
Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increased Mitochondrial Fission In Vitro and Atherosclerosis in High-Fat-Diet-Fed ApoE-/- Mice
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
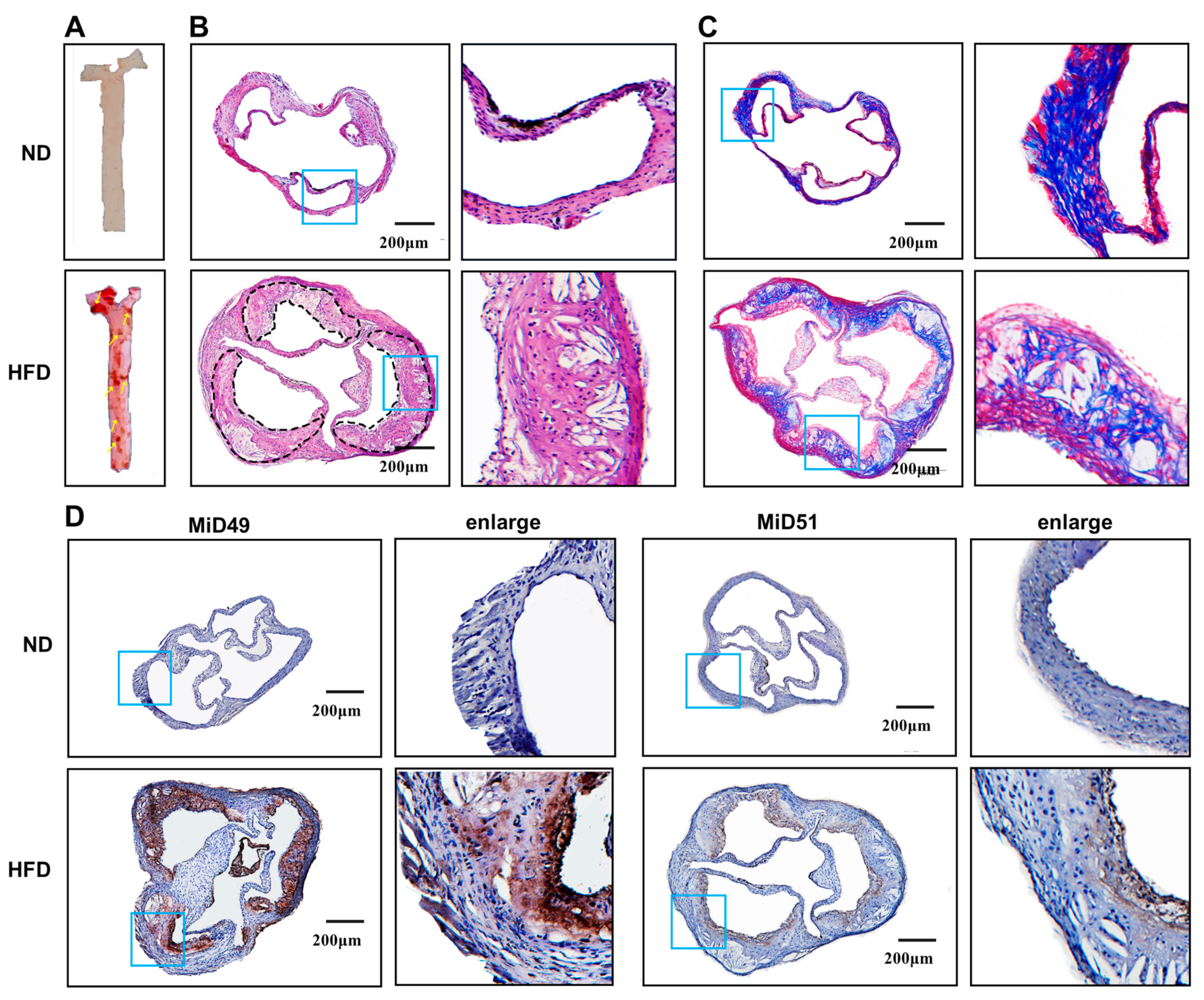
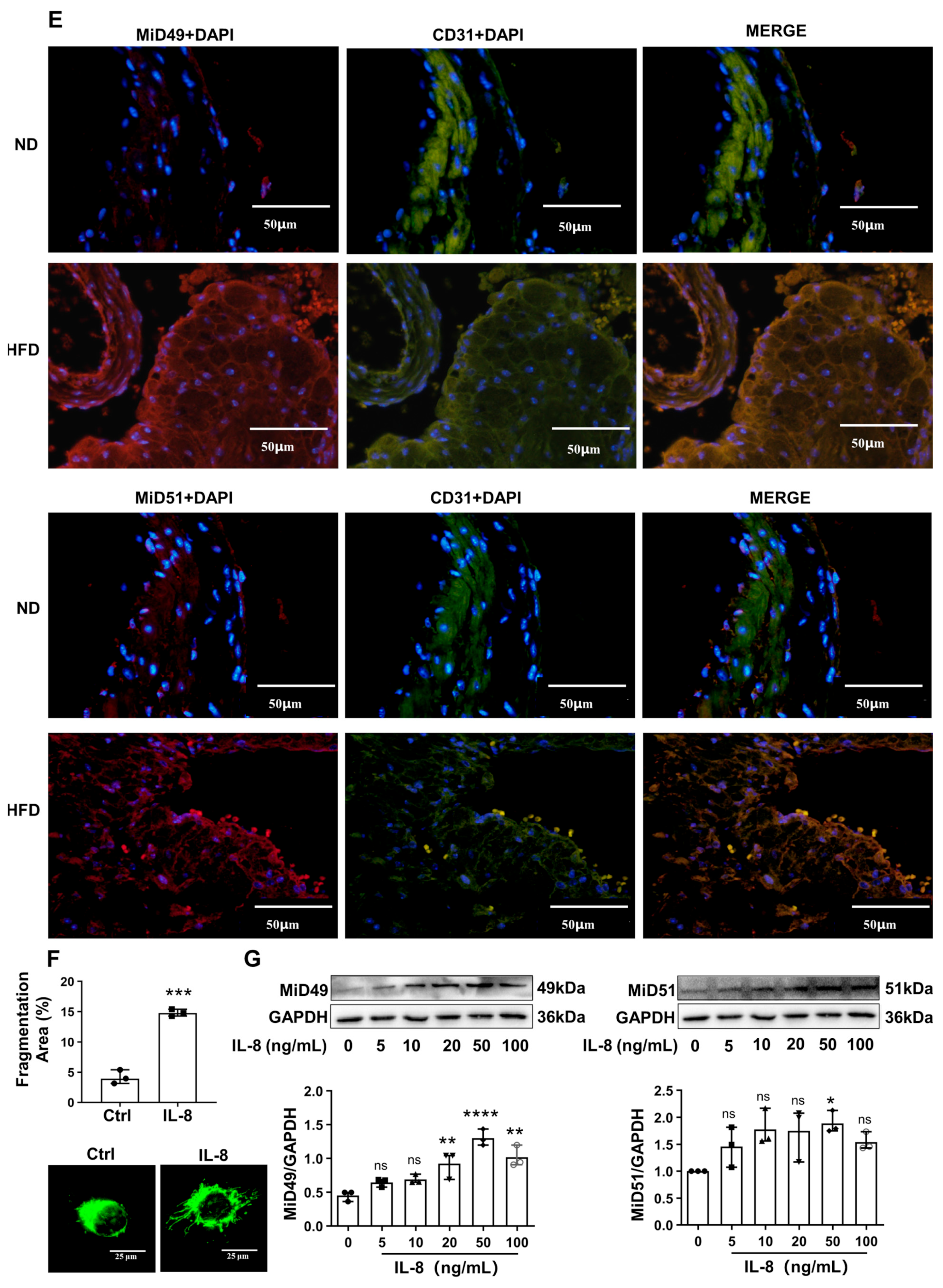
2.1. Increased Expression of MiD49 or MiD51 in Atherosclerotic ECs
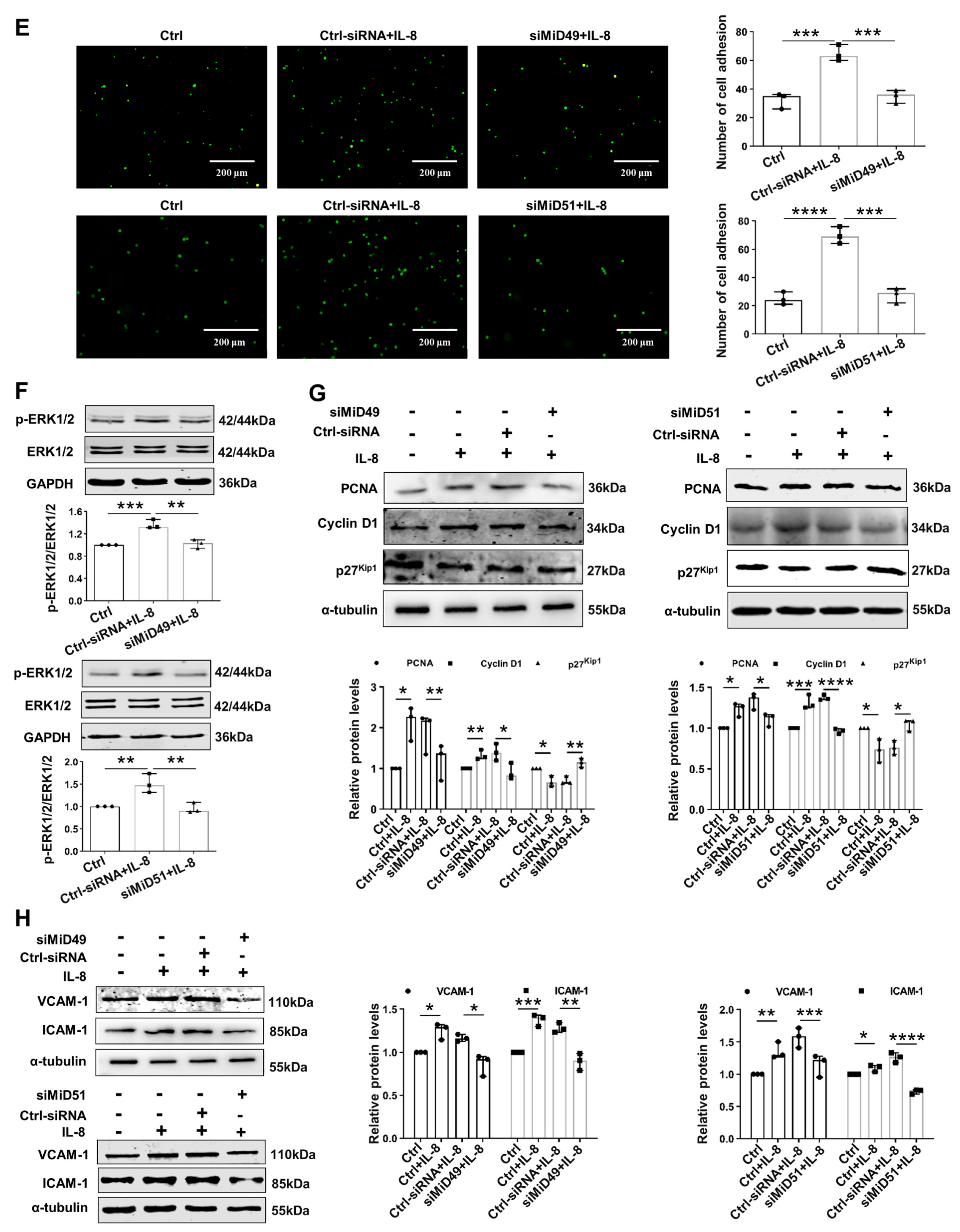
2.2. Silencing MiD49 or MiD51 Inhibited Mitochondrial Fission, Cell Adhesion and Proliferation by Inhibiting Drp1 and Related Proteins
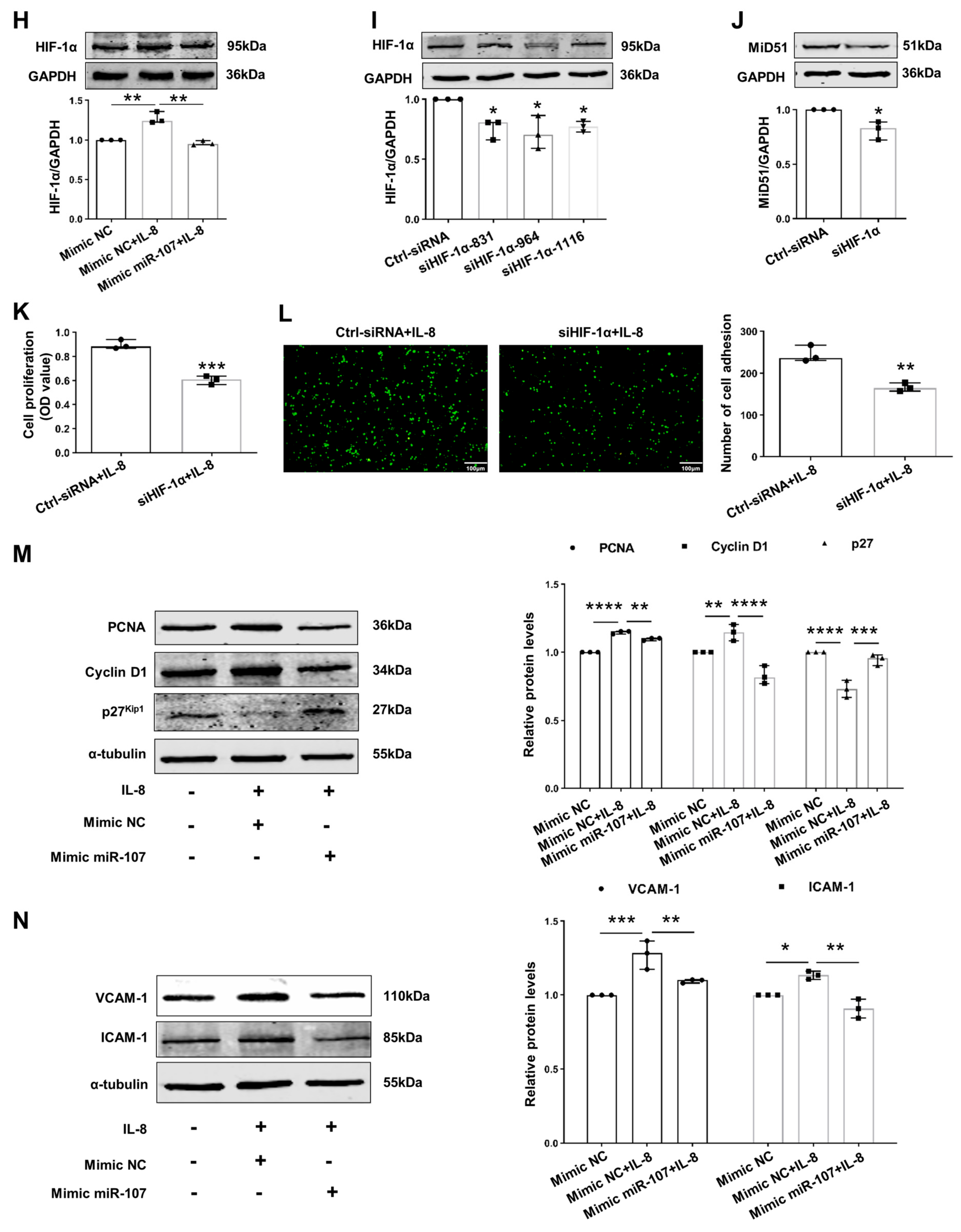
2.3. Downregulation of miR-107 Mediated MiD51 Upregulation by Targeting HIF-1α in IL-8-Induced HUVECs
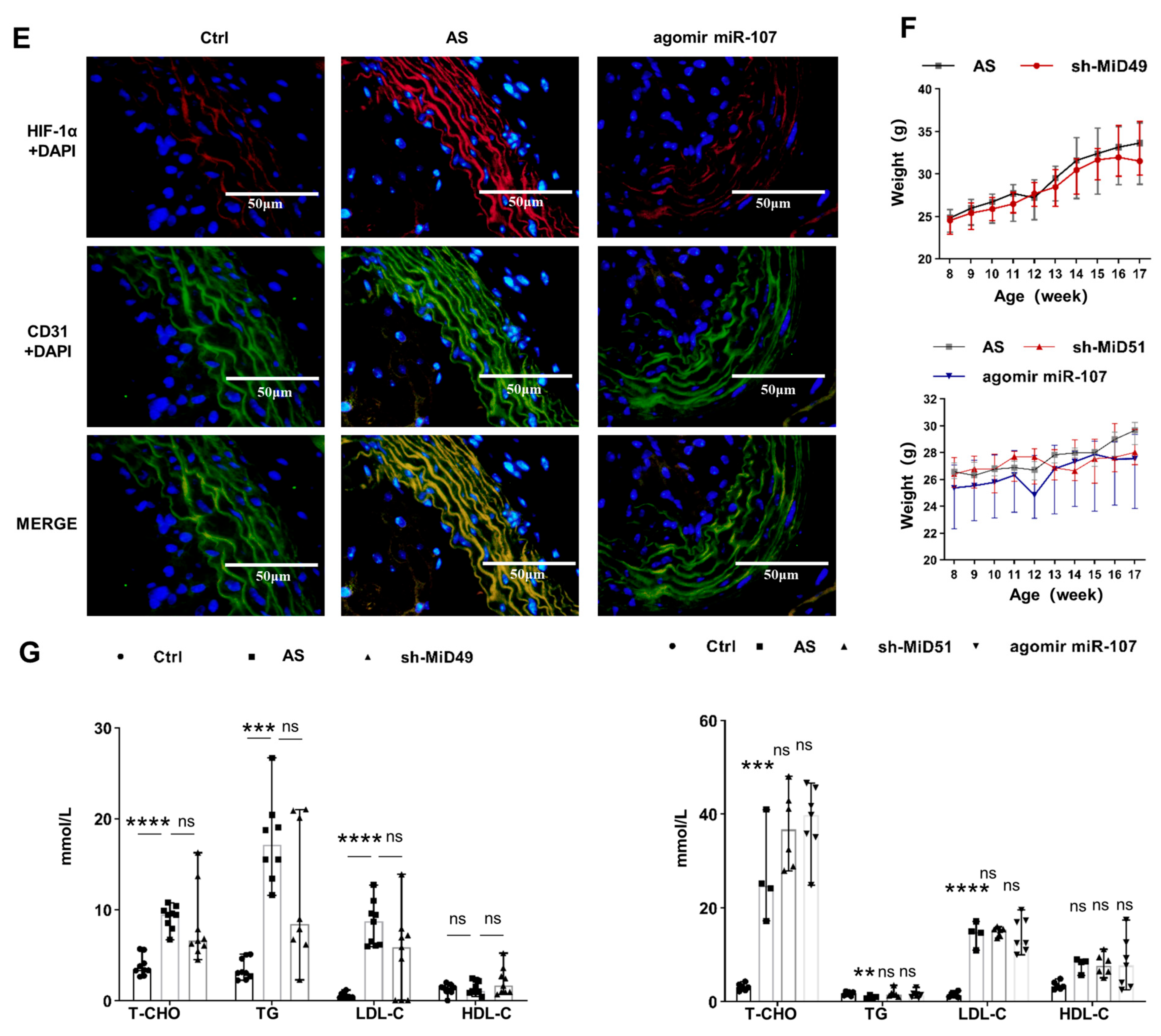
2.4. Potential Therapeutic Implications of MiD49 or miR-107–HIF-1α–MiD51 Pathway in the Formation of the Atherosclerotic Plaque of Mice
3. Discussion
4. Materials and Methods
4.1. Animal Models and Treatment
4.2. Reagents and Antibodies
4.3. Cell Culture
4.4. Immunohistochemistry
4.5. Immunofluorescence Staining
4.6. Cell Transfections
- siMiD51-690 sense: 5′-GGAUGUACGAUCGGGCGAUTT-3′
- siMiD51-690 antisense: 5′-AUCGCCCGAUCGUACAUCCTT-3′
- siMiD51-858 sense: 5′-CCUUCGACACAGAUACAUUTT-3′
- siMiD51-858 antisense: 5′-AAUGUAUCUGUGUCGAAGGTT-3′
- siMiD51-1021 sense: 5′-GCAAGCUGCUGUGGACAUATT-3′
- siMiD51-1021 antisense: 5′-UAUGUCCACAGCAGCUUGCTT-3′
- negative control sense: 5′-UUCUCCGAACGUGUCACGUTT-3′
- negative control antisense: 5′-ACGUGACACGUUCGGAGAATT-3′
- hsa-mir-107 Mimics sense: 5′-AGCAGCAUUGUACAGGGCUAUCA-3′
- hsa-mir-107 Mimics antisense: 5′-AUAGCCCUGUACAAUGCUGCUUU-3′
- negative control sense: 5′-UUCUCCGAACGUGUCAGGUTT-3′
- negative control antisense: 5′-ACGUGACACGUUCGGAGAATT-3′
4.7. Cell Proliferation Assay
4.8. Cell Adhesion Assay
4.9. miR-Binding Luciferase Reporter Assay
4.10. Quantification of Atherosclerotic Lesion Area
4.11. Quantification of Mitochondrial Fragmentation
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Björkegren, J.L.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2020, 178, 2060–2076. [Google Scholar] [CrossRef]
- Samangouei, P.; Crespo-Avilan, G.E.; Cabrera-Fuentes, H.; Hernández-Reséndiz, S.; Ismail, N.I.; Katwadi, K.B.; Boisvert, W.A.; Hausenloy, D.J. MiD49 and MiD51: New mediators of mitochondrial fission and novel targets for cardioprotection. Cond. Med. 2018, 1, 239–246. [Google Scholar]
- Chen, K.-H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension. Circulation 2018, 138, 287–304. [Google Scholar] [CrossRef]
- Shafi, O. Switching of vascular cells towards atherogenesis, and other factors contributing to atherosclerosis: A systematic review. Thromb. J. 2020, 18, 28. [Google Scholar] [CrossRef]
- Shen, L.; Hu, Y.; Lou, J.; Yin, S.; Wang, W.; Wang, Y.; Xia, Y.; Wu, W. CircRNA-0044073 is upregulated in atherosclerosis and increases the proliferation and invasion of cells by targeting miR-107. Mol. Med. Rep. 2019, 19, 3923–3932. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Ji, X.; Gu, J.; Wang, X.; Ding, L.; Zhang, H. microRNA-107 protects against inflammation and endoplasmic reticulum stress of vascular endothelial cells via KRT1-dependent Notch signaling pathway in a mouse model of coronary atherosclerosis. J. Cell. Physiol. 2018, 234, 12029–12041. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Glomset, J.A. The Pathogenesis of Atherosclerosis. N. Engl. J. Med. 1976, 295, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Lee, S.-Y.; Seo, H.-H.; Ham, O.; Lee, C.; Park, J.-H.; Lee, J.; Seung, M.; Yun, I.; Han, S.M.; et al. Regulation of Mitochondrial Morphology by Positive Feedback Interaction Between PKCδ and Drp1 in Vascular Smooth Muscle Cell. J. Cell. Biochem. 2015, 116, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mito-chondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis is an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Kato, J.-Y.; Matsuoka, M.; Strom, D.K.; Sherr, C.J. Regulation of Cyclin D-Dependent Kinase 4 (cdk4) by cdk4-activating kinase. Mol. Cell. Biol. 1994, 14, 2713–2721. [Google Scholar] [CrossRef]
- Cybulsky, M.I.; Gimbrone, M.A. Endothelial Expression of a Mononuclear Leukocyte Adhesion Molecule During Atherogenesis. Science 1991, 251, 788–791. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Y.; Xia, X.; Sun, H.; Gao, J.; Ren, Q.; Zhou, T.; Ma, C.; Xia, J.; Yin, C. Activation of macrophage TBK1-HIF-1α-mediated IL-17/IL-10 signaling by hyperglycemia aggravates the complexity of coronary atherosclerosis: An in vivo and in vitro study. FASEB J. 2021, 35, e21609. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, Y.; Zhu, L.; Yang, K.; Liang, K.; Tan, J.; Yu, B. SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1α and reactive oxygen species. Cell Death Dis. 2021, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhou, L.; Yin, W.; Bai, J.; Liu, R. miR-125a induces apoptosis, metabolism disorder and migrationimpairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int. J. Oncol. 2018, 53, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Mao, Y.-Y.; Chen, Q.; Zhang, B.; Zhang, S.; Wu, H.-J.; Li, Y. Hypoxia-induced ROS promotes mitochondrial fission and cisplatin chemosensitivity via HIF-1α/Mff regulation in head and neck squamous cell carcinoma. Cell. Oncol. 2021, 44, 1167–1181. [Google Scholar] [CrossRef]
- Kuo, C.-W.; Tsai, M.-H.; Lin, T.-K.; Tiao, M.-M.; Wang, P.-W.; Chuang, J.-H.; Chen, S.-D.; Liou, C.-W. mtDNA as a Mediator for Expression of Hypoxia-Inducible Factor 1α and ROS in Hypoxic Neuroblastoma Cells. Int. J. Mol. Sci. 2017, 18, 1220. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Tang, Y.; Wang, X.; Li, Z.; Li, R.; Ti, Z.; Gao, W.; Bai, J.; Lv, Y. Increased mitochondrial fission is critical for hypoxia-induced pancreatic beta cell death. PLoS ONE 2018, 13, e0197266. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Liu, J.; Zhang, J.; Hu, X.; Cui, Y.; Wei, X.; Ma, Y.; Li, X.; Zhao, Y. Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increased Mitochondrial Fission In Vitro and Atherosclerosis in High-Fat-Diet-Fed ApoE-/- Mice. Int. J. Mol. Sci. 2024, 25, 244. https://doi.org/10.3390/ijms25010244
Ren J, Liu J, Zhang J, Hu X, Cui Y, Wei X, Ma Y, Li X, Zhao Y. Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increased Mitochondrial Fission In Vitro and Atherosclerosis in High-Fat-Diet-Fed ApoE-/- Mice. International Journal of Molecular Sciences. 2024; 25(1):244. https://doi.org/10.3390/ijms25010244
Chicago/Turabian StyleRen, Jinyi, Jiaqing Liu, Jiahui Zhang, Xinxin Hu, Ying Cui, Xiaoqing Wei, Yang Ma, Xia Li, and Ying Zhao. 2024. "Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increased Mitochondrial Fission In Vitro and Atherosclerosis in High-Fat-Diet-Fed ApoE-/- Mice" International Journal of Molecular Sciences 25, no. 1: 244. https://doi.org/10.3390/ijms25010244