (n) | 30/162 (18.5%) data missing from 75 patients | 24/185 (13.0%) data missing from 124 patients | 80/314 (25.5%) data missing from 233 patients | 0/4 (0%) data missing from 28 patients | |
| Normal EMG (n) | 0/237 (0%) | 0/309 (0%) | 547/547 (100%) | 32/32 (100%) | |
| Abnormal QST (n) | 130/161 (80.2%) * data missing from 76 patients | 139/182 (76.4%) * data missing from 127 patients | 329/347 (94.8%) data missing from 200 patients | 12/12 (100%) data missing from 20 patients | |
| Abnormal IENFD (n) | 12/12 (100%) * data missing from 225 patients | N/A * | 249/495 (50.3%) data missing from 52 patients | 21/29 (72.4%) data missing from 3 patients | |
| Abnormal QST + abnormal IENFD (n) | 0/12 (0%) * data missing from 225 patients | N/A * | 114/347 (32.9%) data missing from 200 patients | 1/12 (8.3%) data missing from 20 patients | |
| PI-NRS (mean ± SD) | 7.71 ± 1.71 no data missing from patients | 0.13 ± 0.49 no data missing from patients | 6.51 ± 2.20 no data missing from patients | 0.40 ± 0.93 no data missing from patients | |
| Diabetes mellitus | Type 1 (n) Type 2 (n) Unspecified (n) | 37/237 (15.6%) 198/237 (83.5%) 2/237 (0.8%) | 79/309 (25.6%) 229/237 (74.1%) 1/309 (0.3%) | N/A | N/A |
| Hypertension (n) | 137/237 (57.8%) | 155/309 (50.2%) | 78/547 (14.3%) | 1/32 (3.1%) | |
| Hypercholesterolemia (n) | 113/237 (47.7%) | 131/309 (42.4%) | 50/547 (9.1%) | 0/32 (0%) | |
| Cardiovascular diseases (n) | 84/237 (35.4%) | 105/309 (34.0%) | 33/547 (6.0%) | 0/32 (0%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almomani, R.; Sopacua, M.; Marchi, M.; Ślęczkowska, M.; Lindsey, P.; de Greef, B.T.A.; Hoeijmakers, J.G.J.; Salvi, E.; Merkies, I.S.J.; Ferdousi, M.; et al. Genetic Profiling of Sodium Channels in Diabetic Painful and Painless and Idiopathic Painful and Painless Neuropathies. Int. J. Mol. Sci. 2023, 24, 8278. https://doi.org/10.3390/ijms24098278
Almomani R, Sopacua M, Marchi M, Ślęczkowska M, Lindsey P, de Greef BTA, Hoeijmakers JGJ, Salvi E, Merkies ISJ, Ferdousi M, et al. Genetic Profiling of Sodium Channels in Diabetic Painful and Painless and Idiopathic Painful and Painless Neuropathies. International Journal of Molecular Sciences. 2023; 24(9):8278. https://doi.org/10.3390/ijms24098278
Chicago/Turabian StyleAlmomani, Rowida, Maurice Sopacua, Margherita Marchi, Milena Ślęczkowska, Patrick Lindsey, Bianca T. A. de Greef, Janneke G. J. Hoeijmakers, Erika Salvi, Ingemar S. J. Merkies, Maryam Ferdousi, and et al. 2023. "Genetic Profiling of Sodium Channels in Diabetic Painful and Painless and Idiopathic Painful and Painless Neuropathies" International Journal of Molecular Sciences 24, no. 9: 8278. https://doi.org/10.3390/ijms24098278