

Approach for Elucidating the Molecular Mechanism of Epithelial to Mesenchymal Transition in Fibrosis of Asthmatic Airway Remodeling Focusing on Cl− Channels


Abstract
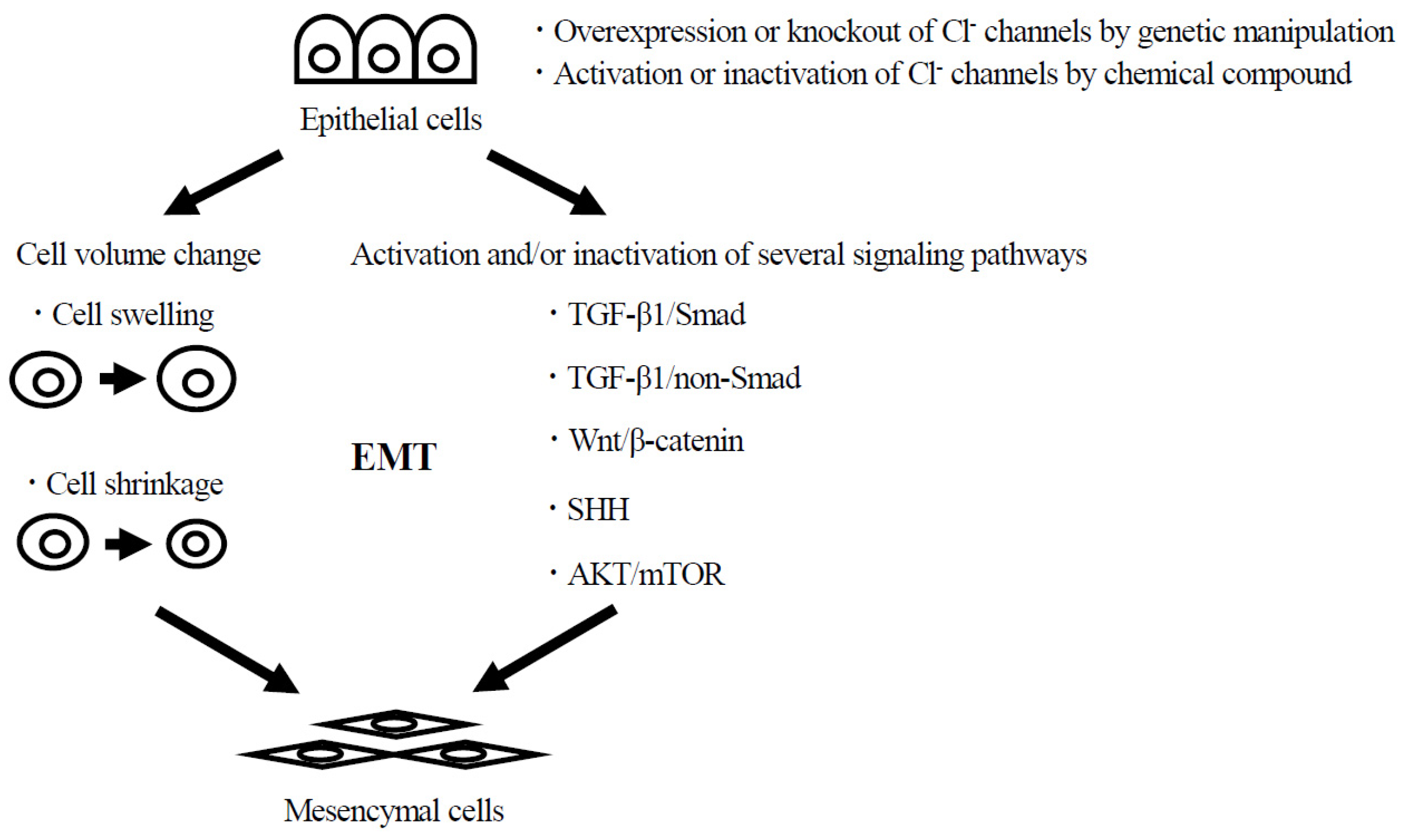
:1. Introduction
2. The Molecular Mechanism of EMT in Fibrosis of Asthmatic Airway Remodeling
2.1. TGF-β Signaling Pathway
2.2. Wnt Signaling Pathway
2.3. Other Signaling Pathways
3. The Roles of Cl− Channels on Morphological Changes Such as Cell Differentiation and Transdifferentiation
4. Relationship between Cl− Channels and EMT That Causes Carcinogenesis, Migration, and Invasion on Various Tissues
5. Relationship between Cl− Channels and EMT That Causes Fibrosis in the Airways and Other Tissues
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Porsbjerg, C.; Melén, E.; Lehtimäki, L.; Shaw, D. Asthma. Lancet 2023, 401, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis. Primers 2015, 1, 15025. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Polosa, R. The mechanisms, diagnosis, and management of severe asthma in adults. Lancet 2006, 368, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.P.; Ferguson, G.; Deniz, Y.; Reisner, C. Uncontrolled asthma: A review of the prevalence, disease burden and options for treatment. Respir. Med. 2006, 100, 1139–1151. [Google Scholar] [CrossRef]
- Elias, J.A.; Zhu, Z.; Chupp, G.; Homer, R.J. Airway remodeling in asthma. J. Clin. Invest. 1999, 104, 1001–1006. [Google Scholar] [CrossRef]
- Ordoñez, C.L.; Khashayar, R.; Wong, H.H.; Ferrando, R.; Wu, R.; Hyde, D.M.; Hotchkiss, J.A.; Zhang, Y.; Novikov, A.; Dolganov, G.; et al. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am. J. Respir. Crit. Care Med. 2001, 163, 517–523. [Google Scholar] [CrossRef]
- Holgate, S.T. Epithelium dysfunction in asthma. J. Allergy Clin. Immunol. 2007, 120, 1233–1244. [Google Scholar] [CrossRef]
- Liesker, J.J.; Ten Hacken, N.H.; Zeinstra-Smith, M.; Rutgers, S.R.; Postma, D.S.; Timens, W. Reticular basement membrane in asthma and COPD: Similar thickness, yet different composition. Int. J. Chron. Obstruct. Pulmon. Dis. 2009, 4, 127–135. [Google Scholar]
- Bourdin, A.; Neveu, D.; Vachier, I.; Paganin, F.; Godard, P.; Chanez, P. Specificity of basement membrane thickening in severe asthma. J. Allergy Clin. Immunol. 2007, 119, 1367–1374. [Google Scholar] [CrossRef]
- Joubert, P.; Hamid, Q. Role of airway smooth muscle in airway remodeling. J. Allergy Clin. Immunol. 2005, 116, 713–716. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.M. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am. J. Respir. Crit. Care Med. 2001, 164, S39–S45. [Google Scholar] [CrossRef] [PubMed]
- Pain, M.; Bermudez, O.; Lacoste, P.; Royer, P.J.; Botturi, K.; Tissot, A.; Brouard, S.; Eickelberg, O.; Magnan, A.; Moss, R.B. Tissue remodelling in chronic bronchial diseases: From the epithelial to mesenchymal phenotype. Eur. Respir. Rev. 2014, 23, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Hackett, T.L. Epithelial–mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- McNiven, M.A. Breaking away: Matrix remodeling from the leading edge. Trends Cell Biol. 2013, 23, 16–21. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Jentsch, T.J. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016, 17, 293–307. [Google Scholar] [CrossRef]
- Okada, Y.; Sabirov, R.Z.; Sato-Numata, K.; Numata, T. Cell Death Induction and Protection by Activation of Ubiquitously Expressed Anion/Cation Channels. Part 1: Roles of VSOR/VRAC in Cell Volume Regulation, Release of Double-Edged Signals and Apoptotic/Necrotic Cell Death. Front. Cell Dev. Biol. 2020, 8, 614040. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.Y.; Wang, G.L.; Zhou, J.G. The ClC-3 Cl− channel in cell volume regulation, proliferation and apoptosis in vascular smooth muscle cells. Trends Pharmacol. Sci. 2006, 27, 290–296. [Google Scholar] [CrossRef]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef] [PubMed]
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Shi, G.P. Calcium-activated chloride channel regulator 1 (CLCA1): More than a regulator of chloride transport and mucus production. World Allergy Organ. J. 2019, 12, 100077. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Kim, A.; Cho, C.H.; Kim, D.; Jung, H.G.; Kim, S.S.; Yoo, J.; Park, J.Y.; Hwang, E.M. TTYH1 and TTYH2 serve as LRRC8A-independent volume-regulated anion channels in cancer cells. Cells 2019, 8, 562. [Google Scholar] [CrossRef]
- Han, Y.E.; Kwon, J.; Won, J.; An, H.; Jang, M.W.; Woo, J.; Lee, J.S.; Park, M.G.; Yoon, B.E.; Lee, S.E.; et al. Tweety-homolog (Ttyh) Family Encodes the Pore-forming Subunits of the Swelling-dependent Volume-regulated Anion Channel (VRACswell) in the Brain. Exp. Neurobiol. 2019, 28, 183–215. [Google Scholar] [CrossRef]
- Okada, Y.; Okada, T.; Sato-Numata, K.; Numata, T. Reexamination of the roles of LRRC8 and TTYH in the molecular identity of volume-sensitive outwardly rectifying anion channel VSOR. J. Physiol. Sci. 2020, 70, S150. [Google Scholar]
- Wang, D.; Wang, H.; Gao, F.; Wang, K.; Dong, F. ClC-3 Promotes Osteogenic Differentiation in MC3T3-E1 Cell after Dynamic Compression. J. Cell Biochem. 2017, 118, 1606–1613. [Google Scholar] [CrossRef]
- Lu, X.; Ding, Y.; Niu, Q.; Xuan, S.; Yang, Y.; Jin, Y.; Wang, H. ClC-3 chloride channel mediates the role of parathyroid hormone [1-34] on osteogenic differentiation of osteoblasts. PLoS ONE 2017, 12, e0176196. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, J.; Wang, Y.; Weng, Z.; Huang, B.; Yu, M.K.; Zhang, X.; Yuan, P.; Zhao, H.; Chan, W.Y.; et al. CFTR-β-catenin interaction regulates mouse embryonic stem cell differentiation and embryonic development. Cell Death Differ. 2017, 24, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Dong, Y.; Zhao, J.; Yin, Y.; Zheng, Y. The CLC-2 Chloride Channel Modulates ECM Synthesis, Differentiation, and Migration of Human Conjunctival Fibroblasts via the PI3K/Akt Signaling Pathway. Int. J. Mol. Sci. 2016, 17, 910. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Edwards, R.; Yang, Y.; Hahn, A.; Folkers, K.; Ding, J.; Padmakumar, V.C.; Cataisson, C.; Suh, K.S.; Yuspa, S.H. CLIC4 regulates TGF-β-dependent myofibroblast differentiation to produce a cancer stroma. Oncogene 2014, 33, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Kakinouchi, K.; Yoshie, S.; Tsuji, S.; Murono, S.; Hazama, A. Dysfunction of Cl− channels promotes epithelial to mesenchymal transition in oral squamous cell carcinoma via activation of Wnt/β-catenin signaling pathway. Biochem. Biophys. Res. Commun. 2021, 28, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Zhang, J.; Zhang, H.; Ma, X.; Zhang, Y.; Li, Y.; Wang, F. CLCA2 overexpression suppresses epithelial-to-mesenchymal transition in cervical cancer cells through inactivation of ERK/JNK/p38-MAPK signaling pathways. BMC Mol. Cell Biol. 2022, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Wang, Y.; Chen, J.J.; Zhang, X.H.; Dong, J.D.; Tsang, L.L.; Huang, X.R.; Cai, Z.; Lan, H.Y.; Jiang, X.H.; et al. Defective CFTR leads to aberrant β-catenin activation and kidney fibrosis. Sci. Rep. 2017, 7, 5233. [Google Scholar] [CrossRef]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef]
- Guarino, M.; Tosoni, A.; Nebuloni, M. Direct contribution of epithelium to organ fibrosis: Epithelial-mesenchymal transition. Hum. Pathol. 2009, 40, 1365–1376. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Border, W.A.; Huang, Y.; Noble, N.A. TGF-beta isoforms in renal fibrogenesis. Kidney Int. 2003, 64, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, S.; Yamauchi, Y.; Kawasaki, S.; Takami, K.; Takizawa, H.; Nagase, T.; Kohyama, T. Simultaneous stimulation with TGF-β1 and TNF-α induces epithelial mesenchymal transition in bronchial epithelial cells. Int. Arch. Allergy Immunol. 2011, 155, 119–128. [Google Scholar] [CrossRef]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Vignola, A.M.; Chanez, P.; Chiappara, G.; Merendino, A.; Pace, E.; Rizzo, A.; la Rocca, A.M.; Bellia, V.; Bonsignore, G.; Bousquet, J. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1997, 156, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Minshall, E.M.; Leung, D.Y.; Martin, R.J.; Song, Y.L.; Cameron, L.; Ernst, P.; Hamid, Q. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 1997, 17, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, T.; Pazdrak, K.; Kalita, M.; Konig, R.; Choudhary, S.; Tian, B.; Boldogh, I.; Brasier, A.R. Systems biology approaches to understanding Epithelial Mesenchymal Transition (EMT) in mucosal remodeling and signaling in asthma. World Allergy Organ. J. 2014, 7, 13. [Google Scholar] [CrossRef]
- Sagara, H.; Okada, T.; Okumura, K.; Ogawa, H.; Ra, C.; Fukuda, T.; Nakao, A. Activation of TGF-beta/Smad2 signaling is associated with airway remodeling in asthma. J. Allergy Clin. Immunol. 2002, 110, 249–254. [Google Scholar] [CrossRef]
- Wang, W.; Yang, Z.; Li, M.; Wang, Z.; Shan, Y.; Qu, Z. Six1 Promotes Epithelial-Mesenchymal Transition in Bronchial Epithelial Cells via the TGFβ1/Smad Signalling Pathway. Int. Arch. Allergy Immunol. 2021, 182, 479–488. [Google Scholar] [CrossRef]
- Liu, W.; Sun, T.; Wang, Y. Integrin αvβ6 mediates epithelial-mesenchymal transition in human bronchial epithelial cells induced by lipopolysaccharides of Pseudomonas aeruginosa via TGF-β1-Smad2/3 signaling pathway. Folia Microbiol. 2020, 65, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Itoigawa, Y.; Harada, N.; Harada, S.; Katsura, Y.; Makino, F.; Ito, J.; Nurwidya, F.; Kato, M.; Takahashi, F.; Atsuta, R.; et al. TWEAK enhances TGF-β-induced epithelial-mesenchymal transition in human bronchial epithelial cells. Respir Res. 2015, 16, 48. [Google Scholar] [CrossRef]
- Doerner, A.M.; Zuraw, B.L. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.C.; Naura, A.S.; Aguilera-Aguirre, L.; Boldogh, I.; Boulares, H.A.; Calhoun, W.J.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibition prevents allergic airway remodeling through PI3K/AKT/GSK3β pathway in mice. PLoS ONE 2013, 8, e57442. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L.; Han, X.; Chen, Y.; Diao, J. Loke zupa decoction attenuates bronchial EMT-mediated airway remodelling in chronic asthma through the PI3K-Akt/HIF-1α signaling pathway. Pharm. Biol. 2023, 1, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Shin, J.M.; Yang, H.W.; Park, I.H. DNMTs Are Involved in TGF-β1-Induced Epithelial-Mesenchymal Transitions in Airway Epithelial Cells. Int. J. Mol. Sci. 2022, 23, 3003. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.J.; Park, D.W.; Seo, J.Y.; Moon, J.Y.; Kim, T.H.; Sohn, J.W.; Shin, D.H.; Yoon, H.J.; Park, S.S.; Kim, S.H. The Wnt/β-catenin signaling pathway regulates the development of airway remodeling in patients with asthma. Exp. Mol. Med. 2015, 47, e198. [Google Scholar] [CrossRef]
- Song, J.; Zhu, X.M.; Wei, Q.Y. MSCs reduce airway remodeling in the lungs of asthmatic rats through the Wnt/β-catenin signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 11199–11211. [Google Scholar]
- Takemaru, K.I.; Moon, R.T. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 2000, 149, 249–254. [Google Scholar] [CrossRef]
- Moheimani, F.; Roth, H.M.; Cross, J.; Reid, A.T.; Shaheen, F.; Warner, S.M.; Hirota, J.A.; Kicic, A.; Hallstrand, T.S.; Kahn, M.; et al. Disruption of β-catenin/CBP signaling inhibits human airway epithelial-mesenchymal transition and repair. Int. J. Biochem. Cell Biol. 2015, 68, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Song, W.; Zhou, L.; Mao, Y.; Hong, W. House dust mite induces Sonic hedgehog signaling that mediates epithelial-mesenchymal transition in human bronchial epithelial cells. Mol. Med. Rep. 2019, 20, 4674–4682. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.N.; Meng, P.; Zou, X.L.; Zhang, M.; Li, H.K.; Yang, H.L.; Li, H.T.; Zhang, T.T. IL-37 protects against airway remodeling by reversing bronchial epithelial-mesenchymal transition via IL-24 signaling pathway in chronic asthma. Respir. Res. 2022, 23, 244. [Google Scholar] [CrossRef]
- Huang, C.; Sun, Y.; Liu, N.; Zhang, Z.; Wang, X.; Lu, D.; Zhou, L.; Zhang, C. IL-27 attenuates airway inflammation and epithelial-mesenchymal transition in allergic asthmatic mice possibly via the RhoA/ROCK signalling pathway. Eur. Cytokine Netw. 2022, 33, 13–24. [Google Scholar] [PubMed]
- Wang, Z.; Li, L.; Wang, C.; Piao, Y.; Jiang, J.; Li, L.; Yan, G.; Piao, H. Recombinant Pyrin Domain Protein Attenuates Airway Inflammation and Alleviates Epithelial-Mesenchymal Transition by Inhibiting Crosstalk Between TGFβ1 and Notch1 Signaling in Chronic Asthmatic Mice. Front. Physiol. 2020, 11, 559470. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zhang, R.; Wang, J.; Li, Y.; Li, F.; Zhang, Y.; Zheng, X.; Shen, Y.; Wang, Y.; Zhou, L. CLC-2 is a positive modulator of oligodendrocyte precursor cell differentiation and myelination. Mol. Med. Rep. 2018, 17, 4515–4523. [Google Scholar] [CrossRef]
- Wang, H.; Mao, Y.; Zhang, B.; Wang, T.; Li, F.; Fu, S.; Xue, Y.; Yang, T.; Wen, X.; Ding, Y.; et al. Chloride channel ClC-3 promotion of osteogenic differentiation through Runx2. J. Cell Biochem. 2010, 111, 49–58. [Google Scholar] [CrossRef]
- Yin, Z.; Tong, Y.; Zhu, H.; Watsky, M.A. ClC-3 is required for LPA-activated Cl- current activity and fibroblast-to-myofibroblast differentiation. Am. J. Physiol. Cell Physiol. 2008, 294, C535–C542. [Google Scholar] [CrossRef]
- Chen, L.; König, B.; Stauber, T. LRRC8 channel activation and reduction in cytosolic chloride concentration during early differentiation of C2C12 myoblasts. Biochem. Biophys. Res. Commun. 2020, 532, 482–488. [Google Scholar] [CrossRef]
- Chen, L.; Becker, T.M.; Koch, U.; Stauber, T. The LRRC8/VRAC anion channel facilitates myogenic differentiation of murine myoblasts by promoting membrane hyperpolarization. J. Biol. Chem. 2019, 294, 14279–14288. [Google Scholar] [CrossRef]
- Li, P.; Singh, J.; Sun, Y.; Ma, X.; Yuan, P. CFTR constrains the differentiation from mouse embryonic stem cells to intestine lineage cells. Biochem. Biophys. Res. Commun. 2019, 510, 322–328. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wu, B.; Ye, W.; Le, D.D.; Sinclair, A.W.; Padovano, V.; Chen, Y.; Li, K.X.; Sit, R.; Tan, M.; et al. Chloride channels regulate differentiation and barrier functions of the mammalian airway. eLife 2020, 14, e53085. [Google Scholar] [CrossRef] [PubMed]
- Centeio, R.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. TMEM16A/F support exocytosis but do not inhibit Notch-mediated goblet cell metaplasia of BCi-NS1.1 human airway epithelium. Front. Physiol. 2023, 14, 1157704. [Google Scholar] [CrossRef] [PubMed]
- Scudieri, P.; Caci, E.; Bruno, S.; Ferrera, L.; Schiavon, M.; Sondo, E.; Tomati, V.; Gianotti, A.; Zegarra-Moran, O.; Pedemonte, N.; et al. Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J. Physiol. 2012, 590, 6141–6155. [Google Scholar] [CrossRef]
- Li, X.; Hu, W.; Zhou, J.; Huang, Y.; Peng, J.; Yuan, Y.; Yu, J.; Zheng, S. CLCA1 suppresses colorectal cancer aggressiveness via inhibition of the Wnt/beta-catenin signaling pathway. Cell Commun. Signal. 2017, 15, 38. [Google Scholar] [CrossRef]
- Qiang, Y.Y.; Li, C.Z.; Sun, R.; Zheng, L.S.; Peng, L.X.; Yang, J.P.; Meng, D.F.; Lang, Y.H.; Mei, Y.; Xie, P.; et al. Along with its favorable prognostic role, CLCA2 inhibits growth and metastasis of nasopharyngeal carcinoma cells via inhibition of FAK/ERK signaling. J. Exp. Clin. Cancer Res. 2018, 37, 34. [Google Scholar] [CrossRef]
- Song, X.; Zhang, S.; Li, S.; Wang, Y.; Zhang, X.; Xue, F. Expression of the CLCA4 Gene in Esophageal Carcinoma and Its Impact on the Biologic Function of Esophageal Carcinoma Cells. J. Oncol. 2021, 2021, 1649344. [Google Scholar] [CrossRef]
- Chen, H.; Liu, Y.; Jiang, C.J.; Chen, Y.M.; Li, H.; Liu, Q.A. Calcium-Activated Chloride Channel A4 (CLCA4) Plays Inhibitory Roles in Invasion and Migration Through Suppressing Epithelial-Mesenchymal Transition via PI3K/AKT Signaling in Colorectal Cancer. Med. Sci. Monit. 2019, 25, 4176–4185. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, M.; Xie, L.K.; Liu, T.; Zou, Z.W.; Li, Y.; Chen, P.; Peng, X.; Ma, C.; Zhang, W.J.; et al. CLCA4 inhibits cell proliferation and invasion of hepatocellular carcinoma by suppressing epithelial-mesenchymal transition via PI3K/AKT signaling. Aging (Albany NY) 2018, 10, 2570–2584. [Google Scholar] [CrossRef]
- Yu, Y.; Walia, V.; Elble, R.C. Loss of CLCA4 promotes epithelial-to-mesenchymal transition in breast cancer cells. PLoS ONE 2013, 8, e83943. [Google Scholar] [CrossRef]
- Xue, W.; Dong, B.; Zhao, Y.; Wang, Y.; Yang, C.; Xie, Y.; Niu, Z.; Zhu, C. Upregulation of TTYH3 promotes epithelial-to-mesenchymal transition through Wnt/β-catenin signaling and inhibits apoptosis in cholangiocarcinoma. Cell. Oncol. 2021, 44, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, J.K.; Bae, Y.; Lee, B.S.; Kim, E.; Cho, C.H.; Ryoo, K.; Yoo, J.; Kim, C.H.; Yi, G.S.; et al. Suppression of 14-3-3gamma-mediated surface expression of ANO1 inhibits cancer progression of glioblastoma cells. Sci. Rep. 2016, 6, 26413. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K.; et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E1026–E1034. [Google Scholar] [CrossRef] [PubMed]
- Shiwarski, D.J.; Shao, C.; Bill, A.; Kim, J.; Xiao, D.; Bertrand, C.A.; Seethala, R.S.; Sano, D.; Myers, J.N.; Ha, P.; et al. To “grow” or “go”: TMEM16A expression as a switch between tumor growth and metastasis in SCCHN. Clin. Cancer Res. 2014, 20, 4673–4688. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cao, Q.H.; Lu, D.J.; Luo, B.; Lu, X.F.; Luo, R.C.; Wang, X.G. TMEM16A overexpression contributes to tumor invasion and poor prognosis of human gastric cancer through TGF-beta signaling. Oncotarget. 2015, 6, 11585–11599. [Google Scholar] [CrossRef]
- Zhang, X.; Li, T.; Han, Y.N.; Ge, M.; Wang, P.; Sun, L.; Liu, H.; Cao, T.; Nie, Y.; Fan, D.; et al. miR-125b Promotes Colorectal Cancer Migration and Invasion by Dual-Targeting CFTR and CGN. Cancers 2021, 13, 5710. [Google Scholar] [CrossRef]
- Zhang, J.T.; Jiang, X.H.; Xie, C.; Cheng, H.; Da, D.J.; Wang, Y.; Fok, K.L.; Zhang, X.H.; Sun, T.T.; Tsang, L.L.; et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim. Biophys. Acta 2013, 1833, 2961–2969. [Google Scholar] [CrossRef]
- Quaresma, M.C.; Pankonien, I.; Clarke, L.A.; Sousa, L.S.; Silva, I.A.L.; Railean, V.; Doušová, T.; Fuxe, J.; Amaral, M.D. Mutant CFTR Drives TWIST1 mediated epithelial-mesenchymal transition. Cell Death Dis. 2020, 11, 920. [Google Scholar] [CrossRef]
- Friard, J.; Corinus, A.; Cougnon, M.; Tauc, M.; Pisani, D.F.; Duranton, C.; Rubera, I. LRRC8/VRAC channels exhibit a noncanonical permeability to glutathione, which modulates epithelial-to-mesenchymal transition (EMT). Cell Death Dis. 2019, 10, 925. [Google Scholar] [CrossRef]
- Yang, S.X.; Zhang, Z.C.; Bai, H.L. ClC-5 alleviates renal fibrosis in unilateral ureteral obstruction mice. Hum. Cell. 2019, 32, 297–305. [Google Scholar] [CrossRef]
- Wang, J.; He, Y.; Yang, G.; Li, N.; Li, M.; Zhang, M. Transient receptor potential canonical 1 channel mediates the mechanical stress-induced epithelial-mesenchymal transition of human bronchial epithelial (16HBE) cells. Int. J. Mol. Med. 2020, 46, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, X.C.; Li, L.; Ma, C.N.; Zhang, Y.J. Effects of TRPC1 on epithelial mesenchymal transition in human airway in chronic obstructive pulmonary disease. Medicine 2017, 96, e8166. [Google Scholar] [CrossRef] [PubMed]
- Pu, Q.; Zhao, Y.; Sun, Y.; Huang, T.; Lin, P.; Zhou, C.; Qin, S.; Singh, B.B.; Wu, M. TRPC1 intensifies house dust mite–induced airway remodeling by facilitating epithelial-to-mesenchymal transition and STAT3/NF-κB signaling. FASEB J. 2019, 33, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Arthur, G.K.; Duffy, S.M.; Roach, K.M.; Hirst, R.A.; Shikotra, A.; Gaillard, E.A.; Bradding, P. KCa3.1 K+ Channel Expression and Function in Human Bronchial Epithelial Cells. PLoS ONE 2015, 10, e0145259. [Google Scholar] [CrossRef]
- Uramoto, H.; Okada, T.; Okada, Y. Protective role of cardiac CFTR activation upon early reperfusion against myocardial infarction. Cell Physiol. Biochem. 2012, 30, 1023–1038. [Google Scholar] [CrossRef]

{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshie, S.; Murono, S.; Hazama, A. Approach for Elucidating the Molecular Mechanism of Epithelial to Mesenchymal Transition in Fibrosis of Asthmatic Airway Remodeling Focusing on Cl− Channels. Int. J. Mol. Sci. 2024, 25, 289. https://doi.org/10.3390/ijms25010289
Yoshie S, Murono S, Hazama A. Approach for Elucidating the Molecular Mechanism of Epithelial to Mesenchymal Transition in Fibrosis of Asthmatic Airway Remodeling Focusing on Cl− Channels. International Journal of Molecular Sciences. 2024; 25(1):289. https://doi.org/10.3390/ijms25010289
Chicago/Turabian StyleYoshie, Susumu, Shigeyuki Murono, and Akihiro Hazama. 2024. "Approach for Elucidating the Molecular Mechanism of Epithelial to Mesenchymal Transition in Fibrosis of Asthmatic Airway Remodeling Focusing on Cl− Channels" International Journal of Molecular Sciences 25, no. 1: 289. https://doi.org/10.3390/ijms25010289
APA StyleYoshie, S., Murono, S., & Hazama, A. (2024). Approach for Elucidating the Molecular Mechanism of Epithelial to Mesenchymal Transition in Fibrosis of Asthmatic Airway Remodeling Focusing on Cl− Channels. International Journal of Molecular Sciences, 25(1), 289. https://doi.org/10.3390/ijms25010289