
Induction of Inflammation Disrupts the Negative Interplay between STING and S1P Axis That Is Observed during Physiological Conditions in the Lung
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
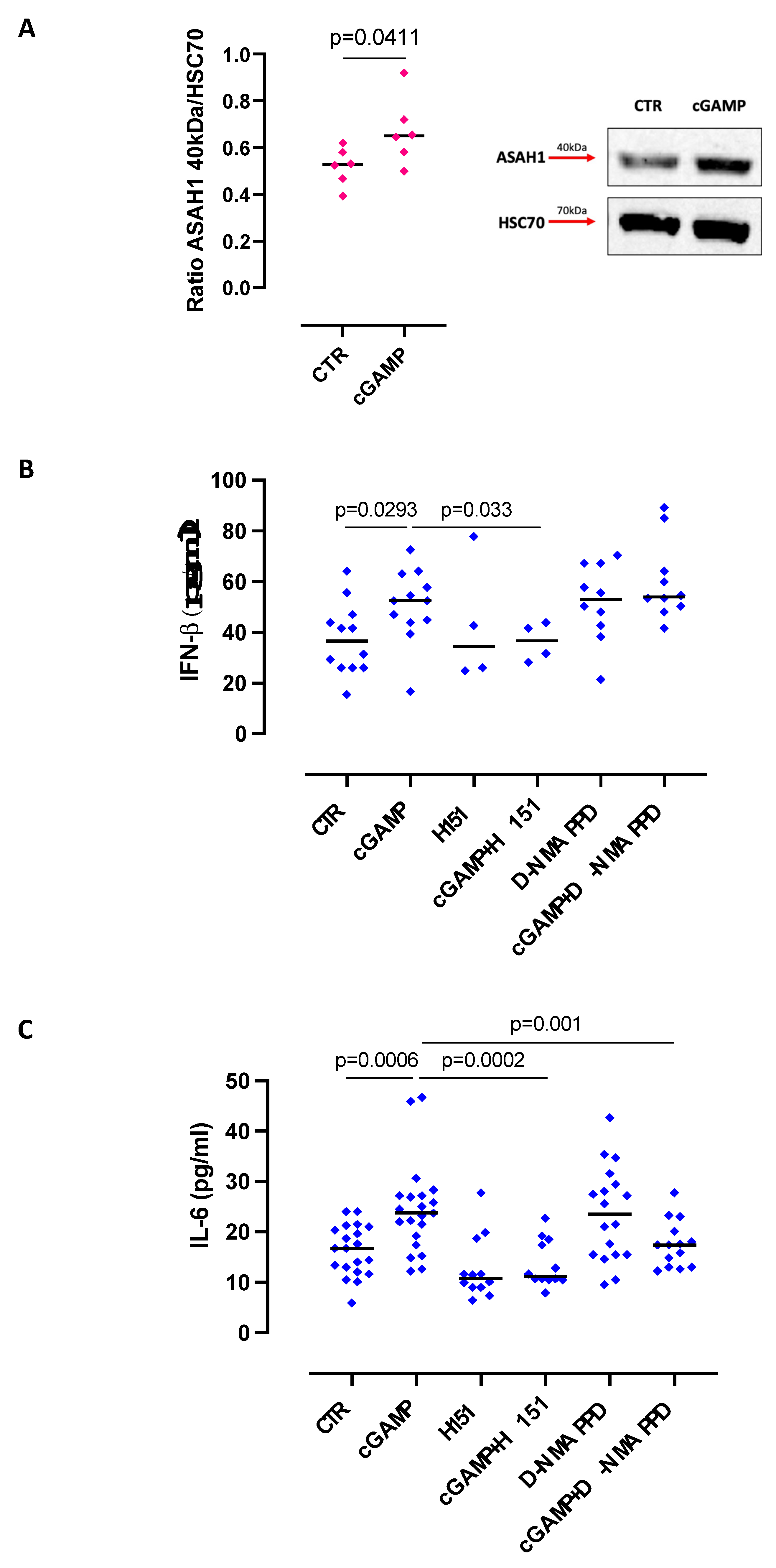
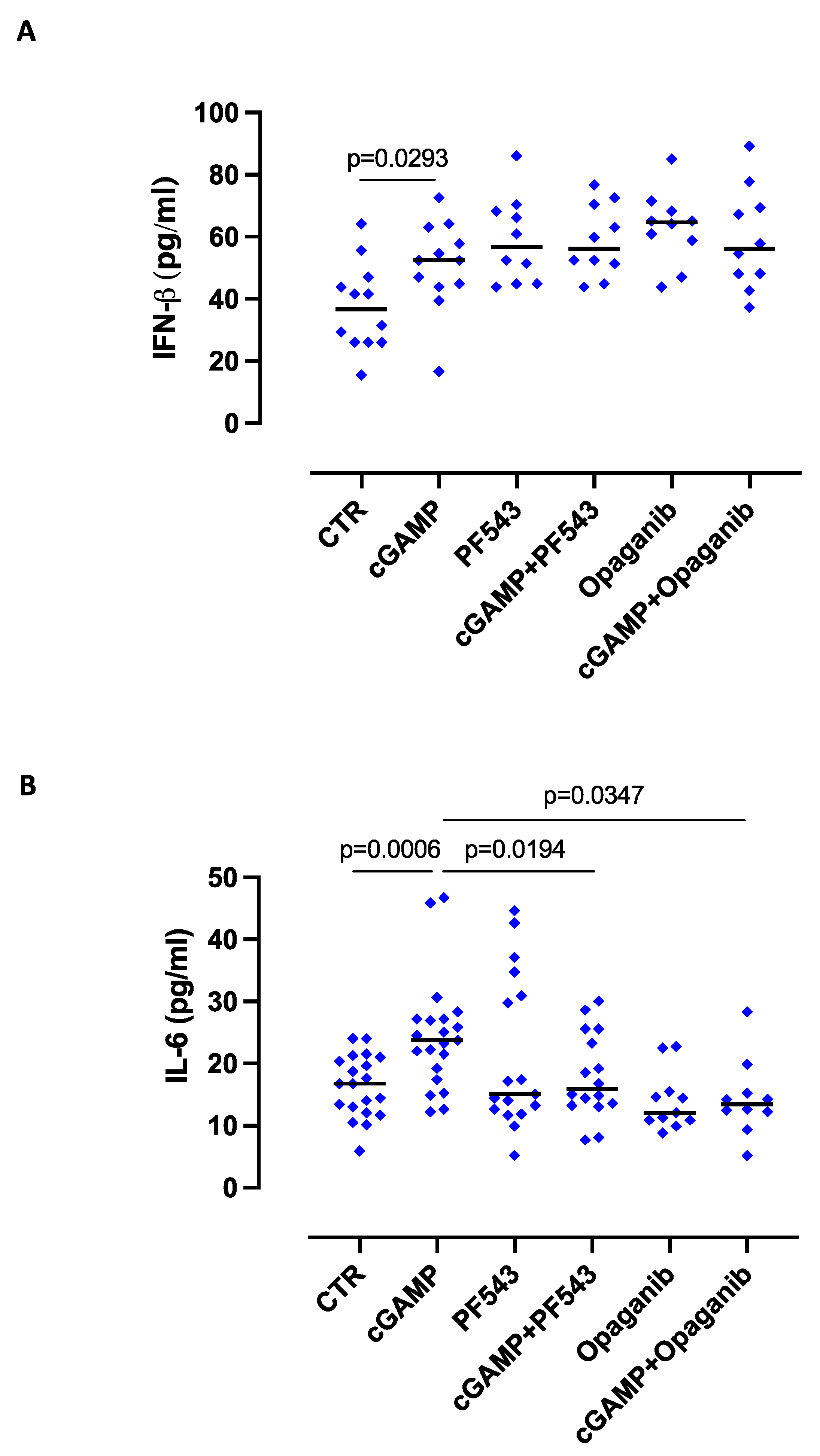
2.1. The Release of IL-6 after STING Activation Is S1P-Dependent in Lung Epithelial Cells
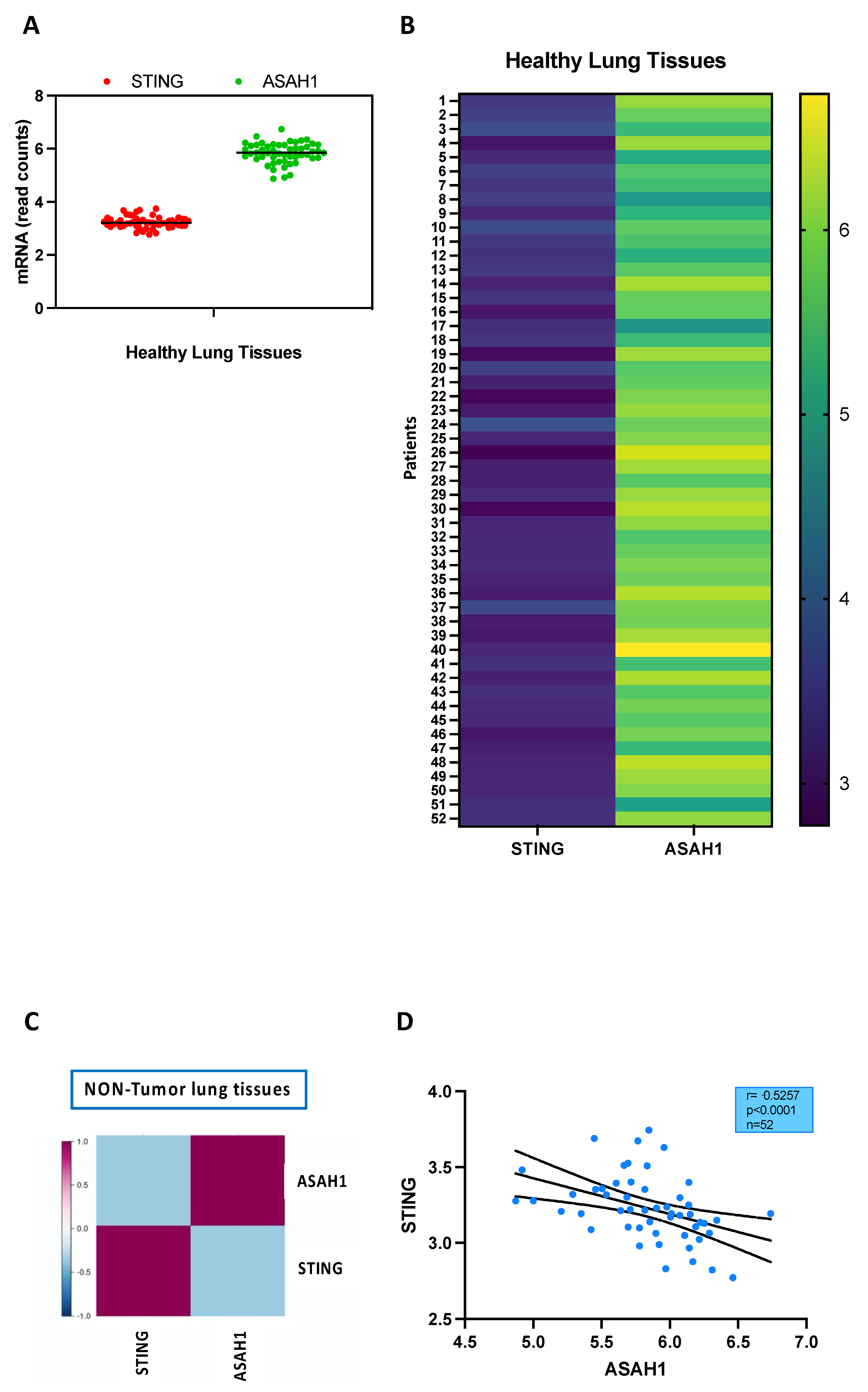
2.2. S1P Axis and STING Pathway Are Not Connected in the Lung during Physiological Conditions
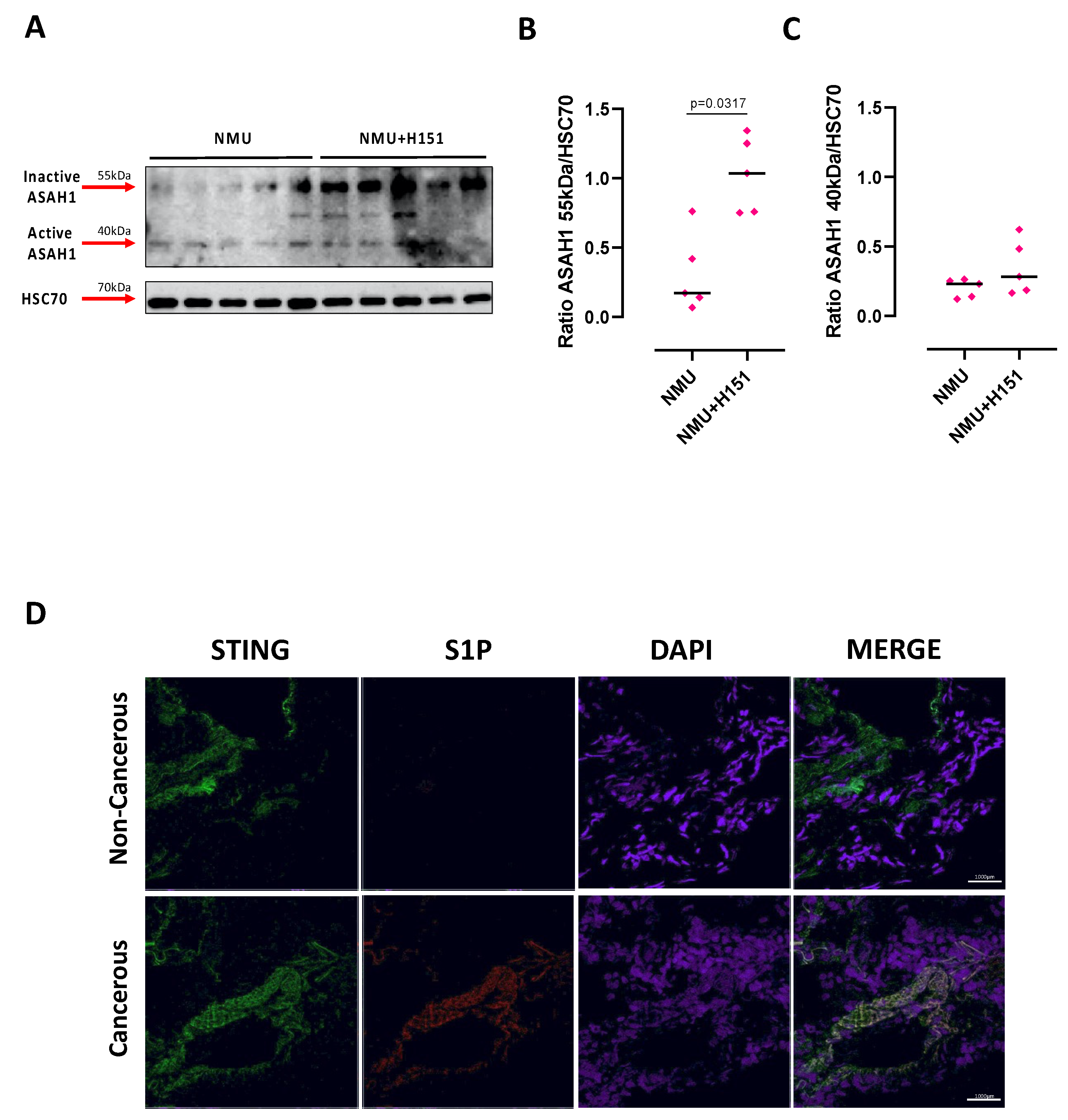
2.3. S1P Axis and STING Pathway Are Interconnected during Pathological Conditions in the Lung
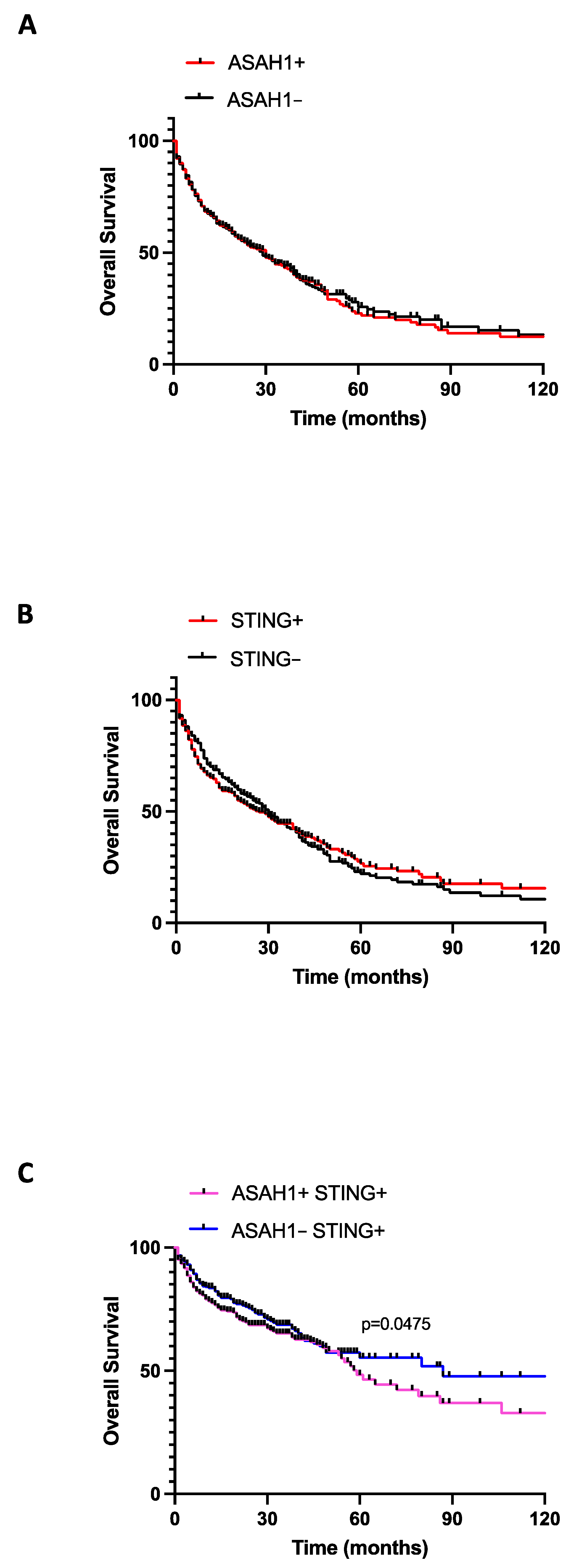
2.4. The Co-Expression of STING and S1P Increased Lung Cancer Mortality
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Cytokine Measurements
4.3. Western Blotting Analysis
4.4. Mouse Model
4.5. Correlation Analysis of Transcripts Expression
4.6. Survival Analysis
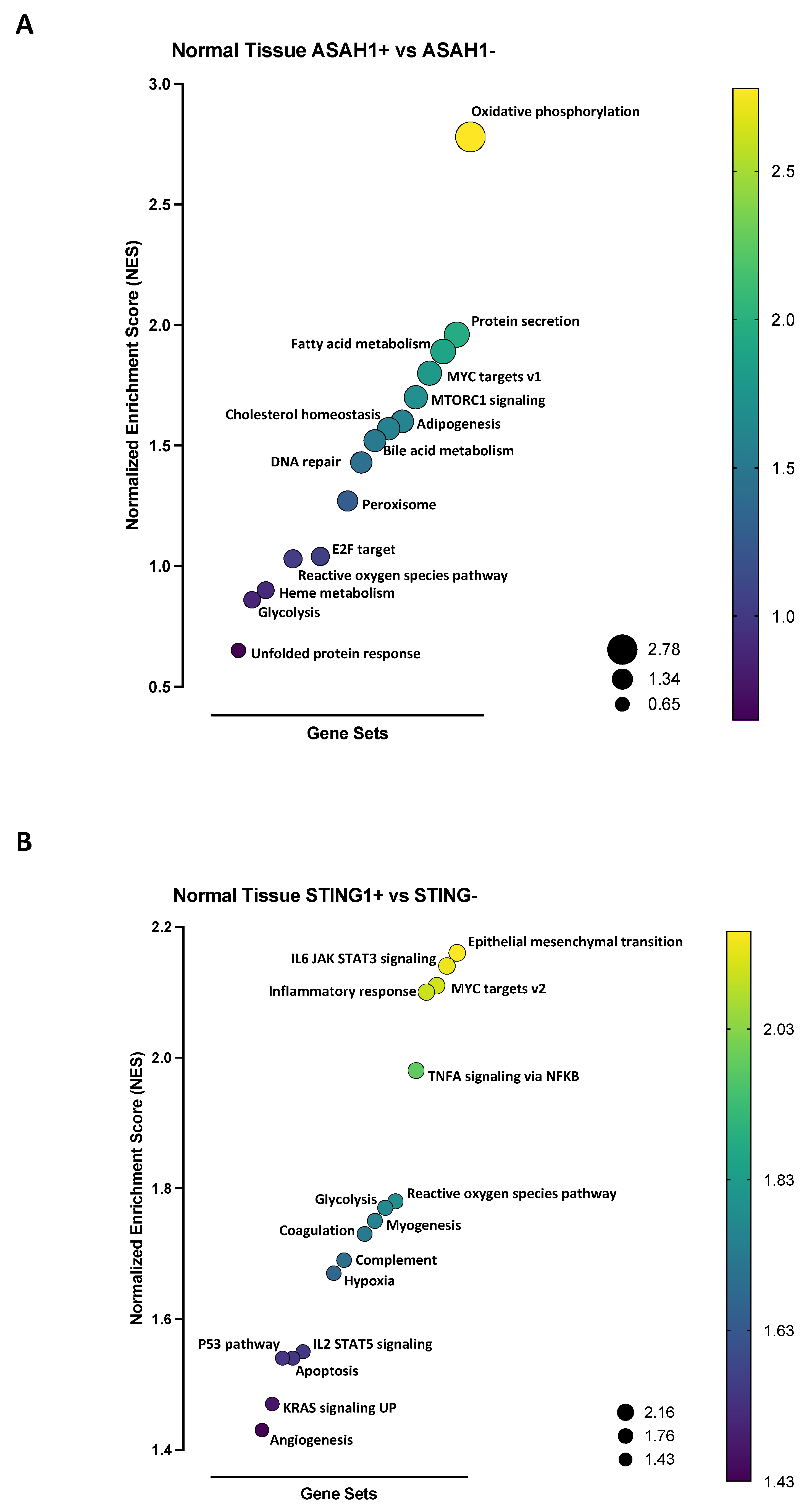
4.7. Gene Set Enrichment Analysis (GSEA)
4.8. Human Samples
4.9. Immunofluorescence Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell. Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Zhang, R.; Kang, R.; Tang, D. STING1 in Different Organelles: Location Dictates Function. Front. Immunol. 2022, 13, 842489. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Chinn, I.K.; Hong, D.; Orange, J.S.; Lupski, J.R.; Mendoza, A.; Pedroza, L.A.; Barber, G.N. Pro-inflammation Associated with a Gain-of-Function Mutation (R284S) in the Innate Immune Sensor STING. Cell. Rep. 2018, 23, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Szego, E.M.; Malz, L.; Bernhardt, N.; Rösen-Wolff, A.; Falkenburger, B.H.; Luksch, H. Constitutively active STING causes neuroinflammation and degeneration of dopaminergic neurons in mice. Elife 2022, 11, e81943. [Google Scholar] [CrossRef]
- Qi, Y.; Wu, Z.; Chen, D.; Zhu, L.; Yang, Y. A role of STING signaling in obesity-induced lung inflammation. Int. J. Obes. 2023, 13, 325–334. [Google Scholar] [CrossRef]
- David, C.; Frémond, M.L. Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI). Cells 2022, 11, 318. [Google Scholar] [CrossRef]
- Hla, T. Physiological and pathological actions of sphingosine 1-phosphate. Semin. Cell. Dev. Biol. 2004, 15, 513–520. [Google Scholar] [CrossRef]
- di Villa Bianca, R.d.; Sorrentino, R.; Sorrentino, R.; Imbimbo, C.; Palmieri, A.; Fusco, F.; Maggi, M.; De Palma, R.; Cirino, G.; Mirone, V. Sphingosine 1-phosphate induces endothelial nitric-oxide synthase activation through phosphorylation in human corpus cavernosum. J. Pharmacol. Exp. Ther. 2006, 316, 703–708. [Google Scholar] [CrossRef]
- Cantalupo, A.; Di Lorenzo, A. S1P Signaling and De Novo Biosynthesis in Blood Pressure Homeostasis. J. Pharmacol. Exp. Ther. 2016, 358, 359–370. [Google Scholar] [CrossRef]
- Roviezzo, F.; Sorrentino, R.; Iacono, V.M.; Brancaleone, V.; Terlizzi, M.; Riemma, M.A.; Bertolino, A.; Rossi, A.; Matteis, M.; Spaziano, G.; et al. Disodium cromoglycate inhibits asthma-like features induced by sphingosine-1-phosphate. Pharmacol. Res. 2016, 113 Pt. A, 626–635. [Google Scholar] [CrossRef]
- Roviezzo, F.; Sorrentino, R.; Terlizzi, M.; Riemma, M.A.; Iacono, V.M.; Rossi, A.; Spaziano, G.; Pinto, A.; D’Agostino, B.; Cirino, G. Toll-Like Receptor 4 Is Essential for the Expression of Sphingosine-1-Phosphate-Dependent Asthma-Like Disease in Mice. Front. Immunol. 2017, 8, 1336. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci. 2018, 109, 3671–3678. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Recent advances in the role of sphingosine 1-phosphate in cancer. FEBS Lett. 2020, 594, 3583–3601. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, M.; Colarusso, C.; Ferraro, G.; Monti, M.C.; Cerqua, I.; Roviezzo, F.; Pinto, A.; Sorrentino, R. Sphingosine-1-Phosphate Contributes to TLR9-Induced TNF-α Release in Lung Tumor Cells. Cell. Physiol. Biochem. 2021, 55, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Akhmetova, K.; Balasov, M.; Chesnokov, I. Drosophila STING protein has a role in lipid metabolism. Elife 2021, 10, e67358. [Google Scholar] [CrossRef] [PubMed]
- Benmerzoug, S.; Rose, S.; Bounab, B.; Gosset, D.; Duneau, L.; Chenuet, P.; Mollet, L.; Le Bert, M.; Lambers, C.; Geleff, S.; et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat. Commun. 2018, 9, 5226. [Google Scholar] [CrossRef]
- Roviezzo, F.; Sorrentino, R.; Bertolino, A.; De Gruttola, L.; Terlizzi, M.; Pinto, A.; Napolitano, M.; Castello, G.; D’Agostino, B.; Ianaro, A.; et al. S1P-induced airway smooth muscle hyperresponsiveness and lung inflammation in vivo: Molecular and cellular mechanisms. Br. J. Pharmacol. 2015, 172, 1882–1893. [Google Scholar] [CrossRef]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef]
- Terlizzi, M.; Di Crescenzo, V.G.; Perillo, G.; Galderisi, A.; Pinto, A.; Sorrentino, R. Pharmacological inhibition of caspase-8 limits lung tumour outgrowth. Br. J. Pharmacol. 2015, 172, 3917–3928. [Google Scholar] [CrossRef]
- Li, Y.; Wilson, H.L.; Kiss-Toth, E. Regulating STING in health and disease. J. Inflamm. 2017, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mirlekar, B.; Johnson, B.M.; Brickey, W.J.; Wrobel, J.A.; Yang, N.; Song, D.; Entwistle, S.; Tan, X.; Deng, M.; et al. STING-induced regulatory B cells compromise NK function in cancer immunity. Nature 2022, 610, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Parveen, F.; Bender, D.; Law, S.H.; Mishra, V.K.; Chen, C.C.; Ke, L.Y. Role of Ceramidases in Sphingolipid Metabolism and Human Diseases. Cells 2019, 8, 1573. [Google Scholar] [CrossRef]
- Terlizzi, M.; Molino, A.; Colarusso, C.; Somma, P.; De Rosa, I.; Troisi, J.; Scala, G.; Salvi, R.; Pinto, A.; Sorrentino, R. Altered lung tissue lipidomic profile in caspase-4 positive non-small cell lung cancer (NSCLC) patients. Oncotarget 2020, 11, 3515–3525. [Google Scholar]
- Terlizzi, M.; Colarusso, C.; Somma, P.; De Rosa, I.; Panico, L.; Pinto, A.; Sorrentino, R. S1P-Induced TNF-α and IL-6 Release from PBMCs Exacerbates Lung Cancer-Associated Inflammation. Cells 2022, 11, 2524. [Google Scholar] [CrossRef]
- Terlizzi, M.; Colarusso, C.; Ferraro, G.; Monti, M.C.; De Rosa, I.; Somma, P.; Salvi, R.; Pinto, A.; Sorrentino, R. Intracellular Sphingosine-1-Phosphate Receptor 3 Contributes to Lung Tumor Cell Proliferation. Cell. Physiol. Biochem. 2021, 55, 539–552. [Google Scholar]
- Joshi, J.C.; Joshi, B.; Rochford, I.; Rayees, S.; Akhter, M.Z.; Baweja, S.; Chava, K.R.; Tauseef, M.; Abdelkarim, H.; Natarajan, V.; et al. SPHK2-Generated S1P in CD11b+ Macrophages Blocks STING to Suppress the Inflammatory Function of Alveolar Macrophages. Cell. Rep. 2020, 30, 4096–4109.e5. [Google Scholar] [CrossRef]
- Al-Asmari, S.S.; Rajapakse, A.; Ullah, T.R.; Pépin, G.; Croft, L.V.; Gantier, M.P. Pharmacological Targeting of STING-Dependent IL-6 Production in Cancer Cells. Front. Cell. Dev. Biol. 2022, 9, 709618. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terlizzi, M.; Colarusso, C.; Falanga, A.; Somma, P.; De Rosa, I.; Panico, L.; Pinto, A.; Maiolino, P.; Sorrentino, R. Induction of Inflammation Disrupts the Negative Interplay between STING and S1P Axis That Is Observed during Physiological Conditions in the Lung. Int. J. Mol. Sci. 2023, 24, 8303. https://doi.org/10.3390/ijms24098303
Terlizzi M, Colarusso C, Falanga A, Somma P, De Rosa I, Panico L, Pinto A, Maiolino P, Sorrentino R. Induction of Inflammation Disrupts the Negative Interplay between STING and S1P Axis That Is Observed during Physiological Conditions in the Lung. International Journal of Molecular Sciences. 2023; 24(9):8303. https://doi.org/10.3390/ijms24098303
Chicago/Turabian StyleTerlizzi, Michela, Chiara Colarusso, Anna Falanga, Pasquale Somma, Ilaria De Rosa, Luigi Panico, Aldo Pinto, Piera Maiolino, and Rosalinda Sorrentino. 2023. "Induction of Inflammation Disrupts the Negative Interplay between STING and S1P Axis That Is Observed during Physiological Conditions in the Lung" International Journal of Molecular Sciences 24, no. 9: 8303. https://doi.org/10.3390/ijms24098303