
Activation and Functions of Col6a1+ Fibroblasts in Colitis-Associated Cancer
 , ,
, ,
Abstract
:1. Introduction
2. Results
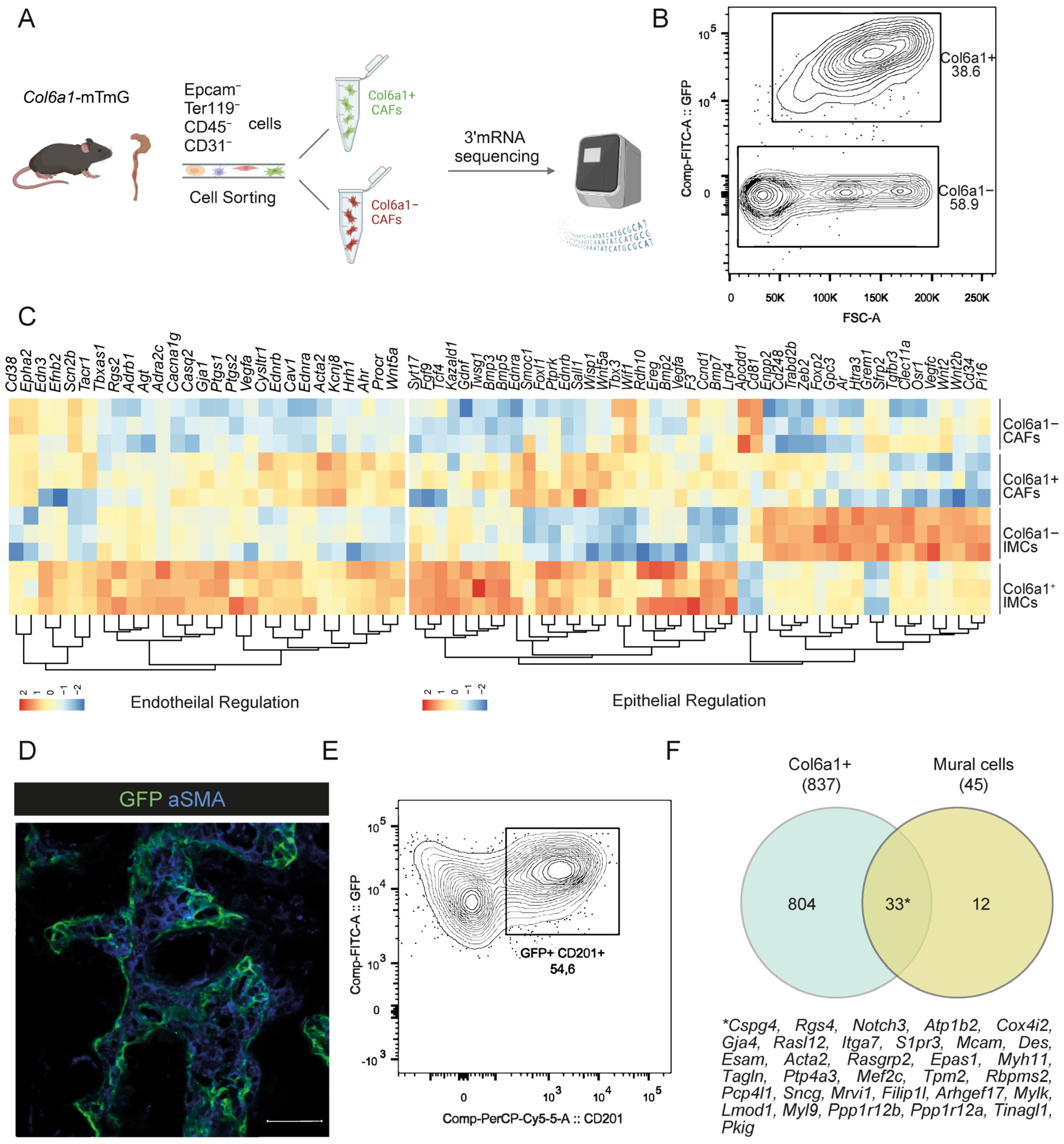
2.1. CAFs Partly Maintain Fibroblast Identities upon AOM/DSS Intestinal Carcinogenesis
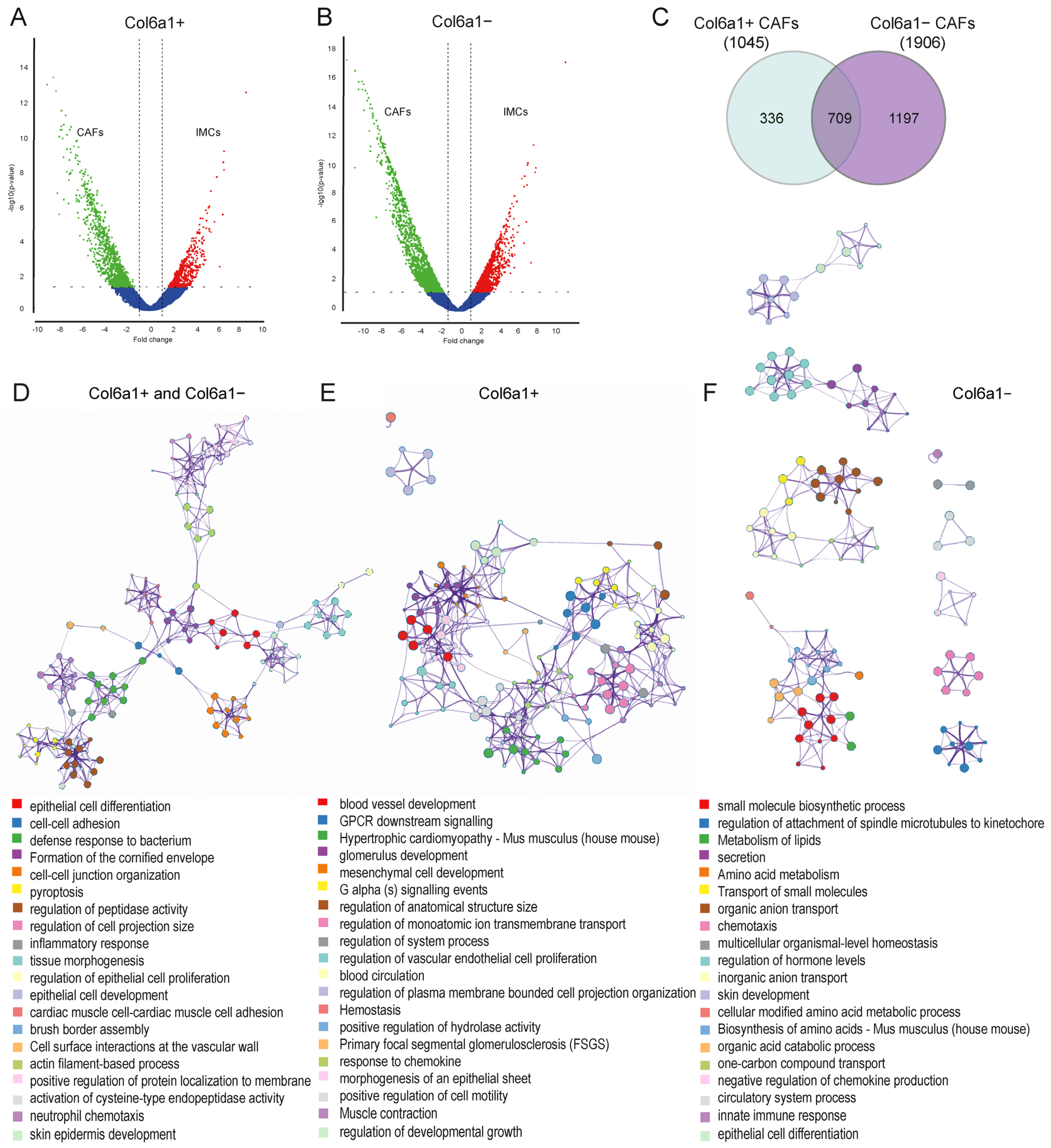
2.2. Col6a1+ and Col6a1− CAFs Are Activated in AOM/DSS Colon Carcinogenesis
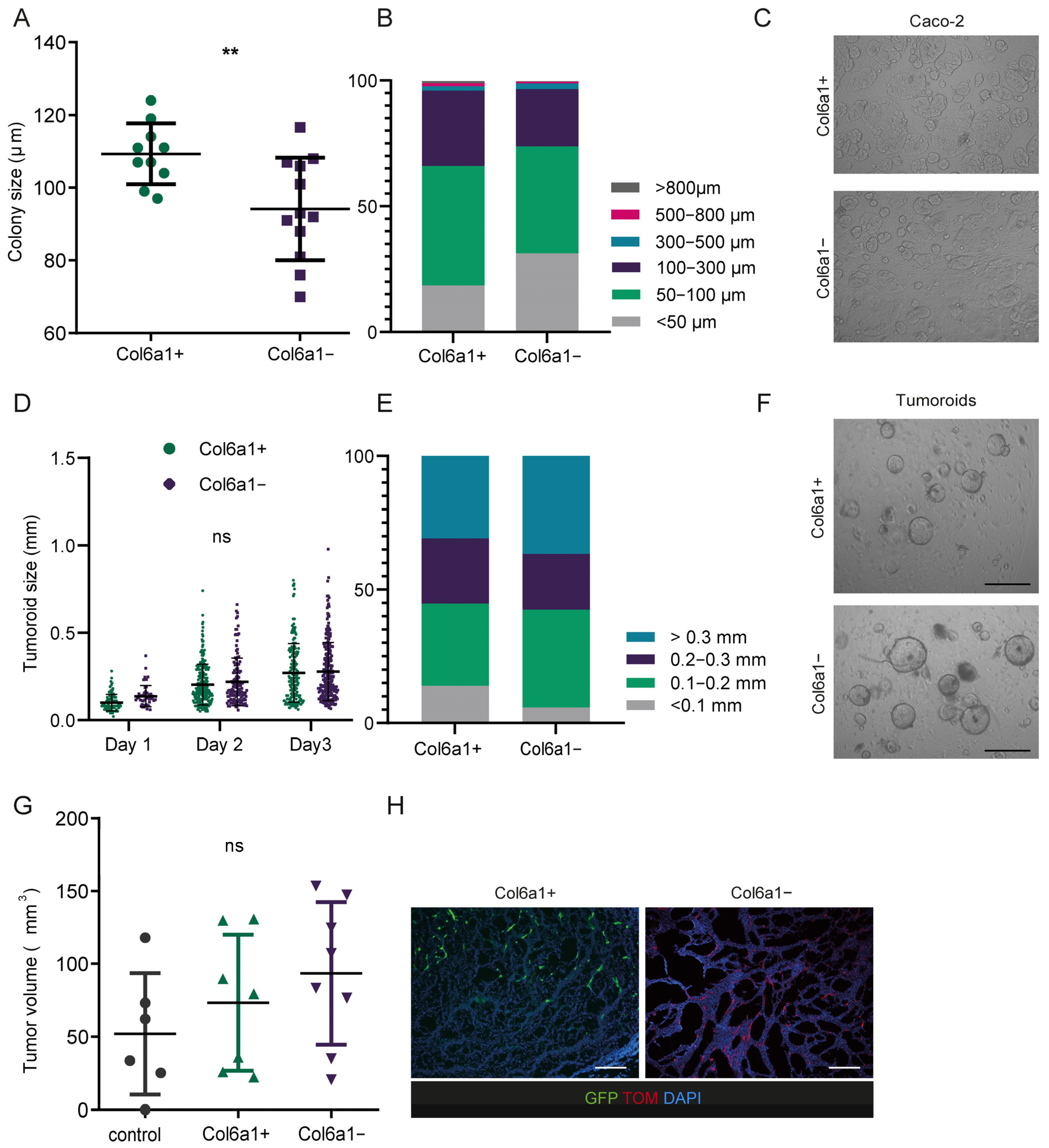
2.3. Col6a1+ and Col6a1− CAFs Support Cancer Cell Growth In Vitro and In Vivo
2.4. Deletion of IL-1R1 or TNFR1 in Col6a1+ IMCs Is not Sufficient to Ameliorate Colitis-Associated Carcinogenesis
3. Discussion
4. Materials and Methods
4.1. Mice and Study Approval
4.2. Induction of Colitis-Associated Cancer
4.3. FACS Analysis and Sorting
4.4. 3′ RNA Sequencing and Analysis
4.5. Isolation and Culture of Primary Mouse Intestinal Mesenchymal Cells
4.6. Tumor Organoids and Co-Culture with IMCs
4.7. Caco-2 Co-Culture Assay
4.8. Allografts
4.9. Immunohistochemistry
4.10. Proteome Profiling
4.11. MIP-2 Elisa
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koliaraki, V.; Prados, A.; Armaka, M.; Kollias, G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. 2020, 21, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, N.; Kraiczy, J.; Shivdasani, R.A. Cellular and molecular architecture of the intestinal stem cell niche. Nat. Cell Biol. 2020, 22, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Gieniec, K.A.; Lannagan, T.R.; Wang, T.; Asai, N.; Mizutani, Y.; Iida, T.; Ando, R.; Thomas, E.M.; Sakai, A.; et al. The Origin and Contribution of Cancer-Associated Fibroblasts in Colorectal Carcinogenesis. Gastroenterology 2022, 162, 890–906. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, G.; Tuveson, D.A. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell 2023, 41, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef]
- Chalkidi, N.; Paraskeva, C.; Koliaraki, V. Fibroblasts in intestinal homeostasis, damage, and repair. Front. Immunol. 2022, 13, 924866. [Google Scholar] [CrossRef]
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 2018, 175, 372–386.e17. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Jackson, M.A.; Korsunsky, I.; Bullers, S.J.; Rue-Albrecht, K.; Christoforidou, Z.; Sathananthan, D.; Thomas, T.; Ravindran, R.; et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med. 2021, 27, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Chen, Z.; Geng, L.; Wang, J.; Liang, H.; Cao, Y.; Chen, H.; Huang, W.; Su, M.; Wang, H.; et al. Mucosal Profiling of Pediatric-Onset Colitis and IBD Reveals Common Pathogenics and Therapeutic Pathways. Cell 2019, 179, 1160–1176.e24. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.S.; Januszyk, M.; Delitto, D.; Yost, K.E.; Griffin, M.; Guo, J.; Guardino, N.; Delitto, A.E.; Chinta, M.; Burcham, A.R.; et al. Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin. Cancer Cell 2022, 40, 1392–1406.e7. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.O.; Hong, Y.; Etlioglu, H.E.; Cho, Y.B.; Pomella, V.; Van den Bosch, B.; Vanhecke, J.; Verbandt, S.; Hong, H.; Min, J.-W.; et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 2020, 52, 594–603. [Google Scholar] [CrossRef]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef]
- Becker, W.R.; Nevins, S.A.; Chen, D.C.; Chiu, R.; Horning, A.M.; Guha, T.K.; Laquindanum, R.; Mills, M.; Chaib, H.; Ladabaum, U.; et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat. Genet. 2022, 54, 985–995. [Google Scholar] [CrossRef]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 2021, 184, 4734–4752.e20. [Google Scholar] [CrossRef]
- Koliaraki, V.; Chalkidi, N.; Henriques, A.; Tzaferis, C.; Polykratis, A.; Waisman, A.; Muller, W.; Hackam, D.J.; Pasparakis, M.; Kollias, G. Innate Sensing through Mesenchymal TLR4/MyD88 Signals Promotes Spontaneous Intestinal Tumorigenesis. Cell Rep. 2019, 26, 536–545.e4. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Pasparakis, M.; Kollias, G. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med. 2015, 212, 2235–2251. [Google Scholar] [CrossRef] [PubMed]
- Pallangyo, C.K.; Ziegler, P.K.; Greten, F.R. IKKβ acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 2015, 212, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F. Cancer: Fibroblasts for all seasons. Nature 2016, 530, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Melissari, M.T.; Henriques, A.; Tzaferis, C.; Prados, A.; Sarris, M.E.; Chalkidi, N.; Mavroeidi, D.; Chouvardas, P.; Grammenoudi, S.; Kollias, G.; et al. Col6a1+/CD201+ mesenchymal cells regulate intestinal morphogenesis and homeostasis. Cell. Mol. Life Sci. 2021, 79, 1. [Google Scholar] [CrossRef] [PubMed]
- Neufert, C.; Becker, C.; Neurath, M.F. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2007, 2, 1998–2004. [Google Scholar] [CrossRef]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Dasgupta, S.; Ghosh, T.; Dhar, J.; Bhuniya, A.; Nandi, P.; Das, A.; Saha, A.; Das, J.; Guha, I.; Banerjee, S.; et al. RGS5-TGFβ-Smad2/3 axis switches pro- to anti-apoptotic signaling in tumor-residing pericytes, assisting tumor growth. Cell Death Differ. 2021, 28, 3052–3076. [Google Scholar] [CrossRef]
- Abdulaal, W.H.; Walker, C.R.; Costello, R.; Redondo-Castro, E.; Mufazalov, I.A.; Papaemmanouil, A.; Rothwell, N.J.; Allan, S.M.; Waisman, A.; Pinteaux, E.; et al. Characterization of a conditional interleukin-1 receptor 1 mouse mutant using the Cre/LoxP system. Eur. J. Immunol. 2016, 46, 912–918. [Google Scholar] [CrossRef]
- Van Hauwermeiren, F.; Armaka, M.; Karagianni, N.; Kranidioti, K.; Vandenbroucke, R.E.; Loges, S.; Van Roy, M.; Staelens, J.; Puimège, L.; Palagani, A.; et al. Safe TNF-based antitumor therapy following p55TNFR reduction in intestinal epithelium. J. Clin. Investig. 2013, 123, 2590–2603. [Google Scholar] [CrossRef]
- Li, S.; Lu, R.; Shu, L.; Chen, Y.; Zhao, J.; Dai, J.; Huang, Q.; Li, X.; Meng, W.; Long, F.; et al. An integrated map of fibroblastic populations in human colon mucosa and cancer tissues. Commun. Biol. 2022, 5, 1326. [Google Scholar] [CrossRef]
- Verginadis, I.I.; Avgousti, H.; Monslow, J.; Skoufos, G.; Chinga, F.; Kim, K.; Leli, N.M.; Karagounis, I.V.; Bell, B.I.; Velalopoulou, A.; et al. A stromal Integrated Stress Response activates perivascular cancer-associated fibroblasts to drive angiogenesis and tumour progression. Nat. Cell Biol. 2022, 24, 940–953. [Google Scholar] [CrossRef]
- Berger, M.; Bergers, G.; Arnold, B.; Hämerling, G.J.; Ganss, R. Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization. Blood 2005, 105, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.B.; Storm, E.E.; Kapoor, V.N.; Chavarria-Smith, J.; Lin, D.L.; Wang, L.; Li, Y.; Kljavin, N.; Ota, N.; Bainbridge, T.W.; et al. IL-1R1-dependent signaling coordinates epithelial regeneration in response to intestinal damage. Sci. Immunol. 2021, 6, eabe8856. [Google Scholar] [CrossRef] [PubMed]
- Armaka, M.; Apostolaki, M.; Jacques, P.; Kontoyiannis, D.L.; Elewaut, D.; Kollias, G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 2008, 205, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Moulos, P.; Hatzis, P. Systematic integration of RNA-Seq statistical algorithms for accurate detection of differential gene expression patterns. Nucleic Acids Res. 2015, 43, e25. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; Da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Core Team, R. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Kollias, G. Isolation of intestinal mesenchymal cells from adult mice. Bio-Protocol 2016, 6, e1940. [Google Scholar] [CrossRef]
- Xue, X.; Shah, Y.M. In vitro organoid culture of primary mouse colon tumors. J. Vis. Exp. 2013, 75, e50210. [Google Scholar]
- Shaker, A.; Swietlicki, E.A.; Wang, L.; Jiang, S.; Onal, B.; Bala, S.; DeSchryver, K.; Newberry, R.; Levin, M.S.; Rubin, D.C. Epimorphin deletion protects mice from inflammation-induced colon carcinogenesis and alters stem cell niche myofibroblast secretion. J. Clin. Investig. 2010, 120, 2081–2093. [Google Scholar] [CrossRef]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Conjugate | Clone/Cat. Number | Company | Use | |
|---|---|---|---|---|---|
| CD45 | A700 | 30-F11 | BioLegend, San Diego, CA, USA. | FC | Lineage-Antibodies |
| CD326 (EpCAM) | APC-efluor780 | G8.8 | eBioscience, Waltham, MA, USA. | FC | |
| Ter119 | APC-efluor780 | TER-119 | eBioscience, Waltham, MA, USA. | FC | |
| CD31 | APC/Fire 750 | 390 | BioLegend, San Diego, CA, USA. | FC | |
| CD201 | PE/Cy5 | eBio1560 | Invitrogen, Waltham, MA, USA | FC | |
| α-SMA | unconjugated | 1A4 | Sigma, St. Louis, MO, USA | IHC | |
| Anti-mouse-IgG | A647 | A-21235 | Invitrogen, Waltham, MA, USA | IHC | Secondary |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chalkidi, N.; Melissari, M.-T.; Henriques, A.; Stavropoulou, A.; Kollias, G.; Koliaraki, V. Activation and Functions of Col6a1+ Fibroblasts in Colitis-Associated Cancer. Int. J. Mol. Sci. 2024, 25, 148. https://doi.org/10.3390/ijms25010148
Chalkidi N, Melissari M-T, Henriques A, Stavropoulou A, Kollias G, Koliaraki V. Activation and Functions of Col6a1+ Fibroblasts in Colitis-Associated Cancer. International Journal of Molecular Sciences. 2024; 25(1):148. https://doi.org/10.3390/ijms25010148
Chicago/Turabian StyleChalkidi, Niki, Maria-Theodora Melissari, Ana Henriques, Athanasia Stavropoulou, George Kollias, and Vasiliki Koliaraki. 2024. "Activation and Functions of Col6a1+ Fibroblasts in Colitis-Associated Cancer" International Journal of Molecular Sciences 25, no. 1: 148. https://doi.org/10.3390/ijms25010148