
Presenilin-1 (PSEN1) Mutations: Clinical Phenotypes beyond Alzheimer’s Disease

Abstract
:1. Introduction
2. Potential Impact of PSEN1 on Other Forms of Neurodegenerative Disease
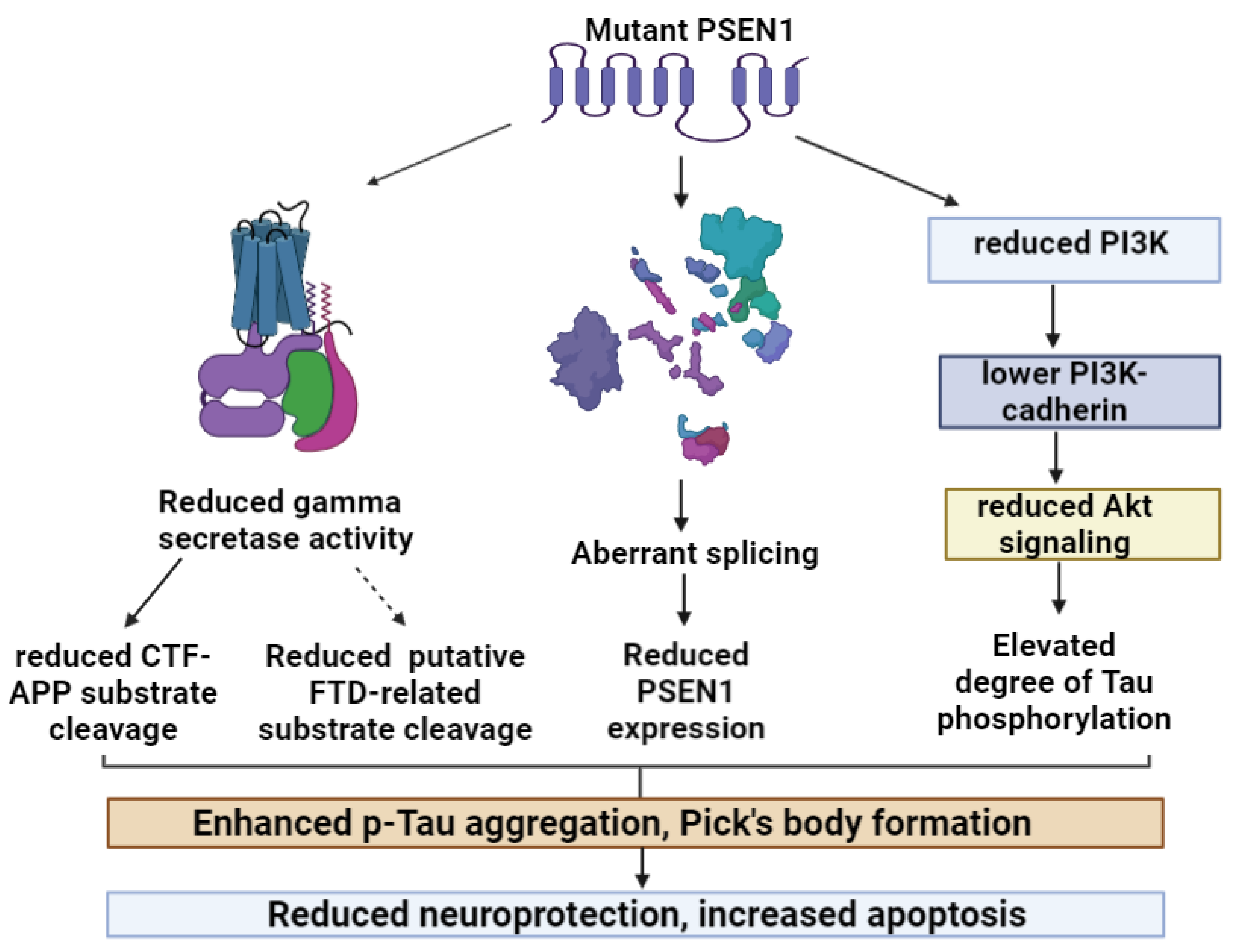
2.1. PSEN1 and Frontotemporal Dysfunctions (FTD; Pick’s Disease)

| Mutation | Initial Diagnosis | Diagnostic Criteria | Imaging | Neuropathology | Revised Diagnosis | Reference |
|---|---|---|---|---|---|---|
| Glu15His | Early onset FTD | NA | NA | NA | NA | [33] |
| Asp40del | bvFTD | Clinical symptoms; imaging | MRI: atrophy involving the lateral frontal lobes | Amyloid deposits in frontal lobes | Frontal variant AD | [34] |
| Leu113Pro | FTD | Lund and Manchester criteria | CT: frontotemporal atrophy; SPECT: hypoperfusion in frontal lobes | AD-like plaques and tangles | May be frontal variant AD | [24,35] |
| Thr122Ala | FTD | NA | NA | NA | NA | [33] |
| Ala137Thr | ||||||
| Met146Val | bvFTD | Lund and Manchester criteria | Frontal temporal atrophy | Co-existence of amyloid plaques, tangles, and Pick’s bodies | AD and Pick’s disease co-existence | [25,26] |
| Gly183Val | FTD | Clinical symptoms, imaging | Severe frontotemporal atrophy | Neocortex: Pick bodies, Tau-positive cytoplasmic neuronal inclusions, and no amyloid plaques | Pick’s disease | [27] |
| Leu226Phe | FTD | Lund and Manchester criteria | frontal atrophy, no hippocampal atrophy, and hypoperfusion in frontal area | Typical AD hallmarks | Frontal variant AD | [29] |
| Met233Leu | FTD | Clinical Consensus Criteria for FTD | PET: hypometabolism in prefrontal, parietal, and temporal cortices | NA | FTD | [31] |
| Pro303Leu | FTD | NA | NA | NA | NA | [33] |
| Arg352dup | FTD | Clinical Consensus Criteria for FTD | Fronto-temporoparietal cortical and right hippocampal atrophy | Ubiquitin-immunoreactive structures | FTD, also carried a GRN splice site mutation | [30] |
| Pro355Ser | FTD/frontal variant AD | Clinical symptoms, imaging | MRI: microbleeds in cortex; FDG-PET: frontotemporal hypometabolism | Suspected Lewy bodies | Frontal variant AD | [36] |
| Val412Ile | FTD | Clinical symptoms, imaging | FDG-PET: hypometabolism in the parietal and frontal cortices | NA | FTD | [32] |
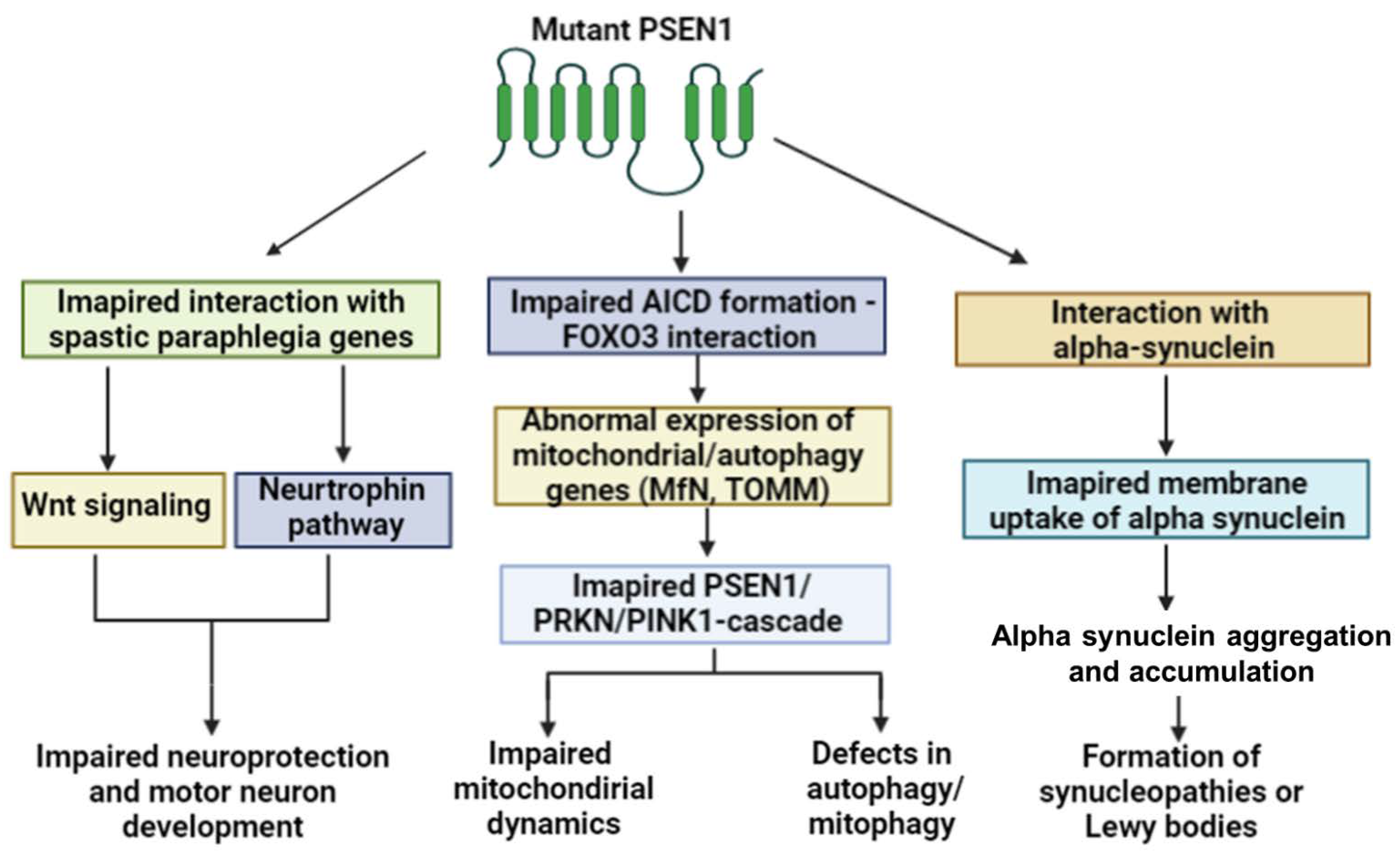
2.2. PSEN1 and Motor Impairment
| Mutation | Initial Diagnosis | Diagnostic Criteria | Imaging | Neuropathology | Revised Diagnosis | Reference |
|---|---|---|---|---|---|---|
| Arg41Ser | l-dopa-responsive early onset Parkinsonism | Imaging and CSF markers | MRI: moderate frontal atrophy; FDG-PET: no amyloid deposits | NA, mild increase in CSF Tau but not amyloid. No amyloid deposits | Early onset Parkinsonism | [50] |
| Gly217Asp | Parkinsonism, dementia | Imaging post-mortem studies | MRI, CT: atrophy of frontal and temporal regions and mild atrophy in cerebellum | Cotton wool plaques, extracellular amyloid fibrils, and tau-immunopositively neurites | Presenile dementia and Parkinsonism | [51] |
| Val272Ala | Parkinsonism, dementia | CERARD, Newcastle | MRI, PET: subcortical-frontal area | Typical AD and Lewy bodies | Subcortical dementia | [52] |
| Glu184Asp | DLB | CERARD | MRI: diffuse cortical atrophy; SPECT: hypoperfusion | Lewy bodies and no amyloid components in astrocytes | AD with Lewy bodies | [56,61] |
| Gly417Ala | Parkinsonism, dementia | NINCDS-ADRDA | MRI: atrophy of parietal and anterior temporal regions and cortex | PiB-PET: diffuse amyloid deposition in several brain areas (cerebellum, frontal, parietal and temporal cortices) | EOAD with Parkinsonism | [64] |
| Gly417Ser | Parkinsonism, AD | Imaging, post-mortem | SPECT: Hypoperfusion in different brain areas | Cotton wool plaques and Lewy bodies | AD with cotton wool plaques | [65] |
| Thr440del | L-DOPA responsive Parkinsonism | Consensus guidelines for DLB | Atrophy of frontal/temporal lobes and brainstem | Neural loss in substania nigra, Lewy bodies, and cotton wool plaques | Variant AD with Lewy bodies | [57] |
2.3. PSEN1 Involvement in Non-Neurodegenerative Disease Phenotypes
3. Atypical Inclusions in the Brain of Patients with PSEN1 Mutations
| Inclusions | Mutation | Amyloid Plaques | Tau | Amyloid Angiopathy | Other Neuropathology | Disease | Reference |
|---|---|---|---|---|---|---|---|
| PBs | Met146Leu | Cortex | Cortex | NA | Pick bodies in upper frontotemporal cortex and dentate gyrus; ballooned neurons. Lewy bodies in amygdala. | AD with Pick pathology | [89] |
| Met146Val | Frontal, parietal cortex | Several brain areas | NA | Co-existence of amyloid plaques, tangles, and Pick’s bodies; cotton wool plaques (CWPs) may also appear. TDP43-positive inclusions also appeared and Lewy bodies were also observed. | AD with Pick pathology | [25,89] | |
| Gly183Val | NA | Neocortex | NA | Neocortex: Pick bodies and Tau-positive cytoplasmic neuronal inclusions. | Pick’s disease | [27] | |
| Ala260Val | Plaque ring around blood vessels; neocortex | Neocortex | NA | Gliosis in cerebral neocortex; Pick-like intraneuronal inclusions in the dentate gyrus. | AD with Pick pathology | [90] | |
| Exon9 deletion | Cortex | Cortex | NA | One case of Pick’s bodies in hippocampus. | AD with Pick pathology | [89] | |
| TDP43 | Gly206Arg | Cortex, striatum and thalamus; cerebellum | Medulla oblongata, dorsal vagal nucleus, substania nigra | Meningeal vasculature | End-stage TDP-43 and alpha synuclein pathology. | EOAD | [93] |
| Arg278Ile | Cortex | NA | Cortex and leptomeninges | Alpha-synuclein and TDP43 pathology in the amygdala. | EOAD | [94] | |
| Gly417Ser | frontal, parietal, and temporal cortices., cerebellum, spinal grey matter | NA | Parietal and anterior temporal regions | CWPs (cortex); Lewy bodies in neocortex; TDP-43 inclusions in the limbic region and temporal cortex. | Spastic Paraparesis, EOAD | [65] | |
| Granulin aggregate | Ala246Glu | Cingulate gyrus, temporal lobe | NA | NA | PGRN aggregates in medial temporal and frontal areas. May co-aggregate with amyloid Gliosis. | Preclinical case | [47] |
| Ubiquitin positive plaques | Glu280Ala | Cerebral cortex, hippocampus, cerebellum, midbrain and basal ganglia | Several brain areas | Ubiquitin–positive plaques surrounded by reactive astrocytes and dystrophic neurites in the cerebellum. | EOAD | [95] | |
| LBs | Pro117Ser | Hippocampus, temporal cortex | cortex | Intracortical vessels | Lewy bodies in the cortex in most patients. | EOAD | [105] |
| Ser132Ala | Hippocampus | Hippocampus | NA | Lewy bodies in neocortex and substania nigra. Neuritic plaques in temporal cortex. | EOAD/DLB | [55] | |
| Ser170Phe | Hippocampus | hippocampus | Several brain areas | Ubiquitin-positive oval intraneuronal inclusions and neuritic plaques in dentate fascia. Lewy bodies in brainstem, limbic areas, and neocortex. | EOAD with Lewy bodies | [101] | |
| Leu174Arg | Isocortex | Allo- and isocortex | NA | Lewy bodies and amygdala and entorhinal cortex. | EOAD | [59] | |
| Glu184Asp | Cortex, cerebellum | Cerebral cortex, amygdala, thalamus, substantia nigra | Leptomeningeal and parenchymal arteries in cortex, brainstem, cerebellum | Lewy bodies and non-amyloid components in plaques and astrocytes. | DLB + primary progressive aphasia | [56] | |
| Leu202Phe | Hippocampus and several brain areas | Several brain areas | Cortical and leptomeningeal blood vessels | Lewy pathology in the amygdala. Gliosis in the cortex. | EOAD | [102] | |
| Met233Val | Cerebral cortex, spinal cord | Cerebral cortex | Leptomeningeal, cerebral, and cerebellar vessels | Lewy bodies in the substantia nigra and cortex. | EOAD Lewy bodies | [60] [106] | |
| Val272Ala | Cortex, thalamus, hypothalamus and mesencephalon, substania nigra | Cortex | NA | Lewy bodies in the cortex and substantia nigra. | Subcortical dementia and parkinsonism | [104] | |
| Ala396Thr | neocortex and basal ganglia | Neocortex, allocortex, substantia nigra, and locus coeruleus | Diffuse plaques and vessels in the entire brain | Lewy bodies in cerebral cortex, caudate nucleus, putamen, hippocampus, and substantia nigra locus coeruleus. Alpha synuclein and Tau may co-aggregate. | AD and DLB | [103] | |
| Ala431Glu | Several brain areas | Several brain areas | NA | Lewy bodies in amygdala, cingulate gyrus, and neocortex in one patient. | AD and DLB | [107] | |
| Thr440fs | cortex, hippocampus, Substantia nigra, pons, medulla | Hippocampus, pons, | Vessels in cerebrum and cortex | Alpha-synuclein-positive Lewy bodies in several brain areas and cotton wool plaques. | AD and DLB | [57] | |
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dewji, N.N. Presenilin structure in mechanisms leading to Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 277–290. [Google Scholar] [CrossRef]
- Chen, Q.; Schubert, D. Presenilin-interacting proteins. Expert Rev. Mol. Med. 2002, 4, 1–18. [Google Scholar] [CrossRef]
- Czech, C.; Tremp, G.; Pradier, L. Presenilins and Alzheimer’s disease: Biological functions and pathogenic mechanisms. Prog. Neurobiol. 2000, 60, 363–384. [Google Scholar] [CrossRef]
- Kelleher, R.J., 3rd; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.; Schmid, B.; Pires, C.; Nielsen, T.T.; Hjermind, L.E.; Nielsen, J.E.; Holst, B.; Hyttel, P.; Freude, K.K. Generation of a gene-corrected isogenic control hiPSC line derived from a familial Alzheimer’s disease patient carrying a L150P mutation in presenilin 1. Stem Cell Res. 2016, 17, 466–469. [Google Scholar] [CrossRef]
- Pires, C.; Schmid, B.; Petræus, C.; Poon, A.; Nimsanor, N.; Nielsen, T.T.; Waldemar, G.; Hjermind, L.E.; Nielsen, J.E.; Hyttel, P.; et al. Generation of a gene-corrected isogenic control cell line from an Alzheimer’s disease patient iPSC line carrying a A79V mutation in PSEN1. Stem Cell Res. 2016, 17, 285–288. [Google Scholar] [CrossRef]
- Konstantinidis, E.; Molisak, A.; Perrin, F.; Streubel-Gallasch, L.; Fayad, S.; Kim, D.Y.; Petri, K.; Aryee, M.J.; Aguilar, X.; György, B.; et al. CRISPR-Cas9 treatment partially restores amyloid-β 42/40 in human fibroblasts with the Alzheimer’s disease PSEN1 M146L mutation. Mol. Ther. Nucleic Acids 2022, 28, 450–461. [Google Scholar] [CrossRef]
- Sierant, M.; Paduszynska, A.; Kazmierczak-Baranska, J.; Nacmias, B.; Sorbi, S.; Bagnoli, S.; Sochacka, E.; Nawrot, B. Specific Silencing of L392VPSEN1Mutant Allele by RNA Interference. Int. J. Alzheimer’s Dis. 2011, 2011, 809218. [Google Scholar] [CrossRef]
- López-García, S.; Jiménez-Bonilla, J.; Delgado, A.L.; Balaguer, P.O.; Ceberio, J.I.; Marraco, I.B.; Rodríguez, E.R.; Sánchez-Juan, P. A Rare PSEN1 (Leu85Pro) Mutation Causing Alzheimer’s Disease in a 29-Year-Old Woman Presenting as Corticobasal Syndrome. J. Alzheimer’s Dis. 2019, 70, 655–658. [Google Scholar] [CrossRef]
- Kim, J.; Bagyinszky, E.; Chang, Y.H.; Choe, G.; Choi, B.-O.; An, S.S.A.; Kim, S. A novel PSEN1 H163P mutation in a patient with early-onset Alzheimer’s disease: Clinical, neuroimaging, and neuropathological findings. Neurosci. Lett. 2012, 530, 109–114. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Q.; Jing, D.; Gao, R.; Zhang, J.; Cui, C.; Qiao, H.; Liang, Z.; Wang, C.; Rosa-Neto, P.; et al. Diagnostic Approach of Early-Onset Dementia with Negative Family History: Implications from Two Cases of Early-Onset Alzheimer’s Disease with De Novo PSEN1 Mutation. J. Alzheimer’s Dis. 2019, 68, 551–558. [Google Scholar] [CrossRef]
- Hooli, B.V.; Kovacs-Vajna, Z.M.; Mullin, K.; Blumenthal, M.A.; Mattheisen, M.; Zhang, C.; Lange, C.; Mohapatra, G.; Bertram, L.; Tanzi, R.E. Rare autosomal copy number variations in early-onset familial Alzheimer’s disease. Mol. Psychiatry 2014, 19, 676–681. [Google Scholar] [CrossRef]
- Roeber, S.; Müller-Sarnowski, F.; Kress, J.; Edbauer, D.; Kuhlmann, T.; Tüttelmann, F.; Schindler, C.; Winter, P.; Arzberger, T.; Müller, U.; et al. Three novel presenilin 1 mutations marking the wide spectrum of age at onset and clinical patterns in familial Alzheimer’s disease. J. Neural Transm. 2015, 122, 1715–1719. [Google Scholar] [CrossRef]
- Colacicco, A.M.; Panza, F.; Basile, A.M.; Solfrizzi, V.; Capurso, C.; D’Introno, A.; Torres, F.; Capurso, S.; Cozza, S.; Flora, R.; et al. F175S change and a novel polymorphism in presenilin-1 gene in late-onset familial Alzheimer’s disease. Eur. Neurol. 2002, 47, 209–213. [Google Scholar] [CrossRef]
- Dermaut, B.; Kumar-Singh, S.; Rademakers, R.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Tau is central in the genetic Alzheimer–frontotemporal dementia spectrum. Trends Genet. 2005, 21, 664–672. [Google Scholar] [CrossRef]
- Larner, A.J.; Doran, M. Genotype-Phenotype Relationships of Presenilin-1 Mutations in Alzheimer’s Disease: An Update. J. Alzheimer’s Dis. 2009, 17, 259–265. [Google Scholar] [CrossRef]
- Bagaria, J.; Bagyinszky, E.; An, S.S.A. Genetics, Functions, and Clinical Impact of Presenilin-1 (PSEN1) Gene. Int. J. Mol. Sci. 2022, 23, 10970. [Google Scholar] [CrossRef]
- Padovani, A.; Premi, E.; Pilotto, A.; Gazzina, S.; Cosseddu, M.; Archetti, S.; Cancelli, V.; Paghera, B.; Borroni, B. Overlap between Frontotemporal Dementia and Alzheimer’s Disease: Cerebrospinal Fluid Pattern and Neuroimaging Study. J. Alzheimer’s Dis. 2013, 36, 49–55. [Google Scholar] [CrossRef]
- Miller, B.L.; Ikonte, C.; Ponton, M.; Levy, M.; Boone, K.; Darby, A.; Berman, N.; Mena, I.; Cummings, J.L. A study of the Lund-Manchester research criteria for frontotemporal dementia: Clinical and single-photon emission CT correlations. Neurology 1997, 48, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Englund, B.; Brun, A.; Gustafson, L.; Passant, U.; Mann, D.; Neary, D.; Snowden, J.S. Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J. Neurol. Neurosurg. Psychiatry 1994, 57, 416–418. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; Van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef] [PubMed]
- Bergmans, B.A.; De Strooper, B. γ-secretases: From cell biology to therapeutic strategies. Lancet Neurol. 2010, 9, 215–226. [Google Scholar] [CrossRef]
- Riudavets, M.A.; Bartoloni, L.; Troncoso, J.C.; Pletnikova, O.; St George-Hyslop, P.; Schultz, M.; Sevlever, G.; Allegri, R.F. Familial dementia with frontotemporal features associated with M146V presenilin-1 mutation. Brain Pathol. 2013, 23, 595–600. [Google Scholar] [CrossRef]
- Schöll, M.; Almkvist, O.; Bogdanovic, N.; Wall, A.; Långström, B.; Viitanen, M.; Nordberg, A. Time Course of Glucose Metabolism in Relation to Cognitive Performance and Postmortem Neuropathology in Met146Val PSEN1 Mutation Carriers. J. Alzheimer’s Dis. 2011, 24, 495–506. [Google Scholar] [CrossRef]
- Dermaut, B.; Kumar-Singh, S.; Engelborghs, S.; Theuns, J.; Rademakers, R.; Saerens, J.; Pickut, B.A.; Peeters, K.; van den Broeck, M.; Vennekens, K.; et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann. Neurol. 2004, 55, 617–626. [Google Scholar] [CrossRef]
- Perrone, F.; Bjerke, M.; Hens, E.; Sieben, A.; Timmers, M.; De Roeck, A.; Vandenberghe, R.; Sleegers, K.; Martin, J.-J.; De Deyn, P.P.; et al. Amyloid-β1–43 cerebrospinal fluid levels and the interpretation of APP, PSEN1 and PSEN2 mutations. Alzheimer’s Res. Ther. 2020, 12, 108. [Google Scholar] [CrossRef]
- Żekanowski, C.; Golan, M.P.; Krzyśko, K.A.; Lipczyńska-Łojkowska, W.; Filipek, S.; Kowalska, A.; Rossa, G.; Pepłońska, B.; Styczyńska, M.; Maruszak, A.; et al. Two novel presenilin 1 gene mutations connected with frontotemporal dementia-like clinical phenotype: Genetic and bioinformatic assessment. Exp. Neurol. 2006, 200, 82–88. [Google Scholar] [CrossRef]
- Boeve, B.F.; Baker, M.; Dickson, D.W.; Parisi, J.E.; Giannini, C.; Josephs, K.A.; Hutton, M.; Pickering-Brown, S.M.; Rademakers, R.; Tang-Wai, D.; et al. Frontotemporal dementia and parkinsonism associated with the IVS1+1G->A mutation in progranulin: A clinicopathologic study. Brain 2006, 129, 3103–3114. [Google Scholar] [CrossRef]
- Mendez, M.F.; McMurtray, A. Frontotemporal Dementia-like Phenotypes Associated With Presenilin-1 Mutations. Am. J. Alzheimer’s Dis. Other Dement. 2006, 21, 281–286. [Google Scholar] [CrossRef]
- Bernardi, L.; Tomaino, C.; Anfossi, M.; Gallo, M.; Geracitano, S.; Costanzo, A.; Colao, R.; Puccio, G.; Frangipane, F.; Curcio, S.A.; et al. Novel PSEN1 and PGRN mutations in early-onset familial frontotemporal dementia. Neurobiol. Aging 2009, 30, 1825–1833. [Google Scholar] [CrossRef]
- Koriath, C.; Kenny, J.; Adamson, G.; Druyeh, R.; Taylor, W.; Beck, J.; Quinn, L.; Mok, T.H.; Dimitriadis, A.; Norsworthy, P.; et al. Predictors for a dementia gene mutation based on gene-panel next-generation sequencing of a large dementia referral series. Mol. Psychiatry 2020, 25, 3399–3412. [Google Scholar] [CrossRef]
- Nygaard, H.B.; Lippa, C.F.; Mehdi, D.; Baehring, J.M. A Novel Presenilin 1 Mutation in Early-Onset Alzheimer’s Disease with Prominent Frontal Features. Am. J. Alzheimer’s Dis. Other Dementiasr. 2014, 29, 433–435. [Google Scholar] [CrossRef]
- Raux, G.; Gantier, R.; Thomas-Anterion, C.; Boulliat, J.; Verpillat, P.; Hannequin, D.; Brice, A.; Frebourg, T.; Campion, D. Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology 2000, 55, 1577–1579. [Google Scholar] [CrossRef]
- Monacelli, F.; Martella, L.; Parodi, M.N.; Odetti, P.; Fanelli, F.; Tabaton, M. Frontal Variant of Alzheimer’s Disease: A Report of a Novel PSEN1 Mutation. J. Alzheimer’s Dis. 2019, 70, 11–15. [Google Scholar] [CrossRef]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef]
- Amtul, Z.; Lewis, P.; Piper, S.; Crook, R.; Baker, M.; Findlay, K.; Singleton, A.; Hogg, M.; Younkin, L.; Younkin, S.G.; et al. A Presenilin 1 Mutation Associated with Familial Frontotemporal Dementia Inhibits γ-Secretase Cleavage of APP and Notch. Neurobiol. Dis. 2002, 9, 269–273. [Google Scholar] [CrossRef]
- Watanabe, H.; Xia, D.; Kanekiyo, T.; Kelleher, R.J., 3rd; Shen, J. Familial Frontotemporal Dementia-Associated Presenilin-1 c.548G>T Mutation Causes Decreased mRNA Expression and Reduced Presenilin Function in Knock-In Mice. J. Neurosci. 2012, 32, 5085–5096. [Google Scholar] [CrossRef]
- Liu, L.; Lauro, B.M.; Wolfe, M.S.; Selkoe, D.J. Hydrophilic loop 1 of Presenilin-1 and the APP GxxxG transmembrane motif regulate γ-secretase function in generating Alzheimer-causing Aβ peptides. J. Biol. Chem. 2021, 296, 100393. [Google Scholar] [CrossRef]
- Evin, G.; Smith, M.J.; Tziotis, A.; McLean, C.; Canterford, L.; Sharples, R.A.; Cappai, R.; Weidemann, A.; Beyreuther, K.; Cotton, R.G.H.; et al. Alternative transcripts of presenilin-1 associated with frontotemporal dementia. Neuroreport 2002, 13, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.E.; Gregory, G.C.; Vickers, J.C.; Brooks, W.S.; Kwok, J.B.; Schofield, P.R.; Kril, J.J.; Halliday, G.M. Positional effects of presenilin-1 mutations on tau phosphorylation in cortical plaques. Neurobiol. Dis. 2004, 15, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Boutajangout, A.; Leroy, K.; Touchet, N.; Authelet, M.; Blanchard, V.; Tremp, G.; Pradier, L.; Brion, J.P. Increased tau phosphorylation but absence of formation of neurofibrillary tangles in mice double transgenic for human tau and Alzheimer mutant (M146L) presenilin-1. Neurosci. Lett. 2002, 318, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Boutajangout, A.; Authelet, M.; Blanchard, V.; Touchet, N.; Tremp, G.; Pradier, L.; Brion, J.-P. Characterisation of cytoskeletal abnormalities in mice transgenic for wild-type human tau and familial Alzheimer’s disease mutants of APP and presenilin-1. Neurobiol. Dis. 2004, 15, 47–60. [Google Scholar] [CrossRef]
- Baki, L.; Shioi, J.; Wen, P.; Shao, Z.; Schwarzman, A.; Gama-Sosa, M.; Neve, R.; Robakis, N.K. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: Effects of FAD mutations. EMBO J. 2004, 23, 2586–2596. [Google Scholar] [CrossRef]
- Shen, J.; Bronson, R.T.; Chen, D.F.; Xia, W.; Selkoe, D.J.; Tonegawa, S. Skeletal and CNS Defects in Presenilin-1-Deficient Mice. Cell 1997, 89, 629–639. [Google Scholar] [CrossRef]
- Gliebus, G.; Rosso, A.; Lippa, C.F. Progranulin and β-Amyloid Distribution: A Case Report of the Brain From Preclinical PS-1 Mutation Carrier. Am. J. Alzheimer’s Dis. Other Dement. 2009, 24, 456–460. [Google Scholar] [CrossRef]
- Tabira, T.; Chui, D.H.; Nakayama, H.; Kuroda, S.; Shibuya, M. Alzheimer’s disease with spastic paresis and cotton wool type plaques. J. Neurosci. Res. 2002, 70, 367–372. [Google Scholar] [CrossRef]
- Chelban, V.; Breza, M.; Szaruga, M.; Vandrovcova, J.; Murphy, D.; Lee, C.J.; Alikhwan, S.; Bourinaris, T.; Vavougios, G.; Ilyas, M.; et al. Spastic paraplegia preceding PSEN1-related familial Alzheimer’s disease. Alzheimer’s Dement. 2021, 13, e12186. [Google Scholar] [CrossRef]
- Gatto, E.M.; Rojas, G.J.; Nemirovsky, S.I.; Da Prat, G.; Persi, G.; Cessarini, M.; Etcheverry, J.L.; Rojas, N.G.; Parisi, V.; Cordoba, M.; et al. A novel mutation in PSEN1 (p.Arg41Ser) in an Argentinian woman with early onset Parkinsonism. Park. Relat. Disord. 2020, 77, 21–25. [Google Scholar] [CrossRef]
- Takao, M.; Ghetti, B.; Hayakawa, I.; Ikeda, E.; Fukuuchi, Y.; Miravalle, L.; Piccardo, P.; Murrell, J.R.; Glazier, B.S.; Koto, A. A novel mutation (G217D) in the Presenilin 1 gene (PSEN1) in a Japanese family: Presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol. 2002, 104, 155–170. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, P.; Xie, F.; Wang, B.; Lin, Z.; Luo, W. A heterozygous de novo PSEN1 mutation in a patient with early-onset parkinsonism. Neurol. Sci. 2022, 43, 1405–1409. [Google Scholar] [CrossRef]
- Checler, F.; Goiran, T.; Da Costa, C.A. Presenilins at the crossroad of a functional interplay between PARK2/PARKIN and PINK1 to control mitophagy: Implication for neurodegenerative diseases. Autophagy 2017, 13, 2004–2005. [Google Scholar] [CrossRef]
- Meeus, B.; Verstraeten, A.; Crosiers, D.; Engelborghs, S.; van den Broeck, M.; Mattheijssens, M.; Peeters, K.; Corsmit, E.; Elinck, E.; Pickut, B.; et al. DLB and PDD: A role for mutations in dementia and Parkinson disease genes? Neurobiol. Aging 2012, 33, 629.e5–629.e18. [Google Scholar] [CrossRef]
- Yokota, O.; Terada, S.; Ishizu, H.; Ujike, H.; Ishihara, T.; Nakashima, H.; Yasuda, M.; Kitamura, Y.; Uéda, K.; Checler, F.; et al. NACP/α-Synuclein, NAC, and β-amyloid pathology of familial Alzheimer’s disease with the E184D presenilin-1 mutation: A clinicopathological study of two autopsy cases. Acta Neuropathol. 2002, 104, 637–648. [Google Scholar] [CrossRef]
- Ishikawa, A.; Piao, Y.-S.; Miyashita, A.; Kuwano, R.; Onodera, O.; Ohtake, H.; Suzuki, M.; Nishizawa, M.; Takahashi, H. A mutantPSEN1 causes dementia with lewy bodies and variant Alzheimer’s disease. Ann. Neurol. 2005, 57, 429–434. [Google Scholar] [CrossRef]
- Tiedt, H.O.; Benjamin, B.; Niedeggen, M.; Lueschow, A. Phenotypic Variability in Autosomal Dominant Familial Alzheimer Disease due to the S170F Mutation of Presenilin-1. Neurodegener. Dis. 2018, 18, 57–68. [Google Scholar] [CrossRef]
- Klünemann, H.H.; Rogaeva, E.; Neumann, M.; Kretzschmar, H.A.; Kandel, M.; Toulina, A.; Sato, C.; Salehi-Rad, S.; Pfister, K.; Klein, H.E.; et al. Novel PS1 mutation in a Bavarian kindred with familial Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2004, 18, 256–258. [Google Scholar]
- Revesz, T.; McLaughlin, J.L.; Rossor, M.N.; Lantos, P.L. Pathology of familial Alzheimer’s disease with Lewy bodies. J. Neural. Transm. Suppl. 1997, 51, 121–135. [Google Scholar] [CrossRef]
- Ryan, N.S.; Nicholas, J.M.; Weston, P.S.J.; Liang, Y.; Lashley, T.; Guerreiro, R.; Adamson, G.; Kenny, J.; Beck, J.; Chavez-Gutierrez, L.; et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: A case series. Lancet Neurol. 2016, 15, 1326–1335. [Google Scholar] [CrossRef]
- Picková, T.; Matěj, R.; Bezdicek, O.; Keller, J.; Van Der Zee, J.; Van Broeckhoven, C.; Cséfalvay, Z.; Rusina, R. Genetic Alzheimer Disease and Sporadic Dementia with Lewy Bodies: A Comorbidity Presenting as Primary Progressive Aphasia. Cogn. Behav. Neurol. 2017, 30, 23–29. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Galasko, D.; Kosaka, K.; Perry, E.K.; Dickson, D.W.; Hansen, L.A.; Salmon, D.P.; Lowe, J.; Mirra, S.S.; Byrne, E.J.; et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology. Neurology 1996, 47, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Moussaud, S.; Zhu, L.; Post, K.L.; Dickson, D.W.; Berezovska, O.; McLean, P.J. Convergence of pathology in dementia with Lewy bodies and Alzheimer’s disease: A role for the novel interaction of alpha-synuclein and presenilin 1 in disease. Brain 2014, 137, 1958–1970. [Google Scholar] [CrossRef]
- Van Giau, V.; Wang, M.J.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. Novel PSEN1 p.Gly417Ala mutation in a Korean patient with early-onset Alzheimer’s disease with parkinsonism. Neurobiol. Aging 2018, 72, 188.e13–188.e17. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Yokota, O.; Haraguchi, T.; Ikeuchi, T.; Zhu, B.; Takenoshita, S.; Terada, S.; Yamada, N. Young adult-onset, very slowly progressive cognitive decline with spastic paraparesis in Alzheimer’s disease with cotton wool plaques due to a novel presenilin1 G417S mutation. Acta Neuropathol. Commun. 2019, 7, 19. [Google Scholar] [CrossRef]
- Li, A.; Peng, Y.; Taiclet, L.M.; Tanzi, R.E. Analysis of hidradenitis suppurativa–linked mutations in four genes and the effects of PSEN1-P242LfsX11 on cytokine and chemokine expression in macrophages. Hum. Mol. Genet. 2019, 28, 1173–1182. [Google Scholar] [CrossRef]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. γ-Secretase Gene Mutations in Familial Acne Inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, M.-H.; Tian, X.; Cheng, H.-T.; Gridley, T.; Shen, J.; Kopan, R. γ-Secretase Functions through Notch Signaling to Maintain Skin Appendages but Is Not Required for Their Patterning or Initial Morphogenesis. Dev. Cell 2004, 7, 731–743. [Google Scholar] [CrossRef]
- Aubin-Houzelstein, G. Notch signaling and the developing hair follicle. Adv. Exp. Med. Biol. 2012, 727, 142–160. [Google Scholar] [CrossRef]
- Ye, Y.; Fortini, M.E. Apoptotic Activities of Wild-Type and Alzheimer’s Disease-Related Mutant Presenilins in Drosophila melanogaster. J. Cell Biol. 1999, 146, 1351–1364. [Google Scholar] [CrossRef]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of Presenilin Genes in Dilated Cardiomyopathy and Heart Failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef]
- Hébert, S.S.; Serneels, L.; Dejaegere, T.; Horré, K.; Dabrowski, M.; Baert, V.; Annaert, W.; Hartmann, D.; De Strooper, B. Coordinated and widespread expression of γ-secretase in vivo: Evidence for size and molecular heterogeneity. Neurobiol. Dis. 2004, 17, 260–272. [Google Scholar] [CrossRef]
- Sandbrink, R.; Masters, C.L.; Beyreuther, K. Beta A4-amyloid protein precursor mRNA isoforms without exon 15 are ubiquitously expressed in rat tissues including brain, but not in neurons. J. Biol. Chem. 1994, 269, 1510–1517. [Google Scholar] [CrossRef]
- Nakajima, M.; Moriizumi, E.; Koseki, H.; Shirasawa, T. Presenilin 1 is essential for cardiac morphogenesis. Dev. Dyn. 2004, 230, 795–799. [Google Scholar] [CrossRef]
- Luxán, G.; D’Amato, G.; MacGrogan, D.; De La Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- Takeda, T.; Asahi, M.; Yamaguchi, O.; Hikoso, S.; Nakayama, H.; Kusakari, Y.; Kawai, M.; Hongo, K.; Higuchi, Y.; Kashiwase, K.; et al. Presenilin 2 regulates the systolic function of heart by modulating Ca2+ signaling. FASEB J. 2005, 19, 2069–2071. [Google Scholar] [CrossRef]
- Song, X.W.; Yuan, Q.N.; Tang, Y.; Cao, M.; Shen, Y.F.; Zeng, Z.Y.; Lei, C.H.; Li, S.; Zhao, X.X.; Yang, Y.J. Conditionally targeted deletion of PSEN1 leads to diastolic heart dysfunction. J. Cell. Physiol. 2018, 233, 1548–1557. [Google Scholar] [CrossRef]
- Song, X.W.; Zhao, F.; Yang, J.; Yuan, Q.N.; Zeng, Z.Y.; Shen, M.; Tang, Y.; Cao, M.; Shen, Y.F.; Li, S.H.; et al. Cardiovascular-specific PSEN1 deletion leads to abnormalities in calcium homeostasis. Cell Biol. Int. 2022, 46, 475–487. [Google Scholar] [CrossRef]
- To, M.D.; Gokgoz, N.; Doyle, T.G.; Donoviel, D.B.; Knight, J.A.; Hyslop, P.S.; Bernstein, A.; Andrulis, I.L. Functional characterization of novel presenilin-2 variants identified in human breast cancers. Oncogene 2006, 25, 3557–3564. [Google Scholar] [CrossRef]
- Kim, Y.C.; Jeong, B.H. Identification of Somatic Mutations in Dementia-related Genes in Cancer Patients. Curr. Alzheimer Res. 2020, 17, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Roperch, J.P.; Alvaro, V.; Prieur, S.; Tuynder, M.; Nemani, M.; Lethrosne, F.; Piouffre, L.; Gendron, M.C.; Israeli, D.; Dausset, J.; et al. Inhibition of presenilin 1 expression is promoted by p53 and p21WAF-1and results in apoptosis and tumor suppression. Nat. Med. 1998, 4, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Amson, R.; Lassalle, J.M.; Halley, H.; Prieur, S.; Lethrosne, F.; Roperch, J.P.; Israeli, D.; Gendron, M.C.; Duyckaerts, C.; Checler, F.; et al. Behavioral alterations associated with apoptosis and down-regulation of presenilin 1 in the brains of p53-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 5346–5350. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wu, P.F.; Ma, J.X.; Liao, M.J.; Xu, L.S.; Xu, M.H.; Yi, L. Presenilin1 exerts antiproliferative effects by repressing the Wnt/β-catenin pathway in glioblastoma. Cell Commun. Signal. 2020, 18, 22. [Google Scholar] [CrossRef]
- Deng, H.; Lv, L.; Li, Y.; Zhang, C.; Meng, F.; Pu, Y.; Xiao, J.; Qian, L.; Zhao, W.; Liu, Q.; et al. The miR-193a-3p regulated PSEN1 gene suppresses the multi-chemoresistance of bladder cancer. Biochim. Biophys. Acta 2015, 1852, 520–528. [Google Scholar] [CrossRef]
- Pan, X.; Zhao, T.; Mu, S.; Li, S. miR-193a Directly Targets PSEN1 and Inhibits Gastric Cancer Cell Growth, the Activation of PI3K/Akt Signaling Pathway, and the Epithelial-to-Mesenchymal Transition. J. Oncol. 2021, 2021, 2804478. [Google Scholar] [CrossRef]
- Dickson, D.W. Pick’s disease: A modern approach. Brain. Pathol. 1998, 8, 339–354. [Google Scholar] [CrossRef]
- Armstrong, R.; Cairns, N.; Lantos, P. The spatial patterns of Pick bodies, Pick cells and Alzheimer’s disease pathology in Pick’s disease. Neuropathology 1999, 19, 64–70. [Google Scholar] [CrossRef]
- Halliday, G.M.; Song, Y.J.; Lepar, G.; Brooks, W.S.; Kwok, J.B.; Kersaitis, C.; Gregory, G.; Shepherd, C.E.; Rahimi, F.; Schofield, P.R.; et al. Pick bodies in a family with presenilin-1 Alzheimer’s disease. Ann. Neurol. 2005, 57, 139–143. [Google Scholar] [CrossRef]
- Ikeda, M.; Sharma, V.; Sumi, S.M.; Rogaeva, E.A.; Poorkaj, P.; Sherrington, R.; Nee, L.; Tsuda, T.; Oda, N.; Watanabe, M.; et al. The Clinical phenotype of two missense mutations in the presenilin I gene in Japanese patients. Ann. Neurol. 1996, 40, 912–917. [Google Scholar] [CrossRef]
- Meneses, A.; Koga, S.; O’Leary, J.; Dickson, D.W.; Bu, G.; Zhao, N. TDP-43 Pathology in Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 84. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef]
- Libard, S.; Giedraitis, V.; Kilander, L.; Ingelsson, M.; Alafuzoff, I. Mixed Pathologies in a Subject with a Novel PSEN1 G206R Mutation. J. Alzheimer’s Dis. 2022, 90, 1601–1614. [Google Scholar] [CrossRef]
- Willumsen, N.; Poole, T.; Nicholas, J.M.; Fox, N.C.; Ryan, N.S.; Lashley, T. Variability in the type and layer distribution of cortical Aβ pathology in familial Alzheimer’s disease. Brain Pathol. 2022, 32, e13009. [Google Scholar] [CrossRef]
- Lemere, C.A.; Lopera, F.; Kosik, K.S.; Lendon, C.L.; Ossa, J.; Saido, T.C.; Yamaguchi, H.; Ruiz, A.; Martinez, A.; Madrigal, L.; et al. The E280A presenilin 1 Alzheimer mutation produces increased Aβ42 deposition and severe cerebellar pathology. Nat. Med. 1996, 2, 1146–1150. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.-P.; Thomas, A.; et al. Dementia with Lewy bodies: An update and outlook. Mol. Neurodegener. 2019, 14, 5. [Google Scholar] [CrossRef]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef]
- Chartier, S.; Duyckaerts, C. Is Lewy pathology in the human nervous system chiefly an indicator of neuronal protection or of toxicity? Cell Tissue Res. 2018, 373, 149–160. [Google Scholar] [CrossRef]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Trojanowsk, J.Q.; Lee, V.M. Lewy Body Pathology in Alzheimer’s Disease. J. Mol. Neurosci. 2001, 17, 225–232. [Google Scholar] [CrossRef]
- Snider, B.J.; Norton, J.; Coats, M.A.; Chakraverty, S.; Hou, C.E.; Jervis, R.; Lendon, C.L.; Goate, A.M.; McKeel, D.W., Jr.; Morris, J.C. Novel Presenilin 1 Mutation (S170F) Causing Alzheimer Disease with Lewy Bodies in the Third Decade of Life. Arch. Neurol. 2005, 62, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.S.; Lashley, T.; Revesz, T.; Dantu, K.; Fox, N.C.; Morris, H.R. Spontaneous ARIA (Amyloid-Related Imaging Abnormalities) and Cerebral Amyloid Angiopathy Related Inflammation in Presenilin 1-Associated Familial Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 44, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Gondim, D.D.; Oblak, A.; Murrell, J.R.; Richardson, R.; Epperson, F.; Ross, O.A.; Ghetti, B. Diffuse Lewy Body Disease and Alzheimer Disease: Neuropathologic Phenotype Associated with the PSEN1 p.A396T Mutation. J. Neuropathol. Exp. Neurol. 2019, 78, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Escrig, A.; Rabano, A.; Guerrero, C.; Simon, J.; Barquero, M.S.; Güell, I.; Ginestal, R.C.; Montero, T.; Orensanz, L. New V272A presenilin 1 mutation with very early onset subcortical dementia and parkinsonism. Eur. J. Neurol. 2004, 11, 663–669. [Google Scholar] [CrossRef]
- Dowjat, W.K.; Kuchna, I.; Wisniewski, T.; Wegiel, J. A novel highly pathogenic Alzheimer presenilin-1 mutation in codon 117 (Pro117Ser): Comparison of clinical, neuropathological and cell culture phenotypes of Pro117Leu and Pro117Ser mutations. J. Alzheimer’s Dis. 2004, 6, 31–43. [Google Scholar] [CrossRef]
- Houlden, H.; Crook, R.; Dolan, R.J.; McLaughlin, J.; Revesz, T.; Hardy, J. A novel presenilin mutation (M233V) causing very early onset Alzheimer’s disease with Lewy bodies. Neurosci. Lett. 2001, 313, 93–95. [Google Scholar] [CrossRef]
- Leverenz, J.B.; Fishel, M.A.; Peskind, E.R.; Montine, T.J.; Nochlin, D.; Steinbart, E.; Raskind, M.A.; Schellenberg, G.D.; Bird, T.D.; Tsuang, D. Lewy body pathology in familial Alzheimer disease: Evidence for disease- and mutation-specific pathologic phenotype. Arch. Neurol. 2006, 63, 370–376. [Google Scholar] [CrossRef]
- Matej, R.; Tesar, A.; Rusina, R. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin. Biochem. 2019, 73, 26–31. [Google Scholar] [CrossRef]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Ramanan, V.K.; Saykin, A.J. Pathways to neurodegeneration: Mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am. J. Neurodegener. Dis. 2013, 2, 145–175. [Google Scholar]
- Vetrivel, K.S.; Zhang, Y.-W.; Xu, H.; Thinakaran, G. Pathological and physiological functions of presenilins. Mol. Neurodegener. 2006, 1, 4. [Google Scholar] [CrossRef]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 178–191. [Google Scholar] [CrossRef]
- Alzheimer’s Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat. Genet. 1995, 11, 219–222. [Google Scholar] [CrossRef]
- Foncin, J.F.; Salmon, D.; Supino-Viterbo, V.; Feldman, R.G.; Macchi, G.; Mariotti, P.; Scoppetta, C.; Caruso, G.; Bruni, A.C. Alzheimer’s presenile dementia transmitted in an extended kindred. Rev. Neurol. 1985, 141, 194–202. [Google Scholar]
- Bergamini, L.; Pinessi, L.; Rainero, I.; Brunetti, E.; Cerrato, P.; Cosentino, L.; Vaula, G.; Bruni, A.C.; Ermio, C.; Gei, G. Familial Alzheimer’s disease. Evidences for clinical and genetic heterogeneity. Acta Neurol. 1991, 13, 534–538. [Google Scholar]
- Yasuda, M.; Maeda, K.; Ikejiri, Y.; Kawamata, T.; Kuroda, S.; Tanaka, C. A novel missense mutation in the presenilin-1 gene in a familial Alzheimer’s disease pedigree with abundant amyloid angiopathy. Neurosci. Lett. 1997, 232, 29–32. [Google Scholar] [CrossRef]
- Janssen, J.C.; Beck, J.A.; Campbell, T.A.; Dickinson, A.; Fox, N.C.; Harvey, R.J.; Houlden, H.; Rossor, M.N.; Collinge, J. Early onset familial Alzheimer’s disease: Mutation frequency in 31 families. Neurology 2003, 60, 235–239. [Google Scholar] [CrossRef]
- Bertram, L. Next Generation Sequencing in Alzheimer’s Disease. Methods Mol. Biol. 2016, 1303, 281–297. [Google Scholar] [CrossRef]
- Bonvicini, C.; Scassellati, C.; Benussi, L.; Di Maria, E.; Maj, C.; Ciani, M.; Fostinelli, S.; Mega, A.; Bocchetta, M.; Lanzi, G.; et al. Next Generation Sequencing Analysis in Early Onset Dementia Patients. J. Alzheimer’s Dis. 2019, 67, 243–256. [Google Scholar] [CrossRef]
- Van Giau, V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 8368. [Google Scholar] [CrossRef] [PubMed]
- An, S.S.A.; Hulme, J.P. Plasma amyloid-beta oligomer and phosphorylated tau: Diagnostic tools for progressive Alzheimer’s disease. Neural Regen. Res. 2023, 18, 2391–2392. [Google Scholar] [CrossRef]
- Ashton, N.J.; Hye, A.; Rajkumar, A.P.; Leuzy, A.; Snowden, S.; Suárez-Calvet, M.; Karikari, T.; Schöll, M.; La Joie, R.; Rabinovici, G.D.; et al. An update on blood-based biomarkers for non-Alzheimer neurodegenerative disorders. Nat. Rev. Neurol. 2020, 16, 265–284. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Mutation |
|---|---|
| FTD or progressive non-fluent aphasia or Pick’s disease | Gln15His; Pro88Arg; Leu113Pro; Thr122Ala; Ser132Ala; Ala137Thr; Met146Val; Met146Leu; Gly183Val; Leu166Pro; Leu226Phe; Met233Leu; Arg278Ile; Pro264Leu Pro303Leu; Arg352dup; Val412Ile |
| ALS | Leu166Pro; Trp203Cys; Ile249Leu |
| PD | Arg41Ser; Leu85Pro; Glu120Lys; Tyr156Cys; Ser170Pro; Gly217Asp; Pro264Leu; Val272Ala; Tyr288His; Tyr389His; Val391Gly; Gly417Ala; Ala434Thr |
| Spastic paraparesis | Ile83_Met84del; Met84Val; Leu85Pro; Glu120Lys; Tyr154Asn; Tyr156_Arg157insIle_Tyr; Leu166Pro; Gln223Arg; Phe237Ile; Val261Leu; Val261Phe; Pro264Leu; Gly266Ser; Arg278Thr; Arg278Lys; Arg278Ser; Glu280Gly;Pro284Ser; Pro284Leu; Tyr288His; Ser290Cys; Thr291Ala; Thr291Pro; Leu381Val; Phe388Ser; Gly417Ser; Leu424Arg; Pro436Gln |
| Ataxia | Pro117Ala; Thr147Pro; Met233Val |
| DLB | Ser132Ala; Ala275Ser; Thr440del |
| Non-neurodegenerative | Pro242fs; Asp333Gly |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Bagyinszky, E.; An, S.S.A. Presenilin-1 (PSEN1) Mutations: Clinical Phenotypes beyond Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 8417. https://doi.org/10.3390/ijms24098417
Yang Y, Bagyinszky E, An SSA. Presenilin-1 (PSEN1) Mutations: Clinical Phenotypes beyond Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(9):8417. https://doi.org/10.3390/ijms24098417
Chicago/Turabian StyleYang, Youngsoon, Eva Bagyinszky, and Seong Soo A. An. 2023. "Presenilin-1 (PSEN1) Mutations: Clinical Phenotypes beyond Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 9: 8417. https://doi.org/10.3390/ijms24098417