
[3+2]-Cycloaddition of Nitrile Imines to Parabanic Acid Derivatives—An Approach to Novel Spiroimidazolidinediones

 , ,
, , 
Abstract
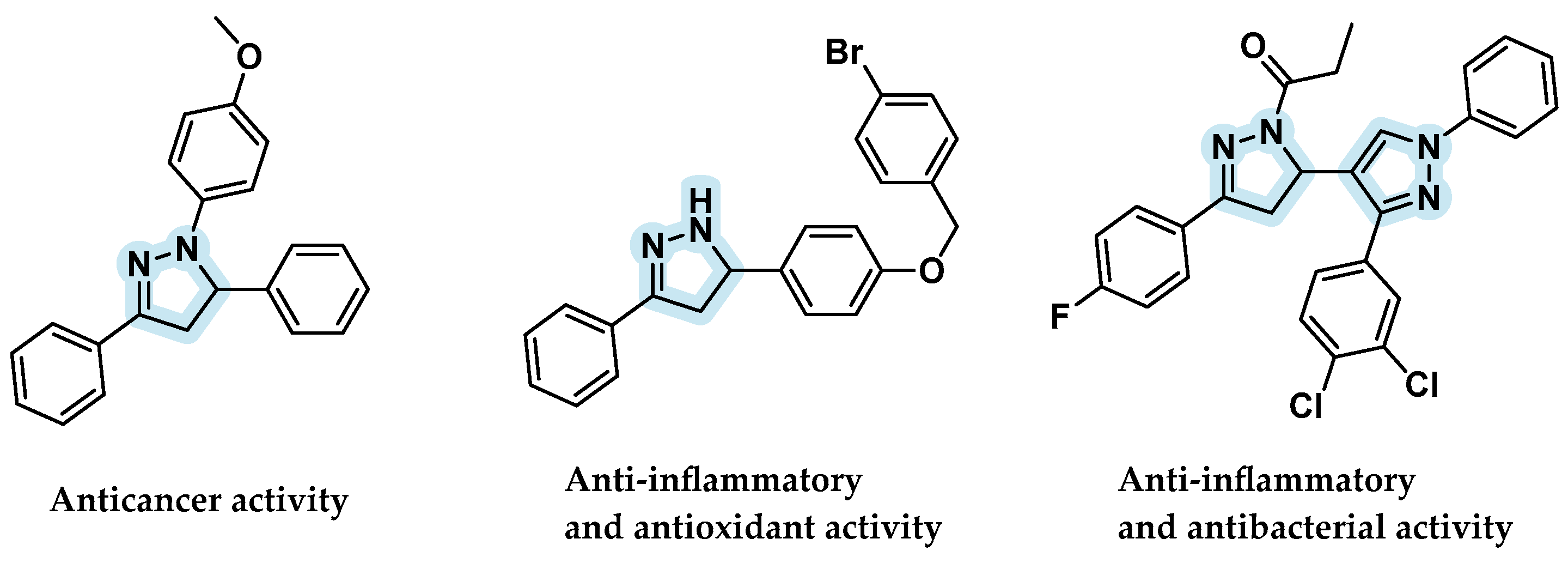
:1. Introduction
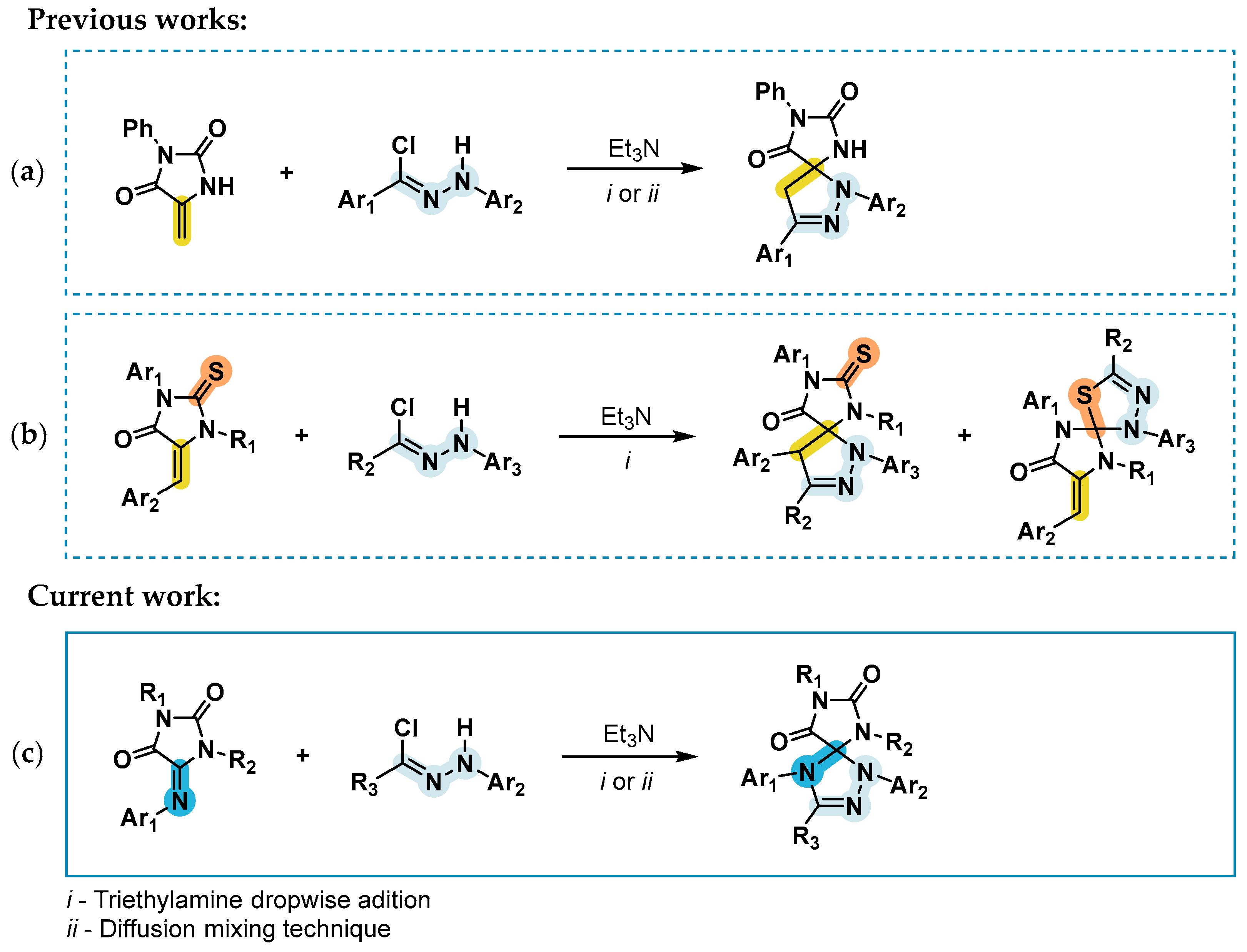
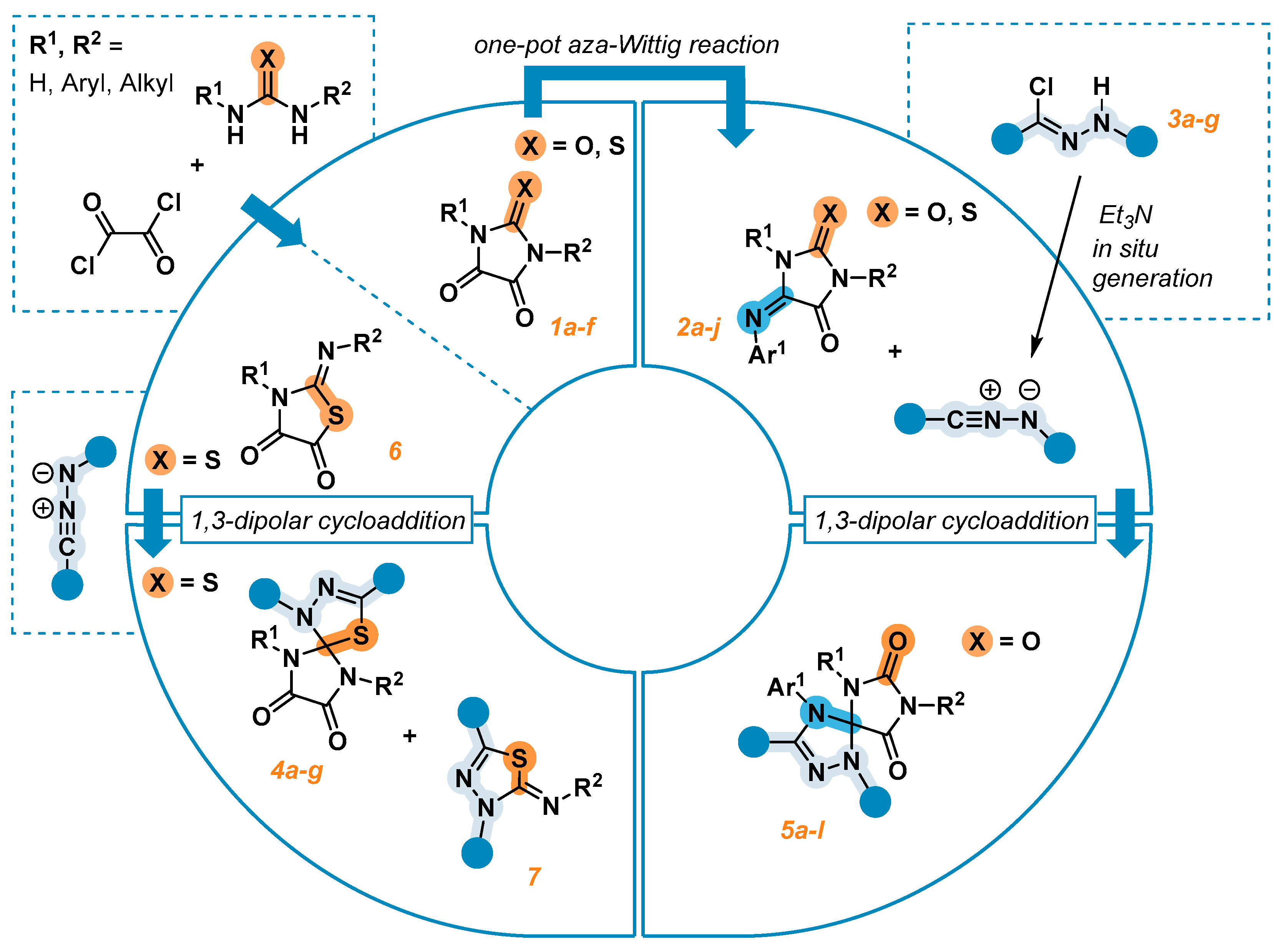
2. Results and Discussion
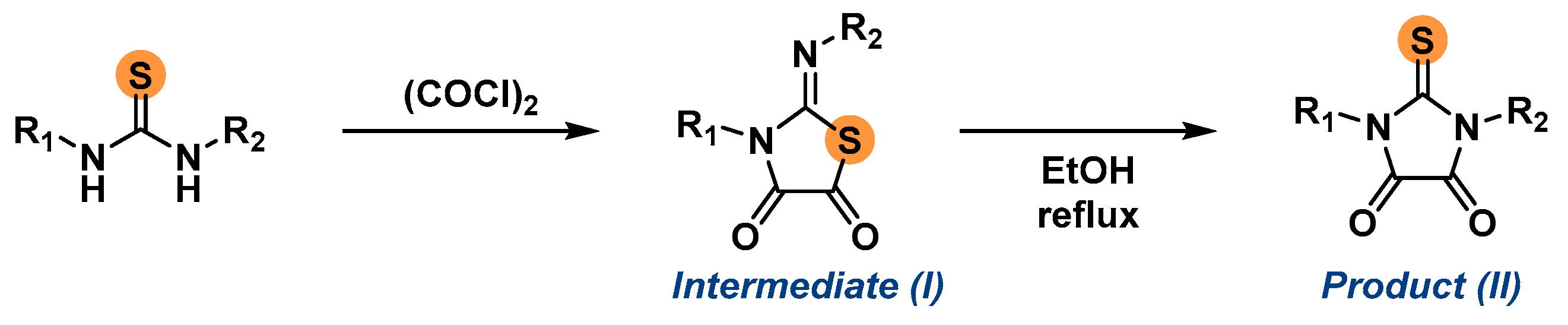
2.1. Synthesis of the Imidazolidine-4,5-Diones 1c–f
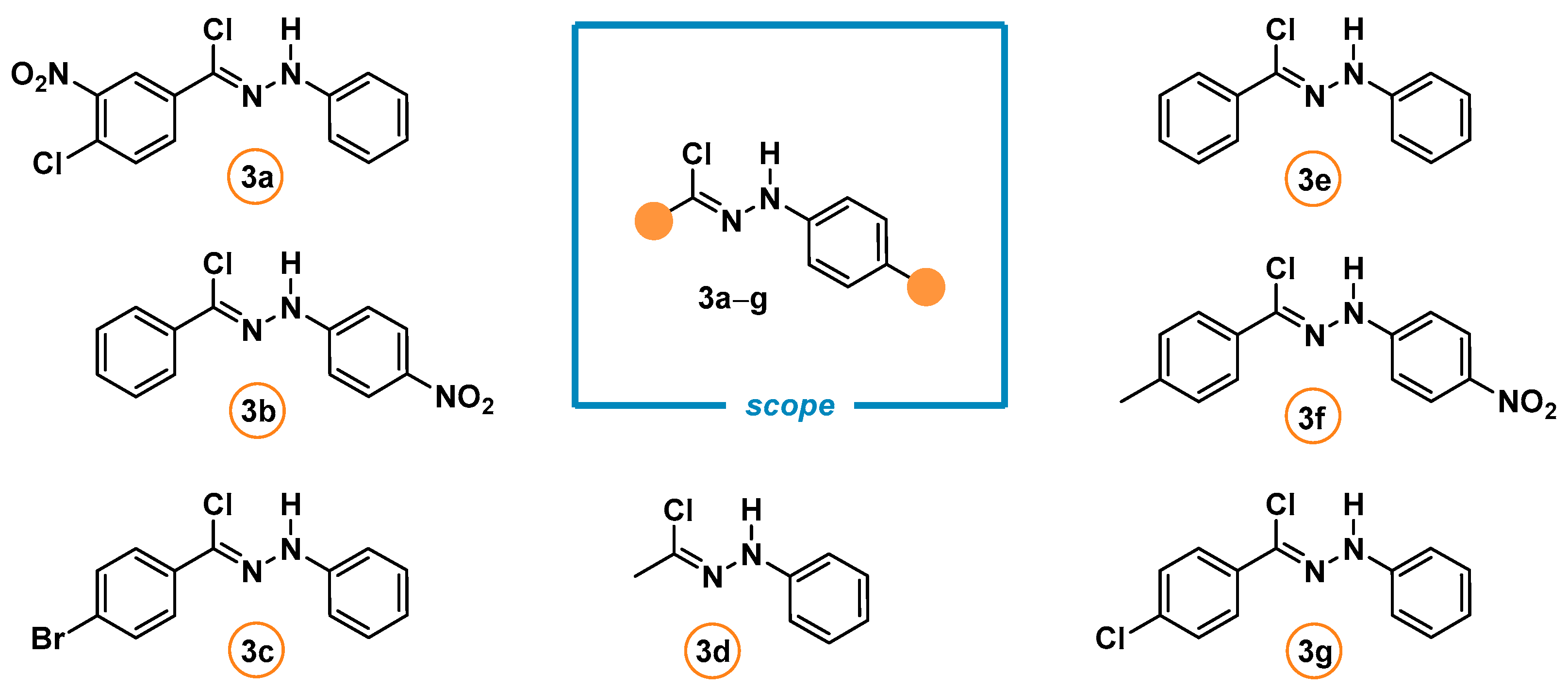
2.2. Synthesis of the 5-Aryliminoimidazolidine-2,4-Diones 2a–l
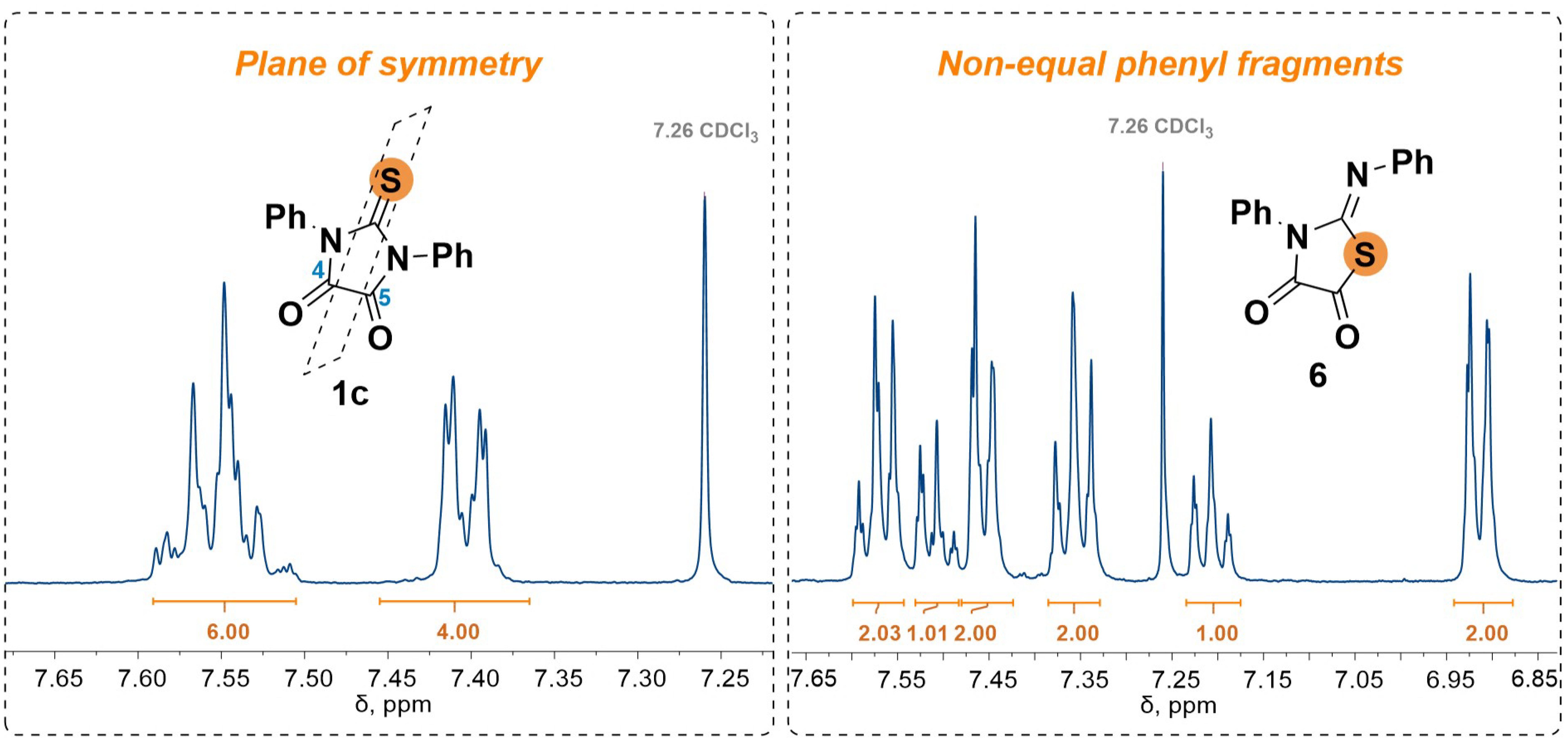
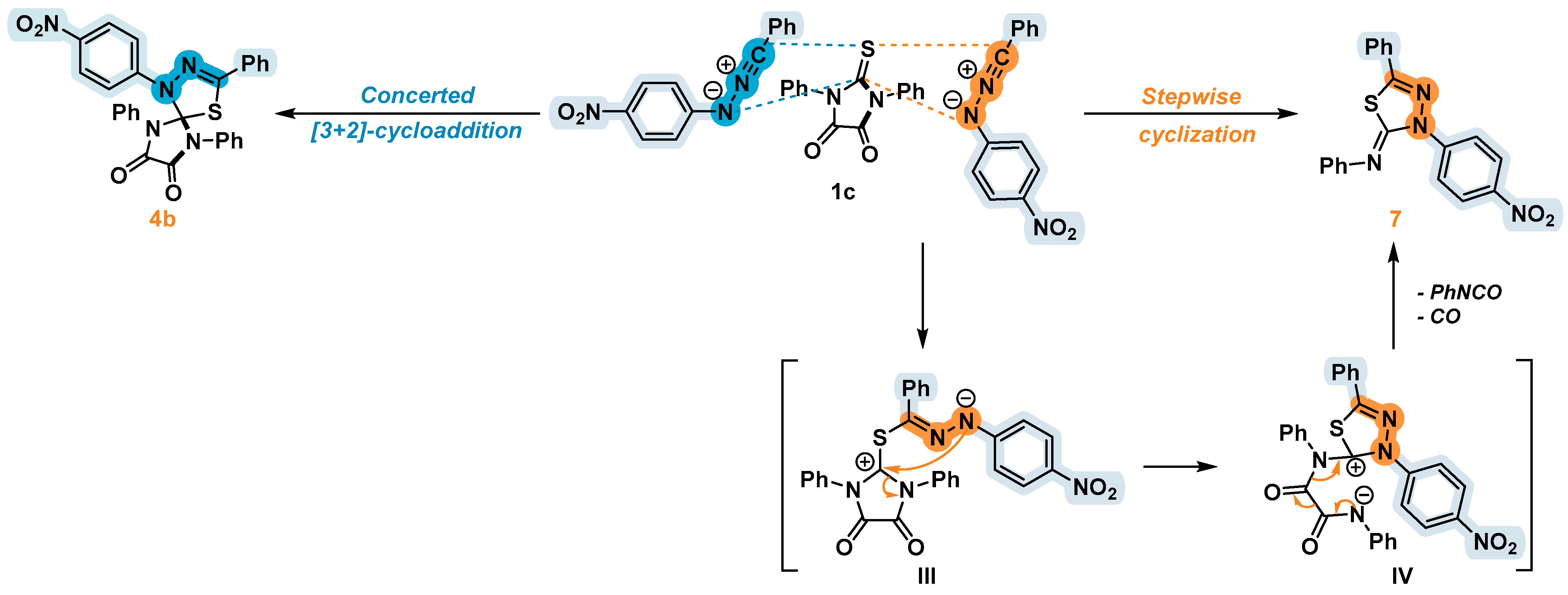
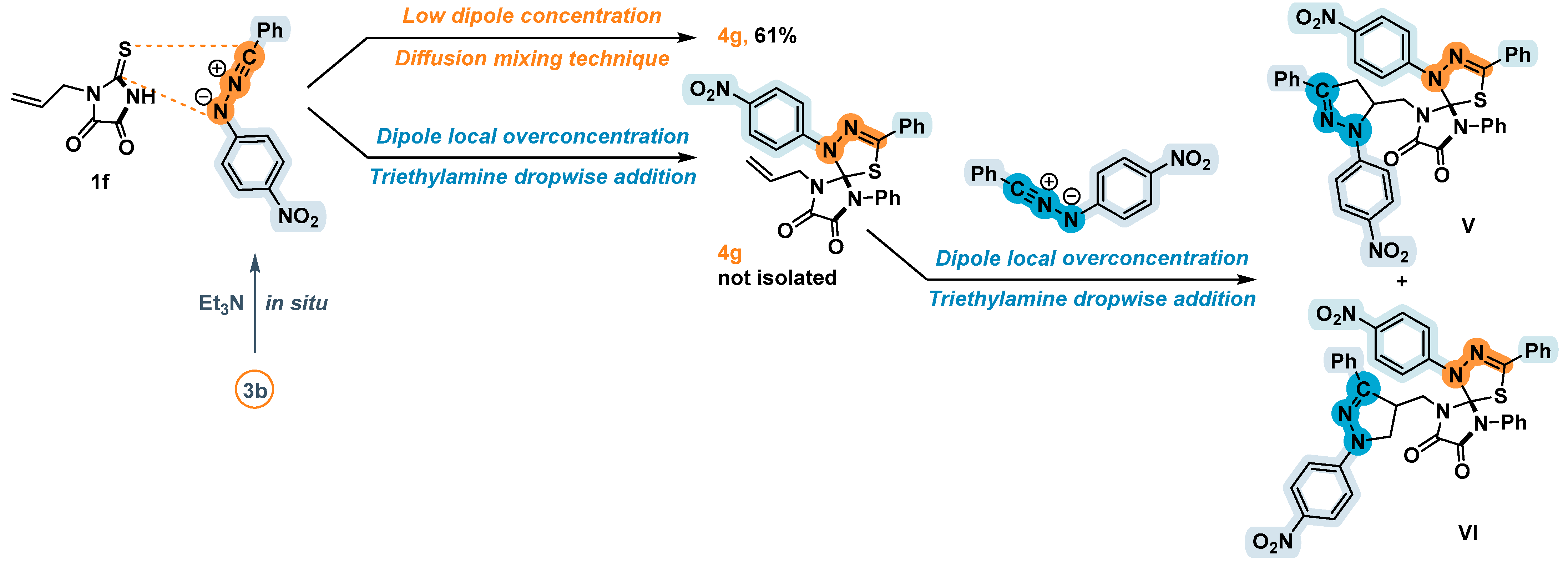
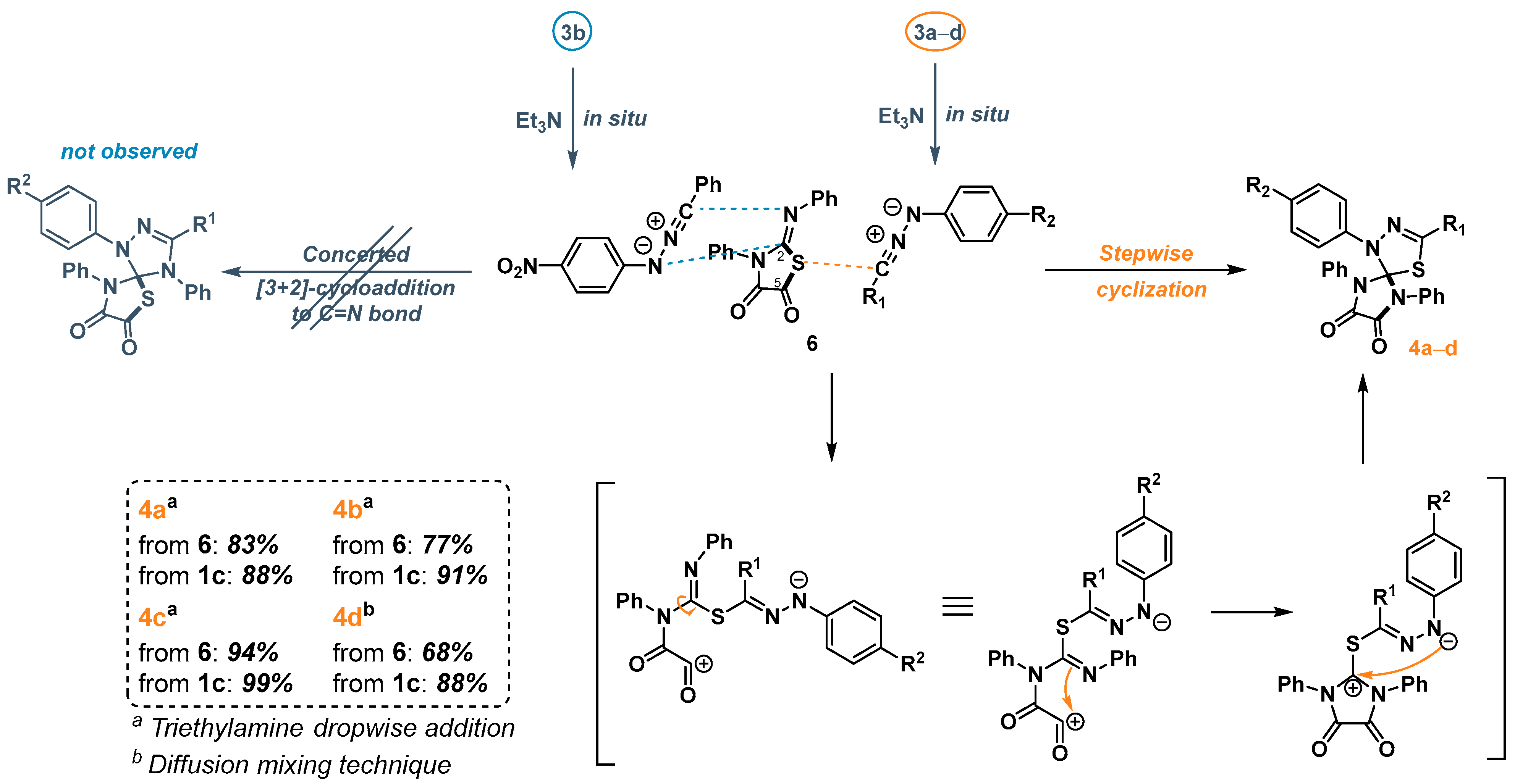
2.3. 1,3-Dipolar Cycloaddition of Nitrile Imines to 2-Thioxoimidazolidine-4,5-Diones 1c–f and 3-Phenyl-2-(Phenylimino)Thiazolidine-4,5-Dione 6
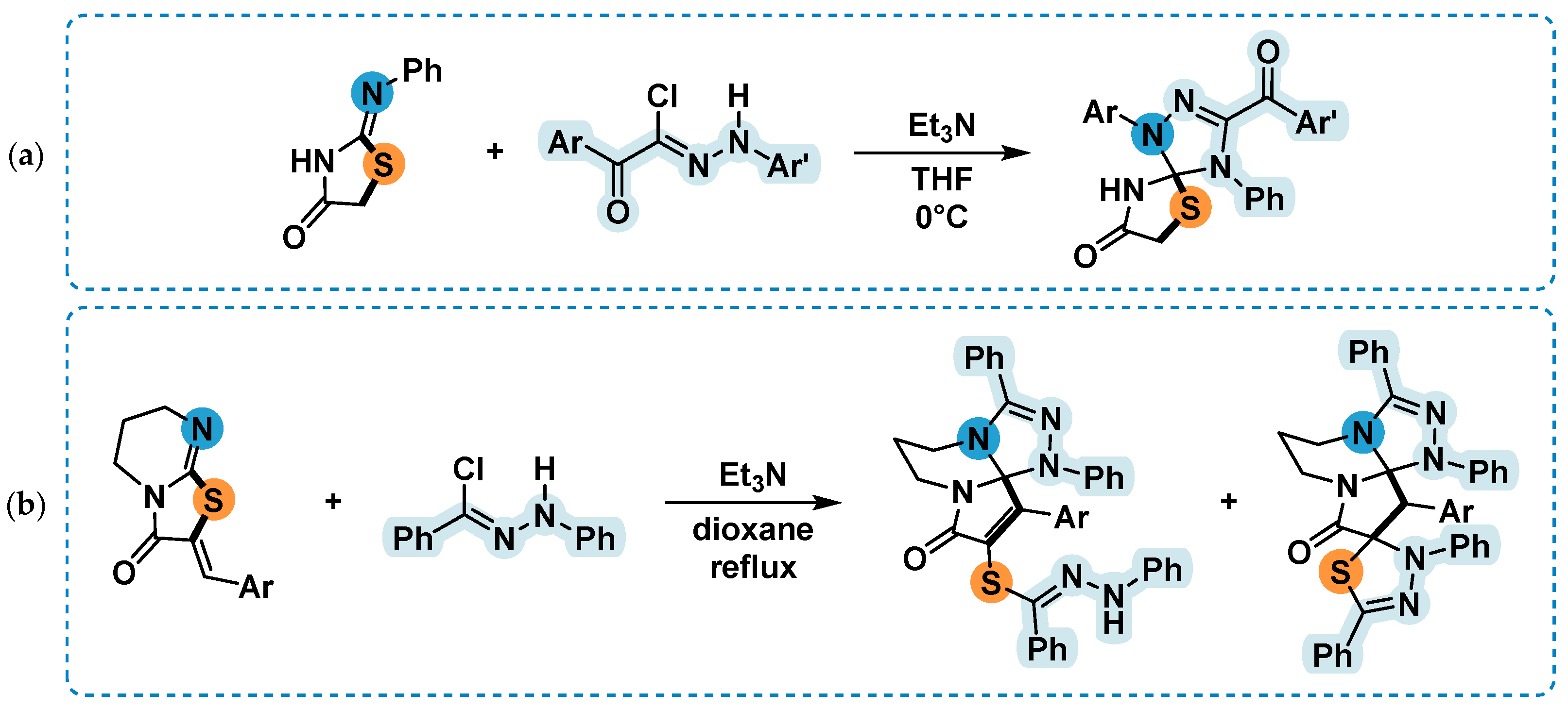
2.4. 1,3-Dipolar Cycloaddition of Nitrile Imines to 5-Arylimino-1,3-Diphenylimidazolidine-2,4,5-Triones
- The presence of substituents with strong mesomeric effects in the aromatic ring of imine 2 decreased the yield of the spiro compound, regardless of whether the substituent was a donor or acceptor. For example, the yield decreased in the sequence 5c (R1 = 4-Br) > 5g (R1 = 4-OMe) > 5b (R1 = 4-NO2) (Table 5).
- The introduction of a donor substituent into the aromatic fragment at the terminal carbon atom of nitrile imine increased the yield of the cycloaddition product. For example, the yields of the products 5c (R1 = 4-Br, R2 = 4-Me-C6H4) and 5g (R1 = 4-OMe, R2 = 4-Me-C6H4) were higher than those of 5h (R1 = 4-Br, R2 = Ph) and 5i (R1 = 4-OMe, R2 = Ph), respectively (Table 5).
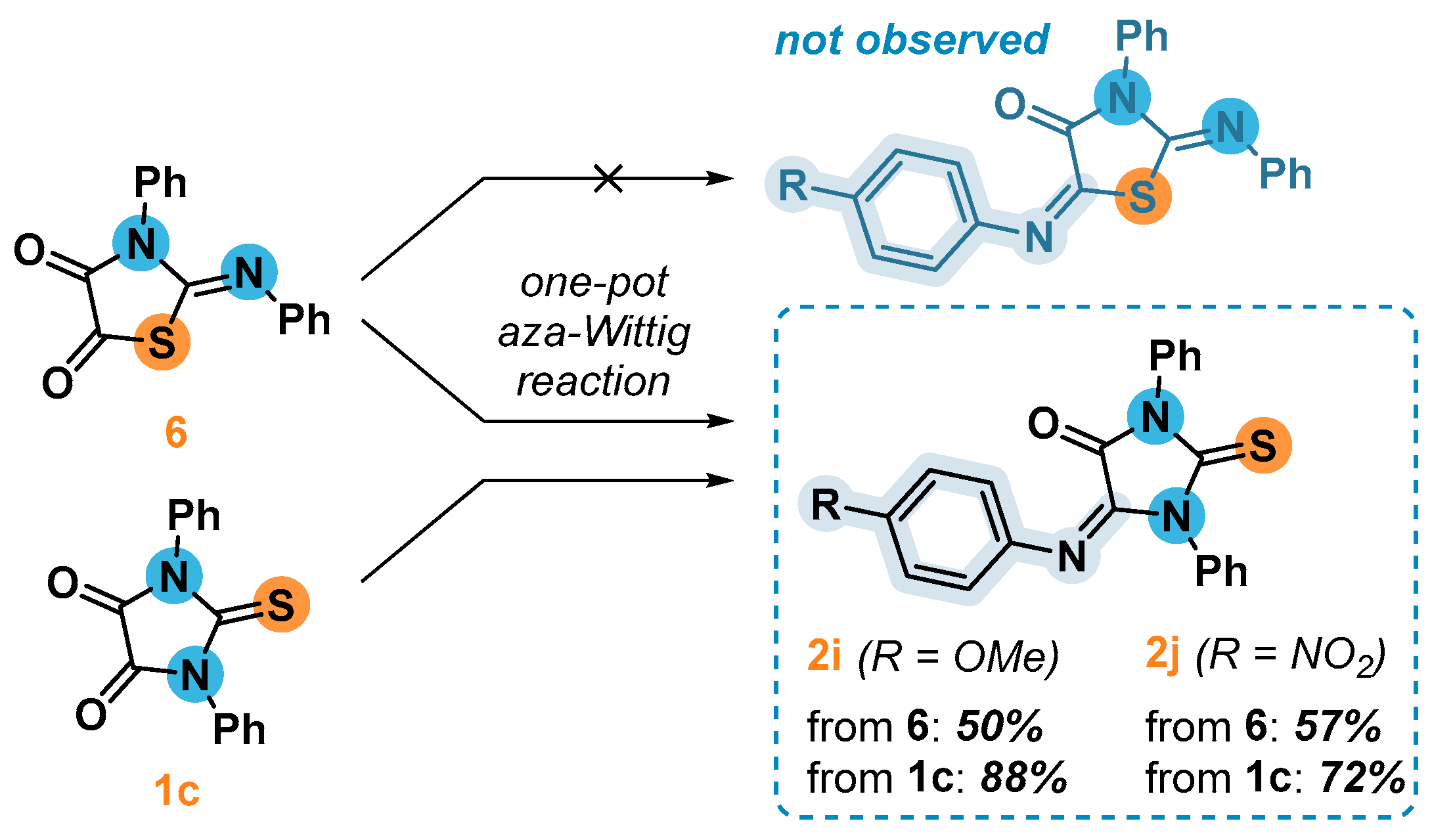
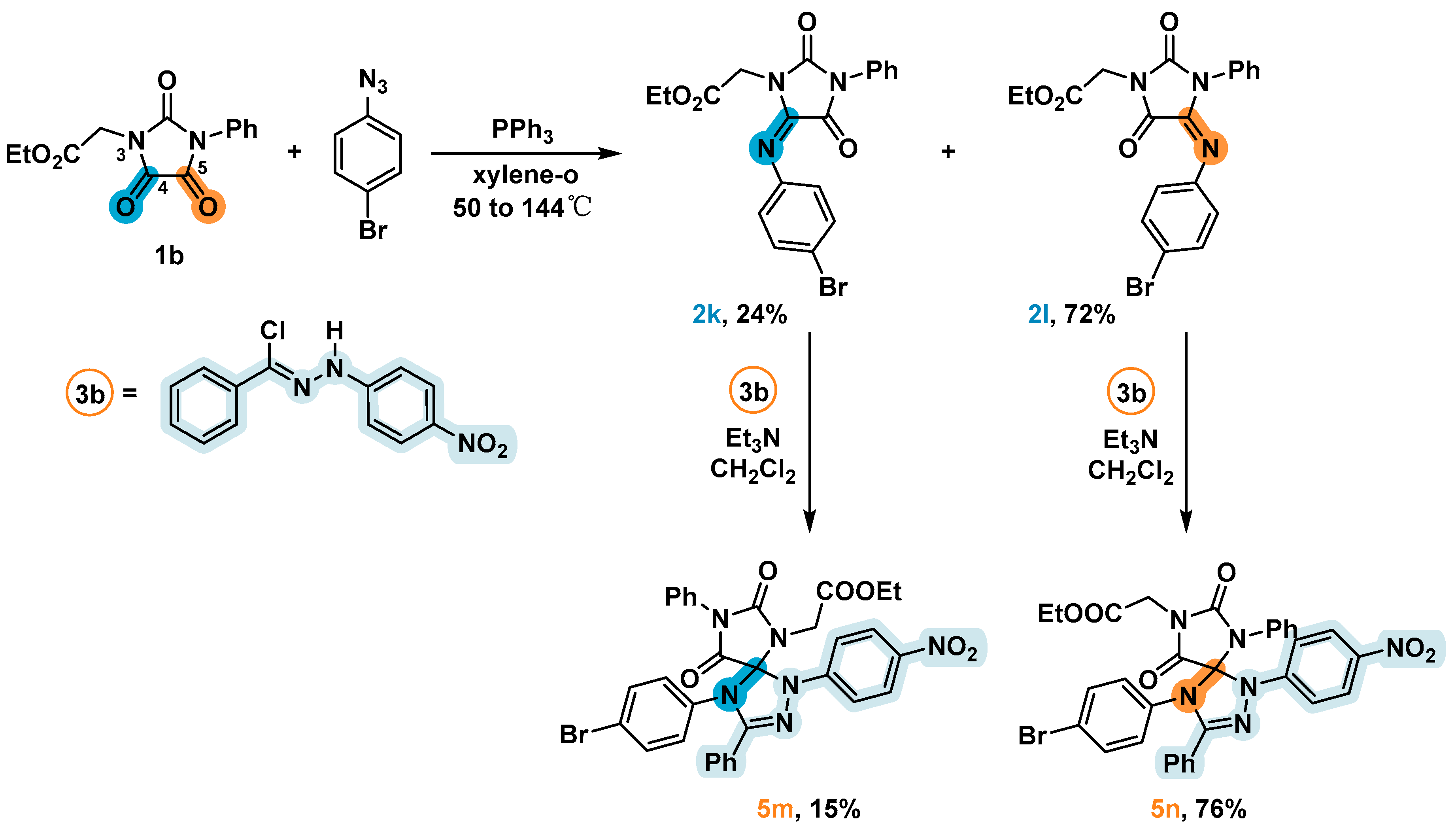
2.5. Synthesis and 1,3-Dipolar Cycloaddition Reactions of the Imines 2k и 2l Obtained from Unsimmetrical Ureas
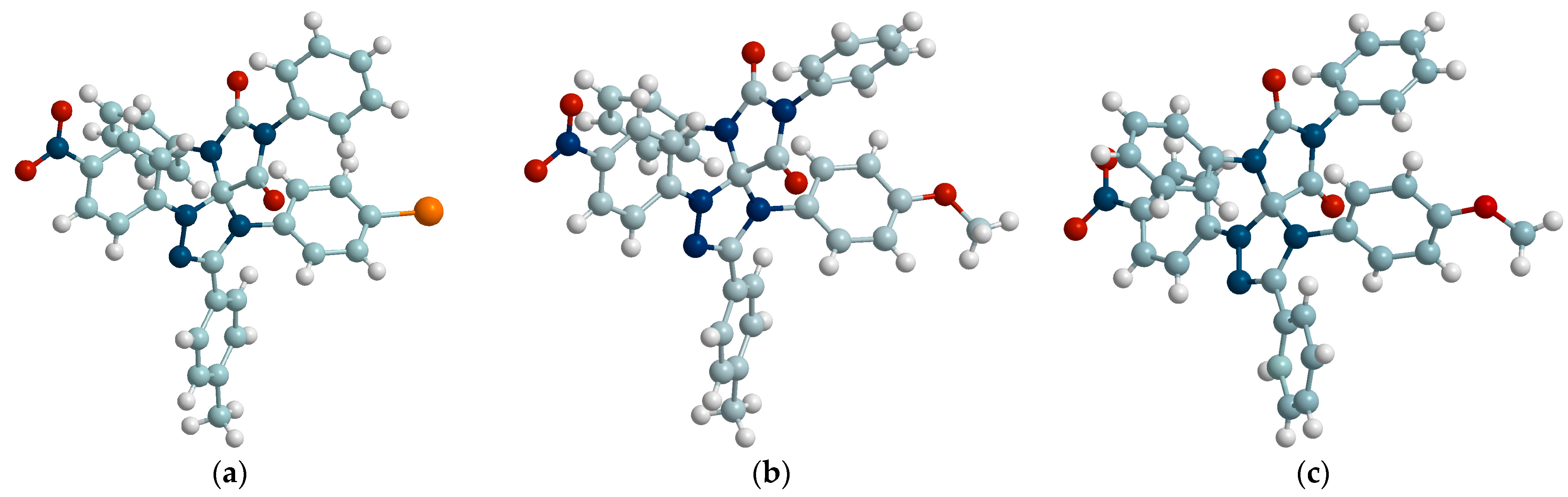
2.6. Biological Evaluation of Selected Spiro-Compounds
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of the Imidazolidine-3,4,5-Triones 1a–b and 3-phenyl-2-(Phenylimino) Thiazolidine-4,5-Dione 6
3.3. General Procedure for the Synthesis of the 2-Thioxoimidazolidine-4,5-Diones 1c–f
3.4. General Procedure for the Synthesis of the 5-Aryliminoimidazolidine-2,4-Diones 2a–l
3.5. General Procedure for the Synthesis of the Spirocyclic Products 4a–g and 5a–n
3.6. Reagents for MTT Test
3.7. Cell Lines and Cytotoxicity Evaluation
3.8. Cytotoxicity
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jamieson, C.; Livingstone, K. The Nitrile Imine 1,3-Dipole; Springer International Publishing: Cham, Switzerland, 2020; ISBN 978-3-030-43480-9. [Google Scholar]
- Wang, Y.; Yan, L.; Yan, Y.; Li, S.; Lu, H.; Liu, J.; Dong, J. Dipolarophile-Controlled Regioselective 1,3-Dipolar Cycloaddition: A Switchable Divergent Access to Functionalized N-Fused Pyrrolidinyl Spirooxindoles. Int. J. Mol. Sci. 2023, 24, 3771. [Google Scholar] [CrossRef] [PubMed]
- Mantzanidou, M.; Pontiki, E.; Hadjipavlou-Litina, D. Pyrazoles and Pyrazolines as Anti-Inflammatory Agents. Molecules 2021, 26, 3439. [Google Scholar] [CrossRef] [PubMed]
- Chalkha, M.; Akhazzane, M.; Moussaid, F.Z.; Daoui, O.; Nakkabi, A.; Bakhouch, M.; Chtita, S.; Elkhattabi, S.; Housseini, A.I.; El Yazidi, M. Design, Synthesis, Characterization, in Vitro Screening, Molecular Docking, 3D-QSAR, and ADME-Tox Investigations of Novel Pyrazole Derivatives as Antimicrobial Agents. New J. Chem. 2022, 46, 2747–2760. [Google Scholar] [CrossRef]
- Viveka, S.; Dinesha; Shama, P.; Nagaraja, G.K.; Ballav, S.; Kerkar, S. Design and Synthesis of Some New Pyrazolyl-Pyrazolines as Potential Anti-Inflammatory, Analgesic and Antibacterial Agents. Eur. J. Med. Chem. 2015, 101, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pan, Y.; Huang, X.; Mao, Z.; Wang, H. A Novel Methodology for Synthesis of Dihydropyrazole Derivatives as Potential Anticancer Agents. Org. Biomol. Chem. 2014, 12, 2028–2032. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Ali, A.; Ali, A.; Thiriveedhi, A.; Bakht, M.A.; Yusuf, M.; Salahuddin; Afzal, O.; Altamimi, A.S.A. Pyrazoline Containing Compounds as Therapeutic Targets for Neurodegenerative Disorders. ACS Omega 2022, 7, 38207–38245. [Google Scholar] [CrossRef] [PubMed]
- Shawali, A.S. Chemoselectivity in 1,3-Dipolar Cycloaddition Reactions of Nitrilimines with Multifunctionalized Dipolarophiles. Curr. Org. Chem. 2014, 18, 598–614. [Google Scholar] [CrossRef]
- Cho, S.; Kim, S.-H.; Shin, D. Recent Applications of Hydantoin and Thiohydantoin in Medicinal Chemistry. Eur. J. Med. Chem. 2019, 164, 517–545. [Google Scholar] [CrossRef]
- Yavari, I.; Taheri, Z.; Sheikhi, S.; Bahemmat, S.; Halvagar, M.R. Synthesis of Thia- and Thioxo-Tetraazaspiro[4.4]Nonenones from Nitrile Imines and Arylidenethiohydantoins. Mol. Divers. 2021, 25, 777–785. [Google Scholar] [CrossRef]
- Filkina, M.E.; Baray, D.N.; Beloglazkina, E.K.; Grishin, Y.K.; Roznyatovsky, V.A.; Kukushkin, M.E. Regioselective Cycloaddition of Nitrile Imines to 5-Methylidene-3-Phenyl-Hydantoin: Synthesis and DFT Calculations. Int. J. Mol. Sci. 2023, 24, 1289. [Google Scholar] [CrossRef]
- Ivanenkov, Y.A.; Kukushkin, M.E.; Beloglazkina, A.A.; Shafikov, R.R.; Barashkin, A.A.; Ayginin, A.A.; Serebryakova, M.S.; Majouga, A.G.; Skvortsov, D.A.; Tafeenko, V.A.; et al. Synthesis and Biological Evaluation of Novel Dispiro-Indolinones with Anticancer Activity. Molecules 2023, 28, 1325. [Google Scholar] [CrossRef] [PubMed]
- Hassaneen, H.M.; Daboun, H.A.; Abdelhadi, H.A.; Abdel-Reheim, N.A. Site Selectivity and Regiochemistry of Nitrilimines. Cycloadditions to 1,3-Diphenyl-2-Thiono-4-Imidazolidinone and Its 5-Phenylmethylene Derivatives. Phosphorus. Sulfur. Silicon Relat. Elem. 1995, 107, 269–273. [Google Scholar] [CrossRef]
- Shybanov, D.E.; Filkina, M.E.; Kukushkin, M.E.; Grishin, Y.K.; Roznyatovsky, V.A.; Zyk, N.V.; Beloglazkina, E.K. Diffusion Mixing with a Volatile Tertiary Amine as a Very Efficient Technique for 1,3-Dipolar Cycloaddition Reactions Proceeding via Dehydrohalogenation of Stable Precursors of Reactive Dipoles. New J. Chem. 2022, 46, 18575–18586. [Google Scholar] [CrossRef]
- Ribeiro, C.J.A.; Nunes, R.C.; Amaral, J.D.; Gonçalves, L.M.; Rodrigues, C.M.P.; Moreira, R.; Santos, M.M.M. Spirotriazoline Oxindoles: A Novel Chemical Scaffold with in Vitro Anticancer Properties. Eur. J. Med. Chem. 2017, 140, 494–509. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.M.; Dawood, K.M.; Khedr, N.A. Regioselective Synthesis of Novel 4,4′-and 5,5′-Bi-(1,2,4-Triazole) Derivatives. J. Chem. Res. 2007, 2007, 472–474. [Google Scholar] [CrossRef]
- Üngören, Ş.H.; Kani, İ.; Günay, A. A Facile Protocol for the Preparation of 5-Alkylidene and 5-Imino Substituted Hydantoins from N,N′-Disubstituted Parabanic Acids. Tetrahedron Lett. 2012, 53, 4758–4762. [Google Scholar] [CrossRef]
- Biltz, H.; Topp, E. Synthesis of Parabanic and Substituted Parabanic Acids. Berichte Dtsch. Chem. Ges. 1913, 46, 1387–1404. [Google Scholar] [CrossRef]
- Stoffel, P.J. The Preparation of Parabanic Acids from 1,1,3-Trisubstituted Ureas via a Hofmann Elimination Reaction. J. Org. Chem. 1964, 29, 2794–2796. [Google Scholar] [CrossRef]
- Ulrich, H.; Sayigh, A.A.R. The Reaction of Oxalyl Chloride with Substituted Ureas and Thioureas. J. Org. Chem. 1965, 30, 2781–2783. [Google Scholar] [CrossRef]
- Watanabe, N.; Hamano, M.; Todaka, S.; Asaeda, T.; Ijuin, H.K.; Matsumoto, M. Diphenylparabanic Acid as a Synthon for the Synthesis of α-Diketones and α-Ketocarboxylic Acids. J. Org. Chem. 2012, 77, 632–639. [Google Scholar] [CrossRef]
- Üngören, Ş.H.; Albayrak, S.; Günay, A.; Yurtseven, L.; Yurttaş, N. A New Method for the Preparation of 5-Acylidene and 5-Imino Substituted Rhodanine Derivatives and Their Antioxidant and Antimicrobial Activities. Tetrahedron 2015, 71, 4312–4323. [Google Scholar] [CrossRef]
- Yavari, I.; Taheri, Z.; Naeimabadi, M.; Bahemmat, S.; Halvagar, M.R. A Convenient Synthesis of Tetrasubstituted Pyrazoles from Nitrile Imines and 2-(Thioxothiazolidin-5-Ylidene)Acetates. Synlett 2018, 29, 918–921. [Google Scholar] [CrossRef]
- Ghasempour, L.; Asghari, S.; Tajbakhsh, M.; Mohseni, M. Preparation of New Spiropyrazole, Pyrazole and Hydantoin Derivatives and Investigation of Their Antioxidant and Antibacterial Activities. Chem. Biodivers. 2021, 18, e2100197. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Q.; Zhao, Q.; Yu, W.; Chang, J. Synthesis of 2-Imino-1,3,4-Thiadiazoles from Hydrazides and Isothiocyanates via Sequential Oxidation and P(NMe2)3-Mediated Annulation Reactions. Org. Lett. 2020, 22, 4378–4382. [Google Scholar] [CrossRef] [PubMed]
- Dalloul, H.M. Synthesis of Spiro-Heterocycles with the Thiazolidinone Moiety from Nitrilimines. J. Chin. Chem. Soc. 2009, 56, 196–201. [Google Scholar] [CrossRef]
- Liu, B.; Li, X.F.; Liu, H.C.; Yu, X.Y. Unexpected Nitrilimine Cycloaddition of Thiazolo[3,2-a]Pyrimidine Derivatives. Tetrahedron Lett. 2013, 54, 6952–6954. [Google Scholar] [CrossRef]
- Houk, K.N.; Sims, J.; Watts, C.R.; Luskus, L.J. Origin of Reactivity, Regioselectivity, and Periselectivity in 1,3-Dipolar Cycloadditions. J. Am. Chem. Soc. 1973, 95, 7301–7315. [Google Scholar] [CrossRef]
- Bojilova, A.; Rodios, N.A.; Tsoleridis, C.A.; Alexandrou, N.E. The Reaction of 1-(N-phenacylidene)Amino-1,2,3-triazoles with Diphenylnitrilimine. J. Heterocycl. Chem. 1990, 27, 735–738. [Google Scholar] [CrossRef]
- Shybanov, D.E.; Kukushkin, M.E.; Hrytseniuk, Y.S.; Grishin, Y.K.; Roznyatovsky, V.A.; Tafeenko, V.A.; Skvortsov, D.A.; Zyk, N.V.; Beloglazkina, E.K. [4+2]-Cycloaddition to 5-Methylidene-Hydantoins and 5-Methylidene-2-Thiohydantoins in the Synthesis of Spiro-2-Chalcogenimidazolones. Int. J. Mol. Sci. 2023, 24, 5037. [Google Scholar] [CrossRef]
- Tietze, L.F.; Eicher, T. Reaktionen und Synthesen Im Organisch-Chemischen Praktikum und Forschungslaboratorium; Thieme Verlag: Stuttgart, Germany, 1991; ISBN 3527308741. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound № | X | R1 | R2 | Yield a, % |
| 1a b | O | Ph | Ph | 99 |
| 1b b | O | Ph | CH2COOEt | 99 |
| 1c c | S | Ph | Ph | 98 |
| 1d c | S | 4-EtO-C6H4 | CH2COOEt | 93 |
| 1e c | S | 4-MeO-C6H4 | CH2COOEt | 77 |
| 1f c | S | All | H | 47 |
 | |||
|---|---|---|---|
| Compound № | X | R3 | Yield a, % |
| 2a | O | 4-Cl | 79 |
| 2b | O | 2-Cl | 60 |
| 2c | O | 4-Br | 60 |
| 2d | O | 2-Br | 76 |
| 2e | O | 4-Me | 84 |
| 2f | O | 4-OMe | 65 |
| 2g | O | 4-NO2 | 75 |
| 2h | S | 4-Br | 82 |
| 2i | S | 4-OMe | 88 |
| 2j | S | 4-NO2 | 72 |
 | ||||||
|---|---|---|---|---|---|---|
| Compound № | R1 | R2 | R3 | R4 | Diffusion Mixing | Dropwise Addition |
| Yield a, % | Yield a, % | |||||
| 4a | Ph | Ph | 3-NO2-4-Cl-C6H3 | H | 96 | 88 |
| 4b | Ph | Ph | Ph | 4-NO2 | 44 | 91 |
| 4c | Ph | Ph | 4-Br-C6H4 | H | 99 | 99 |
| 4d | Ph | Ph | Me | H | 88 | 74 |
| 4e | 4-EtO-C6H4 | CH2COOEt | 4-Br-C6H4 | H | 77 | 49 |
| 4f | 4-MeO-C6H4 | CH2COOEt | Ph | 4-NO2 | 93 | 94 |
| 4g | All | H | Ph | 4-NO2 | 61 | - b |
| Entry | Solvent | T | Yield a of 4b, % | Yield a of 7, % |
|---|---|---|---|---|
| 1 | CH3OH | rt | 60 | - |
| 2 | CH3CN | rt | 75 | <1 b |
| 3 | CHCl3 | −17 °C | 49 | 32 |
| 4 | CHCl3 | rt | 44 | 35 |
| 5 | (CH3)2CO | rt | 34 | 45 |
 | |||||
|---|---|---|---|---|---|
| Compound № | R1 | R2 | R3 | Diffusion Mixing | Dropwise Addition |
| Yield a, % | Yield a, % | ||||
| 5a | 4-Br | Ph | H | 71 | 62 |
| 5b | 4-NO2 | 4-Me-C6H4 | NO2 | 40 | 13 |
| 5c | 4-Br | 4-Me-C6H4 | NO2 | 90 | 85 |
| 5d | 2-Br | 4-Me-C6H4 | NO2 | 84 | 83 |
| 5e | 2-Cl | 4-Me-C6H4 | NO2 | 87 | 81 |
| 5f | 4-Me | 4-Me-C6H4 | NO2 | 71 | 88 |
| 5g | 4-OMe | 4-Me-C6H4 | NO2 | 52 | 38 |
| 5h | 4-Br | Ph | NO2 | 62 | 50 |
| 5i | 4-OMe | Ph | NO2 | 60 | 50 |
| 5j | 4-Me | 4-Cl-C6H4 | H | 57 | 93 |
| 5k | 4-NO2 | Me | H | 88 | 59 |
| 5l | 4-Br | Me | H | 73 | 57 |
| Compound № | IC50, µM | R1 | R2 | R3 | R4 | Structure Type |
|---|---|---|---|---|---|---|
| 4a | 24.75 ± 0.01 | Ph | Ph | 3-NO2-4-Cl-C6H3 | H |  |
| 4b | 35.04 ± 0.06 | Ph | Ph | Ph | 4-NO2 | |
| 4e | 48.51 ± 0.03 | 4-EtO-C6H4 | CH2COOEt | 4-Br-C6H4 | H | |
| 4f | 29.61 ± 0.08 | 4-MeO-C6H4 | CH2COOEt | Ph | 4-NO2 | |
| 5c | 40.99 ± 0.01 | 4-Br | 4-Me-C6H4 | NO2 | - |  |
| 5f | 43.41 ± 0.01 | 4-Me | 4-Me-C6H4 | NO2 | - | |
| 5j | 35.07 ± 0.01 | 4-Me | 4-Cl-C6H4 | H | - | |
| Reference | IC50, µM | Cell line | R1 | R2 | Structure type | |
| [12] | 1.2–3.4 | LNCaP | Ar/CH3CHPh | Cl/Br |  | |
| [30] | 4–30 | HEK293T | H/Me | H/Me |  | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsova, J.V.; Tkachenko, V.T.; Petrovskaya, L.M.; Filkina, M.E.; Shybanov, D.E.; Grishin, Y.K.; Roznyatovsky, V.A.; Tafeenko, V.A.; Pestretsova, A.S.; Yakovleva, V.A.; et al. [3+2]-Cycloaddition of Nitrile Imines to Parabanic Acid Derivatives—An Approach to Novel Spiroimidazolidinediones. Int. J. Mol. Sci. 2024, 25, 18. https://doi.org/10.3390/ijms25010018
Kuznetsova JV, Tkachenko VT, Petrovskaya LM, Filkina ME, Shybanov DE, Grishin YK, Roznyatovsky VA, Tafeenko VA, Pestretsova AS, Yakovleva VA, et al. [3+2]-Cycloaddition of Nitrile Imines to Parabanic Acid Derivatives—An Approach to Novel Spiroimidazolidinediones. International Journal of Molecular Sciences. 2024; 25(1):18. https://doi.org/10.3390/ijms25010018
Chicago/Turabian StyleKuznetsova, Juliana V., Varvara T. Tkachenko, Lada M. Petrovskaya, Maria E. Filkina, Dmitry E. Shybanov, Yuri K. Grishin, Vitaly A. Roznyatovsky, Viktor A. Tafeenko, Anna S. Pestretsova, Vera A. Yakovleva, and et al. 2024. "[3+2]-Cycloaddition of Nitrile Imines to Parabanic Acid Derivatives—An Approach to Novel Spiroimidazolidinediones" International Journal of Molecular Sciences 25, no. 1: 18. https://doi.org/10.3390/ijms25010018
APA StyleKuznetsova, J. V., Tkachenko, V. T., Petrovskaya, L. M., Filkina, M. E., Shybanov, D. E., Grishin, Y. K., Roznyatovsky, V. A., Tafeenko, V. A., Pestretsova, A. S., Yakovleva, V. A., Pokrovsky, V. S., Kukushkin, M. E., & Beloglazkina, E. K. (2024). [3+2]-Cycloaddition of Nitrile Imines to Parabanic Acid Derivatives—An Approach to Novel Spiroimidazolidinediones. International Journal of Molecular Sciences, 25(1), 18. https://doi.org/10.3390/ijms25010018