
Epigenetic Regulation of EMP/EMT-Dependent Fibrosis



Abstract
1. Introduction
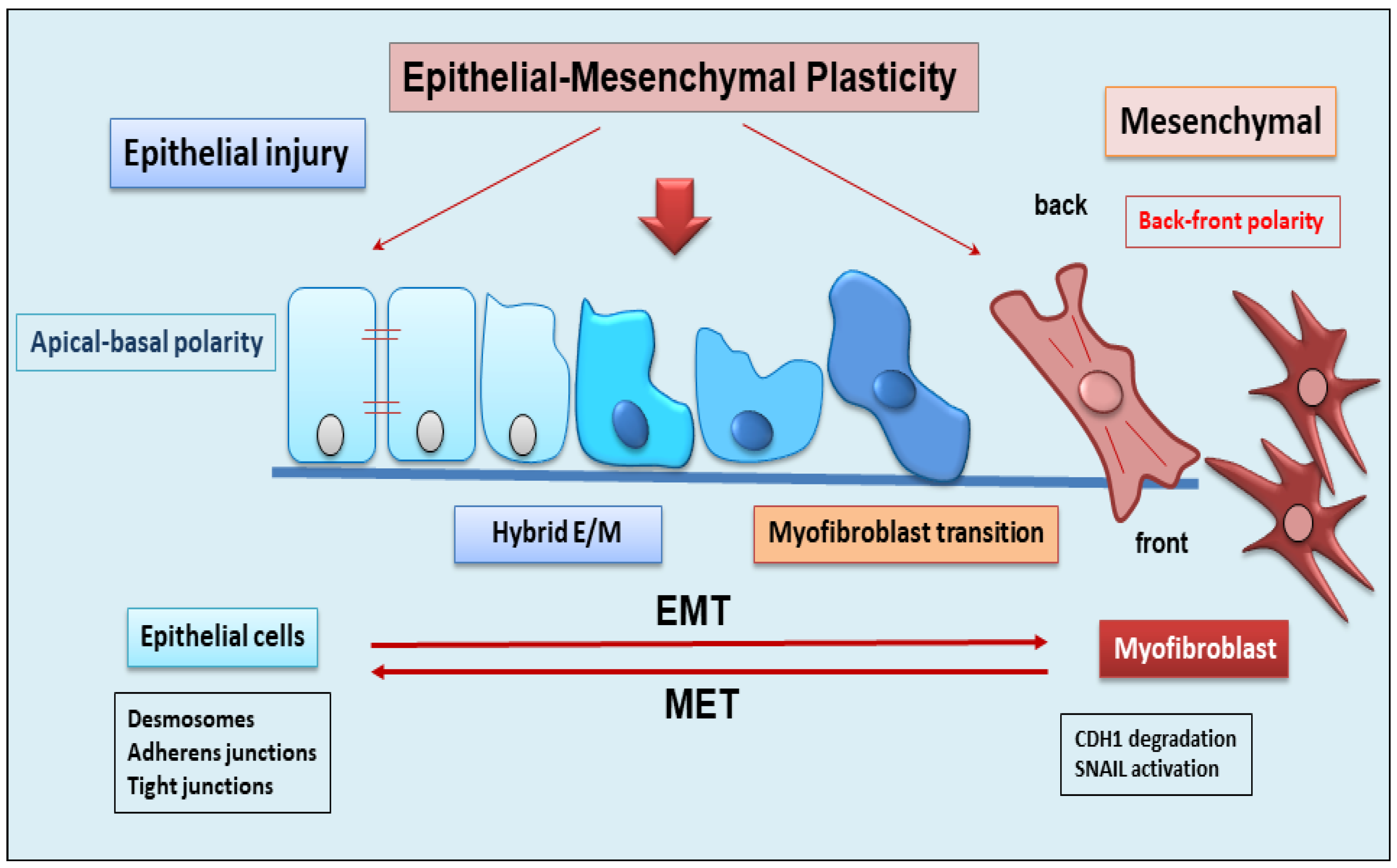
2. The Dynamic Balance between EMT and EMP
3. Role of EMP/EMT in Organ Fibrosis
Contribution of Epithelium to the Fibrotic Organ Process via EMT Activation
4. Main Epigenetic Mechanisms
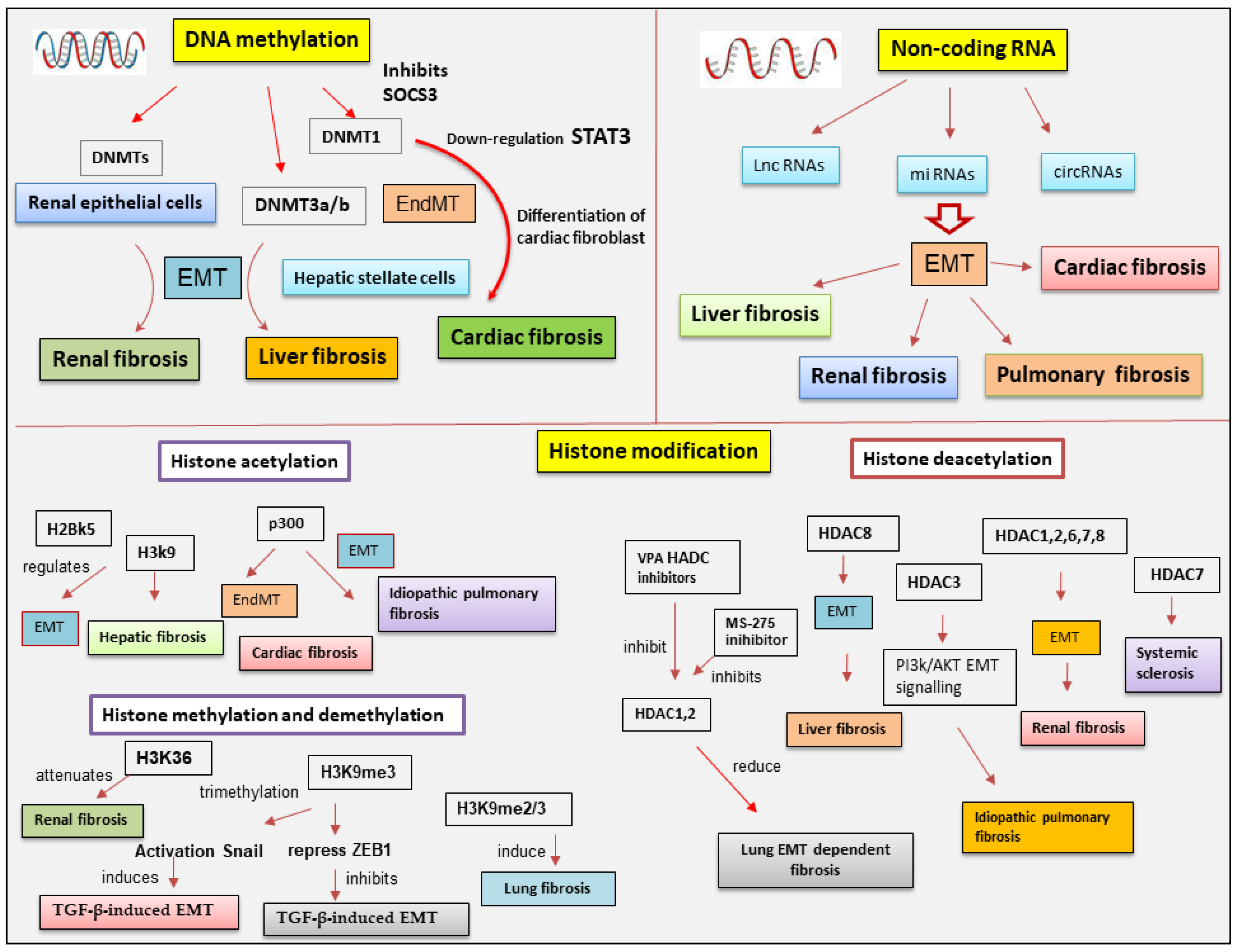
5. Epigenetics Regulation of EMP/EMT-Dependent Fibrosis
5.1. DNA Methylation in EMT-Dependent Fibrosis
5.2. The Involvement of Histone Modifications in EMT-Dependent Fibrosis
5.2.1. Histone Acetylation and Deacetylation
Class I Histone Deacetylase Involvement in EMT-Dependent Fibrosis
Class II Histone Deacetylases in EMT-Dependent Fibrosis
5.3. Histone Methylation and Demethylation Affects EMT-Dependent Fibrosis
5.4. Epigenetic Involvement of ncRNAs in EMT-Related Fibrosis
5.4.1. MiRNAs in EMT-Dependent Fibrosis
5.4.2. CircRNA Regulation of Fibrosis Correlated to EMT
5.4.3. LncRNA Regulation of EMT Related to Fibrosis
6. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z.; Iredale, J.; Friedman, S.L. Scraping fibrosis: Expressway to the core of fibrosis. Nat. Med. 2011, 17, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Grundtman, C.; Mayerl, C.; Wimpissinger, T.F.; Feichtinger, J.; Zelger, B.; Sgonc, R.; Wolfram, D. The immunology of fibrosis. Annu. Rev. Immunol. 2013, 31, 107–135. [Google Scholar] [CrossRef] [PubMed]
- Hnderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Subhadarshini, S.; Markus, J.; Sahoo, S.; Jolly, M.K. Dynamics of Epithelial-Mesenchymal Plasticity: What Have Single-Cell Investigations Elucidated so Far? ACS Omega 2023, 8, 11665–11673. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Wu, X.Q.; Zhang, D.D.; Wang, Y.N.; Guo, Y.; Li, P.; Xiong, Q.; Zhao, Y.Y. Deciphering the cellular mechanisms underlying fibrosis-associated diseases and therapeutic avenues. Pharmacol. Res. 2021, 163, 105316. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Ribatti, D.; Lisi, S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in TGFβ-Dependent EMT. Biomolecules 2021, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S. Towards a Unified Approach in Autoimmune Fibrotic Signalling Pathways. Int. J. Mol. Sci. 2023, 24, 9060. [Google Scholar] [CrossRef]
- Povero, D.; Busletta, C.; Novo, E.; di Bonzo, L.V.; Cannito, S.; Paternostro, C.; Parola, M. Liver fibrosis: A dynamic and potentially reversible process. Histol. Histopathol. 2010, 25, 1075–1091. [Google Scholar]
- Liu, Y.; Wen, D.; Ho, C.; Yu, L.; Zheng, D.; O’Reilly, S.; Gao, Y.; Li, Q.; Zhang, Y. Epigenetics as a versatile regulator of fibrosis. J. Transl. Med. 2023, 21, 164. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Györfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Xue, T.; Qiu, X.; Liu, H.; Gan, C.; Tan, Z.; Xie, Y.; Wang, Y.; Ye, T. Epigenetic regulation in fibrosis progress. Pharmacol. Res. 2021, 173, 105910. [Google Scholar] [CrossRef]
- Wang, X.C.; Song, K.; Tu, B.; Sun, H.; Zhou, Y.; Xu, S.S.; Lu, D.; Sha, J.M.; Tao, H. New aspects of the epigenetic regulation of EMT related to pulmonary fibrosis. Eur. J. Pharmacol. 2023, 956, 175959. [Google Scholar] [CrossRef]
- Mobley, R.J.; Abell, A.N. Controlling Epithelial to Mesenchymal Transition through Acetylation of Histone H2BK5. J. Nat. Sci. 2017, 3, e432. [Google Scholar]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. EMT: 2016. Cell 2013, 166, 21–45. [Google Scholar] [CrossRef]
- Bhatia, S.; Monkman, J.; Toh, A.K.L.; Nagaraj, S.H.; Thompson, E.W. Targeting epithelial-mesenchymal plasticity in cancer: Clinical and preclinical advances in therapy and monitoring. Biochem. J. 2017, 474, 3269–3306. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular plasticity in cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Arnoux, V.; Nassour, M.; L’Helgoualc’h, A.; Hipskind, R.A.; Savagner, P. Erk5 controls Slug expression and keratinocyte activation during wound healing. Mol. Biol. Cell 2018, 19, 4738–4749. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; duBois, R.M.; Borok, V. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc. Am. Thorac. Soc. 2006, 3, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Pei, D.; Shu, X.; Gassama-Diagne, A.; Thiery, J.P. Mesenchymal-epithelial Transition in Development and Reprogramming. Nat. Cell Biol. 2019, 21, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Haerinck, J.; Goossens, S.; Berx, G. The epithelial–mesenchymal plasticity landscape: Principles of design and mechanisms of regulation. Nat. Rev. Genet. 2023, 24, 590–609. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Qiu, Z.; Wu, Y. Tackle Epithelial-Mesenchymal Transition with Epigenetic Drugs in Cancer. Front. Pharmacol. 2020, 11, 596239. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Liu, Q.L.; Luo, M.; Huang, C.; Chen, H.N.; Zhou, Z.G. Epigenetic Regulation of Epithelial to Mesenchymal Transition in the Cancer Metastatic Cascade: Implications for Cancer Therapy. Front. Oncol. 2021, 11, 657546. [Google Scholar] [CrossRef] [PubMed]
- Verstappe, J.; Berx, G. A role for partial epithelial-to-mesenchymal transition in enabling stemness in homeostasis and cancer. Semin. Cancer Biol. 2023, 90, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Lin, Y.; Shimizu-Hirota, R.; Hanada, S.; Neilson, E.G.; Greenson, J.K.; Weiss, S.J. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol. Cell. Biol. 2011, 31, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Karin, D.; Koyama, Y.; Brenner, D.; Kisseleva, T. The characteristics of activated portal fibroblasts/myofibroblasts in liver fibrosis. Differentiation 2016, 92, 84–92. [Google Scholar] [CrossRef]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Xie, G.; Swiderska, M.; Choi, S.S.; Karaca, G.; Kruger, L.; Premont, R.; Yang, L.; Syn, W.K.; Metzger, D.; et al. Smoothened is a master regulator of adult liver repair. J. Clin. Investig. 2013, 123, 2380–2394. [Google Scholar] [CrossRef]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial-Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef]
- Yang, Z.C.; Yi, M.J.; Ran, N.; Wang, C.; Fu, P.; Feng, X.Y.; Xu, L.; Qu, Z.H. Transforming growth factor-beta1 induces bronchial epithelial cells to mesenchymal transition by activating the snail pathway and promotes airway remodeling in asthma. Mol. Med. Rep. 2013, 8, 1663–1668. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Hirai, S.; Tanaka, Y.; Sumi, T.; Miyajima, M.; Mishina, T.; Yamada, G.; Otsuka, M.; Hasegawa, T.; Kojima, T.; et al. Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial-mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab. Investig. 2017, 97, 232–242. [Google Scholar] [CrossRef]
- Liu, L.; Sun, Q.; Davis, F.; Mao, J.; Zhao, H.; Ma, D. Epithelial-mesenchymal transition in organ fibrosis development: Current understanding and treatment strategies. Burn. Trauma 2022, 10, tkac011. [Google Scholar] [CrossRef]
- Zolak, J.S.; Jagirdar, R.; Surolia, R.; Karki, S.; Oliva, O.; Hock, T.; Guroji, P.; Ding, Q.; Liu, R.M.; Bolisetty, S.; et al. Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am. J. Pathol. 2013, 182, 1239–1247. [Google Scholar] [CrossRef]
- Luo, G.H.; Lu, Y.P.; Yang, L.; Song, J.; Shi, Y.J.; Li, Y.P. Epithelial to mesenchymal transformation in tubular epithelial cells undergoing anoxia. Transpl. Proc. 2008, 40, 2800–2803. [Google Scholar] [CrossRef]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef]
- Sheng, L.; Zhuang, S. New Insights Into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Front. Physiol. 2020, 11, 569322. [Google Scholar] [CrossRef] [PubMed]
- Park Kim, J.; Lee, Y.J.; Bae, S.U.; Lee, H.W. Inflammatory bowel disease-associated intestinal fibrosis. J. Pathol. Transl. Med. 2023, 57, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Wenxiu, J.; Mingyue, Y.; Fei, H.; Yuxin, L.; Mengyao, W.; Chenyang, L.; Jia, S.; Hong, Z.; Shih, D.Q.; Targan, S.R.; et al. Effect and mechanism of TL1A expression on epithelial-mesenchymal transition during chronic colitis-related intestinal fibrosis. Mediat. Inflamm. 2021, 2021, 1–21. [Google Scholar] [CrossRef]
- Li, M.; Luan, F.; Zhao, Y.; Hao, H.; Zhou, Y.; Han, W.; Fu, X. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp. Biol. Med. 2016, 241, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Masia, D.; Gisbert-Ferrandiz, L.; Bauset, C.; Coll, S.; Mamie, C.; Scharl, M.; Esplugues, J.V.; Alós, R.; Navarro, F.; Cosín-Roger, J.; et al. Succinate activates EMT in intestinal epithelial cells through SUCNR1: A novel protagonist in fistula development. Cell 2020, 9, 1104. [Google Scholar] [CrossRef]
- Blom, J.N.; Feng, Q. Cardiac repair by epicardial EMT: Current targets and a potential role for the primary cilium. Pharmacol. Ther. 2018, 186, 114–129. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.; Kamal, F.; Robbins, J.; Yutzey, K.; Blaxall, B. Cardiac fibrosis: The fibroblast awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Singh, M.K. New Insights into Hippo/YAP Signalling in Fibrotic Diseases. Cells 2022, 11, 2065. [Google Scholar] [CrossRef] [PubMed]
- Aharonov, A.; Shakked, A.; Umanski, K.; Savidor, A.; Genzelinakh, A.; Kain, D.; Lendengolts, D.; Revach, O.Y.; Morikawa, Y.; Dong, J.; et al. ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat. Cell Biol. 2020, 22, 1346–1356. [Google Scholar] [CrossRef]
- Ling, J.; Cai, Z.; Jin, W.; Zhuang, X.; Kan, L.; Wang, F.; Ye, X. Silencing of c-Ski aug-ments TGF-b1-induced epithelial-mesenchymal transition in cardiomyocyte H9C2 cells. Cardiol. J. 2019, 26, 66–76. [Google Scholar] [CrossRef]
- Bookman, A.A.M.; Shen, H.; Cook, R.J.; Bailey, D.; McComb, R.J.; Rutka, J.A.; Slomovic, A.R.; Caffery, B. Whole stimulated salivary flow: Correlation with the pathology of inflammation and damage in minor salivary gland biopsy specimens from patients with primary Sjögren’s syndrome but not patients with sicca. Arthritis Rheumatol. 2011, 63, 2014–2020. [Google Scholar] [CrossRef]
- Llamas-Gutierrez, F.J.; Reyes, E.; Martínez, B.; Hernández-Molina, G. Histopathological environment besides the focus score in Sjögren’s syndrome. Int. J. Rheum. Dis. 2014, 17, 898–903. [Google Scholar] [CrossRef]
- Altrieth, A.L.; O’Keefe, K.J.; Gellatly, V.A.; Tavarez, J.R.; Feminella, S.M.; Moskwa, N.L.; Cordi, C.V.; Turrieta, J.C.; Nelson, D.A.; Larsen, M. Identifying fibrogenic cells following salivary gland obstructive injury. Front. Cell Dev. Biol. 2023, 11, 1190386. [Google Scholar] [CrossRef]
- Hall, B.E.; Zheng, C.; Swaim, W.D.; Cho, A.; Nagineni, C.N.; Eckhaus, M.A.; Flanders, K.C.; Ambudkar, I.S.; Baum, B.J.; Kulkarni, A.B. Conditional overexpression of TGF-beta1 disrupts mouse salivary gland development and function. Lab. Investig. 2010, 90, 543–555. [Google Scholar] [CrossRef]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Ribatti, D.; Lisi, S. TGFβ1-Smad canonical and -Erk noncanonical pathways participate in interleukin-17-induced epithelial-mesenchymal transition in Sjögren’s syndrome. Lab. Investig. 2020, 100, 824–836. [Google Scholar] [CrossRef]
- Ciechomska, M.; O’Reilly, S. Epigenetic Modulation as a Therapeutic Prospect for Treatment of Autoimmune Rheumatic Diseases. Mediat. Inflamm. 2016, 2016, 9607946. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Van Emburgh, B.O.; Robertson, K.D. DNA methylation in development and human disease. Mutat. Res. 2008, 647, 30–38. [Google Scholar] [CrossRef]
- Del Castillo Falconi, V.M.; Torres-Arciga, K.; Matus-Ortega, G.; Díaz-Chávez, J.; Herre-ra, L.A. DNA Methyltransferases: From Evolution to Clinical Applications. Int. J. Mol. Sci. 2022, 23, 8994. [Google Scholar] [CrossRef]
- Liang, Y.; He, L.; Yuan, H.; Jin, Y.; Yao, Y. Association between RUNX3 promoter methylation and non-small cell lung cancer: A meta-analysis. J. Thorac. Dis. 2014, 6, 694–705. [Google Scholar] [PubMed]
- Zhang, X.; Hu, M.; Lyu, X.; Li, C.; Thannickal, V.J.; Sanders, Y.Y. DNA methylation regulated gene expression in organ fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2389–2397. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Schiessel, H. The nucleosome: From structure to function through physics. Curr. Opin. Struct. Biol. 2019, 56, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Hergeth, S.P.; Schneider, R. The H1 linker histones: Multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015, 16, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes 2021, 12, 1596. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Liu, M.; Jiang, J.; Han, Y.; Shi, M.; Li, X.; Wang, Y.; Dong, Z.; Yang, C. Functional Characterization of the Lysine-Specific Histone Demethylases Family in Soybean. Plants 2022, 11, 1398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ramlee, M.K.; Brunmeir, R.; Villanueva, C.J.; Halperin, D.; Xu, F. Dynamic and distinct histone modifications modulate the expression of key adipogenesis regulatory genes. Cell Cycle 2012, 11, 4310–4322. [Google Scholar] [CrossRef]
- Rougeulle, C.; Chaumeil, J.; Sarma, K.; Allis, C.D.; Reinberg, D.; Avner, P.; Heard, E. Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X chromosome. Mol. Cell. Biol. 2004, 24, 5475–5484. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Litt, M.; Felsenfeld, G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 2005, 19, 1885–1893. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Lowndes, N.F.; Toh, G.W. DNA repair: The importance of phosphorylating histone H2AX. Curr. Biol. 2005, 15, R99–R102. [Google Scholar] [CrossRef]
- Lau, A.T.; Lee, S.Y.; Xu, Y.M.; Zheng, D.; Cho, Y.Y.; Zhu, F.; Kim, H.G.; Li, S.Q.; Zhang, Z.; Bode, A.M.; et al. Phosphorylation of histone H2B serine 32 is linked to cell transformation. J. Biol. Chem. 2011, 286, 26628–26637. [Google Scholar] [CrossRef]
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2012, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, biogenesis, and their evolving role in animal development and disease. Vet. Pathol. 2014, 54, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, W.; Claret, F.X. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics 2017, 12, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Dasaradhi, P.V.; Mohmmed, A.; Malhotra, P.; Bhatnagar, R.K.; Mukherjee, S.K. RNA interference: Biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 2003, 67, 657–685. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.N.; Li, Y.; Xia, S.Q.; Zhang, Y.Y.; Zheng, J.H.; Li, W. PIWI Proteins and PIWI-Interacting RNA: Emerging Roles in Cancer. Cell. Physiol. Biochem. 2017, 44, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczyk, M.; Wrzesinski, J. Long Non-Coding RNA Epigenetics. Int. J. Mol. Sci. 2021, 22, 6166. [Google Scholar] [CrossRef]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in di-verse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional classification and experimental dissection of long noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Aliperti, V.; Skonieczna, J.; Cerase, A. Long Non-Coding RNA (lncRNA) Roles in Cell Biology, Neurodevelopment and Neurological Disorders. Non-Coding RNA 2021, 7, 36. [Google Scholar] [CrossRef]
- Huang, S.; Yang, B.; Chen, B.J.; Bliim, N.; Ueberham, U.; Arendt, T.; Janitz, M. The emerging role of circular RNAs in transcriptome regulation. Genomics 2017, 109, 401–407. [Google Scholar] [CrossRef]
- Brasier, A.R.; Qiao, D.; Zhao, Y. The Hexosamine Biosynthetic Pathway Links Innate Inflammation with Epithelial-Mesenchymal Plasticity in Airway Remodeling. Front. Pharmacol. 2021, 12, 808735. [Google Scholar] [CrossRef]
- Galle, E.; Thienpont, B.; Cappuyns, S.; Venken, T.; Busschaert, P.; Van Haele, M.; Van Cutsem, E.; Roskams, T.; van Pelt, J.; Verslype, C.; et al. DNA methylation-driven EMT is a common mechanism of resistance to various therapeutic agents in cancer. Clin. Epigenetics 2020, 12, 27. [Google Scholar] [CrossRef]
- Marrs, J.A.; Andersson-Fisone, C.; Jeong, M.C.; Cohen-Gould, L.; Zurzolo, C.; Nabi, I.R.; Rodriguez-Boulan, E.; Nelson, W.J. Plasticity in epithelial cell phenotype: Modulation by expression of different cadherin cell adhesion molecules. J. Cell Biol. 1995, 129, 507–519. [Google Scholar] [CrossRef]
- Shenoy, S. CDH1 (E-Cadherin) mutation and gastric cancer: Genetics, molecu-larmechanisms and guidelines for management. Cancer Manag. Res. 2019, 11, 10477–10486. [Google Scholar] [CrossRef]
- Bücker, L.; Lehmann, U. CDH1 (E-cadherin) Gene Methylation in Human Breast Cancer: Critical Appraisal of a Long and Twisted Story. Cancers 2022, 14, 4377. [Google Scholar] [CrossRef]
- Kandimalla, R.; van Tilborg, A.A.; Zwarthoff, E.C. DNA methylation-based biomarkers in bladder cancer. Nat. Rev. Urol. 2013, 10, 327–335. [Google Scholar] [CrossRef]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Müller, G.A.; Kalbacher, H.; Salant, D.J.; Müller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef]
- Chang, Y.W.; Singh, K.P. Arsenic induces fibrogenic changes in human kidney epithelial cells potentially through epigenetic alterations in DNA methylation. J. Cell. Physiol. 2019, 234, 4713–4725. [Google Scholar] [CrossRef]
- Tao, H.; Shi, P.; Zhao, X.D.; Xuan, H.Y.; Gong, W.H.; Ding, X.S. DNMT1 deregulation of SOCS3 axis drives cardiac fibroblast activation in diabetic cardiac fibrosis. J. Cell. Physiol. 2021, 236, 3481–3494. [Google Scholar] [CrossRef]
- Dees, C.; Pötter, S.; Zhang, Y.; Bergmann, C.; Zhou, X.; Luber, M.; Wohlfahrt, T.; Karouzakis, E.; Ramming, A.; Gelse, K.; et al. TGF-β-induced epigenetic deregulation of SOCS3 facilitates STAT3 signalling to promote fibrosis. J. Clin. Investig. 2020, 130, 2347–2363. [Google Scholar] [CrossRef]
- Liu, R.; Li, Y.; Zheng, Q.; Ding, M.; Zhou, H.; Li, X. Epigenetic modification in liver fibrosis: Promising therapeutic direction with significant challenges ahead. Acta Pharm. Sin. B 2023, in press. [CrossRef]
- Yu, F.; Lu, Z.; Chen, B.; Wu, X.; Dong, P.; Zheng, J. Salvianolic acid B-induced microRNA-152 inhibits liver fibrosis by attenuating DNMT1-mediated Patched1 methylation. J. Cell. Mol. Med. 2015, 19, 2617–2632. [Google Scholar] [CrossRef]
- Avci, E.; Sarvari, P.; Savai, R.; Seeger, W.; Pullamsetti, S.S. Epigenetic Mechanisms in Parenchymal Lung Diseases: Bystanders or Therapeutic Targets? Int. J. Mol. Sci. 2022, 23, 546. [Google Scholar] [CrossRef]
- Mo, Y.; Zhang, Y.; Wan, R.; Jiang, M.; Xu, Y.; Zhang, Q. miR-21 mediates nickel nanoparticle-induced pulmonary injury and fibrosis. Nanotoxicology 2020, 14, 1175–1197. [Google Scholar] [CrossRef]
- Wu, C.H.; Tang, S.C.; Wang, P.H.; Lee, H.; Ko, J.L. Nickel-induced epithelial mesenchymal transition by reactive oxygen species generation and E-cadherin promoter hypermethylation. J. Biol. Chem. 2012, 287, 25292–25302. [Google Scholar] [CrossRef]
- Ning, L.; Rui, X.; Bo, W.; Qing, G. The critical roles of histone deacetylase 3 in the pathogenesis of solid organ injury. Cell Death Dis. 2021, 12, 734. [Google Scholar] [CrossRef]
- Ghoneim, M.; Fuchs, H.; Musselman, C. Histone tail conformations: A fuzzy affair with DNA. Trends Biochem. Sci. 2021, 46, 564–578. [Google Scholar] [CrossRef]
- Rubio, K.; Molina-Herrera, A.; Pérez-González, A.; Hernández-Galdámez, H.V.; Pi-ña-Vázquez, C.; Araujo-Ramos, T.; Singh, I. EP300 as a Molecular Integrator of Fibrotic Transcriptional Programs. Int. J. Mol. Sci. 2023, 24, 12302. [Google Scholar] [CrossRef]
- Lim, Y.; Jeong, A.; Kwon, D.H.; Lee, Y.U.; Kim, Y.K.; Ahn, Y.; Kook, T.; Park, W.J.; Kook, H. P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation. Int. J. Mol. Sci. 2021, 22, 9944. [Google Scholar] [CrossRef]
- Chu, L.; Xie, D.; Xu, D. Epigenetic Regulation of Fibroblasts and Crosstalk between Cardiomyocytes and Non-Myocyte Cells in Cardiac Fibrosis. Biomolecules 2023, 13, 1382. [Google Scholar] [CrossRef]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Claveria-Cabello, A.; Colyn, L.; Arechederra, M.; Urman, J.M.; Berasain, C.; Avila, M.A.; Fernandez-Barrena, M.G. Epigenetics in Liver Fibrosis: Could HDACs be a Therapeutic Target? Cells 2020, 9, 2321. [Google Scholar] [CrossRef]
- Huang, S.K.; Scruggs, A.M.; Donaghy, J.; Horowitz, J.C.; Zaslona, Z.; Przybranowski, S.; White, E.S.; Peters-Golden, M. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 2013, 4, e621. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Hagood, J.S.; Liu, H.; Zhang, W.; Ambalavanan, N.; Thannickal, V.J. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibro-sis in mice. Eur. Respir. J. 2014, 43, 1448–1458. [Google Scholar] [CrossRef]
- Wu, M.Z.; Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chang, C.C.; Teng, S.C.; Wu, K.J. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef]
- Chen, L.; Alam, A.; Pac-Soo, A.; Chen, Q.; Shang, Y.; Zhao, H.; Yao, S.; Ma, D. Pretreatment with valproic acid alleviates pulmonary fibrosis through epithelial-mesenchymal transition inhibition in vitro and in vivo. Lab. Investig. 2021, 101, 1166–1175. [Google Scholar] [CrossRef]
- Korfei, M.; Skwarna, S.; Henneke, I.; MacKenzie, B.; Klymenko, O.; Saito, S.; Ruppert, C.; von der Beck, D.; Mahavadi, P.; Klepetko, W.; et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 2015, 70, 1022–1032. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Barter, M.J.; Pybus, L.; Litherland, G.J.; Rowan, A.D.; Clark, I.M.; Edwards, D.R.; Cawston, T.E.; Young, D.A. HDAC-mediated control of ERK- and PI3K-dependent TGF-beta-induced extracellular matrix-regulating genes. Matrix Biol. 2010, 29, 602–612. [Google Scholar] [CrossRef]
- Kamio, K.; Azuma, A.; Usuki, J.; Matsuda, K.; Inomata, M.; Nishijima, N.; Itakura, S.; Hayashi, H.; Kashiwada, T.; Kokuho, N.; et al. XPLN is modulated by HDAC inhibitors and negatively regulates SPARC expression by targeting mTORC2 in human lung fibroblasts. Pulm. Pharmacol. Ther. 2017, 44, 61–69. [Google Scholar] [CrossRef]
- Zheng, Q.; Lei, Y.; Hui, S.; Tong, M.; Liang, L. HDAC3 promotes pulmonary fibrosis by activating NOTCH1 and STAT1 signalling and up-regulating inflammasome components AIM2 and ASC. Cytokine 2022, 153, 155842. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Sakle, N.S.; Dehghan, M.H.G.; Shinde, D.B. Histone deacetylases as targets for multiple diseases. Mini Rev. Med. Chem. 2013, 13, 1005–1026. [Google Scholar] [CrossRef]
- Xiong, R.; Geng, B.; Jiang, W.; Hu, Y.; Hu, Z.; Hao, B.; Li, N.; Geng, Q. Histone deacetylase 3 deletion in alveolar type 2 ep-ithelial cells prevents bleomycin-induced pulmonary fibrosis. Clin. Epigenetics 2023, 15, 182. [Google Scholar] [CrossRef]
- Chen, F.; Gao, Q.; Zhang, L.; Ding, Y.; Wang, H.; Cao, W. Inhibiting HDAC3 (Histone Deacetylase 3) Aberration and the Resultant Nrf2 (Nuclear Factor Erythroid-Derived 2-Related Factor-2) Repression Mitigates Pulmonary Fibrosis. Hypertension 2021, 78, e15–e25. [Google Scholar] [CrossRef]
- Jeong, S.H.; Son, E.S.; Lee, Y.E.; Kyung, S.Y.; Park, J.W.; Kim, S.H. Histone deacetylase 3 promotes alveolar epitheli-al–mesenchymal transition and fibroblast migration under hypoxic conditions. Exp. Mol. Med. 2022, 54, 922–931. [Google Scholar] [CrossRef]
- Dai, Q.; Liu, J.; Du, Y.; Hao, X.; Ying, J.; Tan, Y.; He, L.Q.; Wang, W.M.; Chen, N. Histone deacetylase inhibitors attenuate P-aIgA1-induced cell proliferation and extracellular matrix synthesis in human renal mesangial cells in vitro. Acta Pharmacol. Sin. 2016, 37, 228–234. [Google Scholar] [CrossRef]
- Choi, S.Y.; Kee, H.J.; Kurz, T.; Hansen, F.K.; Ryu, Y.; Kim, G.R.; Lin, M.Q.; Jin, L.; Piao, Z.H.; Jeong, M.H. Class I HDACs specifically regulate E-cadherin expression in human renal epithelial cells. J. Cell. Mol. Med. 2016, 20, 2289–2298. [Google Scholar] [CrossRef]
- Ma, P.; Pan, H.; Montgomery, R.L.; Olson, E.N.; Schultz, R.M. Compensatory functions of histone deacetylase 1 (HDAC1) and HDAC2 regulate transcription and apoptosis during mouse oocyte development. Proc. Natl. Acad. Sci. USA 2012, 109, E481–E489. [Google Scholar] [CrossRef]
- Yang, M.; Chen, G.; Zhang, X.; Guo, Y.; Yu, Y.; Tian, L.; Chang, S.; Chen, Z.K. Inhibition of class I HDACs attenuates renal interstitial fibrosis in a murine model. Pharmacol. Res. 2019, 142, 192–204. [Google Scholar] [CrossRef]
- Chen, F.; Gao, Q.; Wei, A.; Chen, X.; Shi, Y.; Wang, H.; Cao, W. Histone Deacetylase 3 Aberration Inhibits Klotho Transcription and Promotes Renal Fibrosis. Cell Death Differ. 2021, 28, 1001–1012. [Google Scholar] [CrossRef]
- Liu, L.; Lin, W.; Zhang, Q.; Cao, W.; Liu, Z. TGF-β induces miR-30d down-regulation and podocyte injury through Smad2/3 and HDAC3-associated transcriptional repression. J. Mol. Med. 2016, 94, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yin, S.; Bi, F.; Liu, L.; Qin, T.; Wang, H.; Cao, W. TIMAP repression by TGFβ and HDAC3-associated Smad signalling regulates macrophage M2 phenotypic phagocytosis. J. Mol. Med. 2017, 95, 273–285. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, J.; Tolbert, E.; Zhao, T.C.; Bayliss, G.; Zhuang, S. Identification of histone deacetylase 8 as a novel therapeutic target for renal fibrosis. FASEB J. 2020, 34, 7295–7310. [Google Scholar] [CrossRef]
- Shan, B.; Yao, T.P.; Nguyen, H.T.; Zhuo, Y.; Levy, D.R.; Klingsberg, R.C.; Tao, H.; Palmer, M.L.; Holder, K.N.; Lasky, J.A. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008, 283, 21065–21073. [Google Scholar] [CrossRef]
- Gu, S.; Liu, Y.; Zhu, B.; Ding, K.; Yao, T.P.; Chen, F.; Zhan, L.; Xu, P.; Ehrlich, M.; Liang, T.; et al. Loss of α-tubulin acetylation is associated with TGF-β-induced epithelial-mesenchymal transition. J. Biol. Chem. 2016, 291, 5396–5405. [Google Scholar] [CrossRef]
- Choi, S.Y.; Piao, Z.H.; Jin, L.; Kim, J.H.; Kim, G.R.; Ryu, Y.; Lin, M.Q.; Kim, H.S.; Kee, H.J.; Jeong, M.H. Piceatannol attenuates renal fibrosis induced by unilateral ureteral obstruction via downregulation of histone deacetylase 4/5 or p38-MAPK signalling. PLoS ONE 2016, 11, e0167340. [Google Scholar] [CrossRef]
- Liu, L.; Liu, F.; Guan, Y.; Zou, J.; Zhang, C.; Xiong, C.; Zhao, T.C.; Bayliss, G.; Li, X.; Zhuang, S. Critical roles ofSMYD2 lysine methyltransferase in mediating renal fibroblast activation and kidney fibrosis. FASEB J. 2021, 35, e21715. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.X.; Yu, C.; Nath, K.A.; Zhuang, S.; Li, X. Targeting lysine-specific demethylase 1A inhibits renal epithelial-mesenchymal transition and attenuates renal fibrosis. FASEB J. 2022, 36, e22122. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, Z.; Dong, X.; Zhao, N.; Liu, Y.; Wang, C.; Chen, J. Schisandrin B inhibits TGF-β1-induced epithelial-mesenchymal transition in human A549 cells through epigenetic silencing of ZEB1. Exp. Lung Res. 2019, 45, 157–166. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., 3rd. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Liu, T.; Xu, P.; Ke, S.; Dong, H.; Zhan, M.; Hu, Q.; Li, J. Histone methyltransferase SETDB1 inhibits TGF-β-induced epithelial-mesenchymal transition in pulmonary fibrosis by regulating SNAI1 expression and the ferroptosis signalling pathway. Arch. Biochem. Biophys. 2022, 715, 109087. [Google Scholar] [CrossRef]
- Nagaraja, S.S.; Subramanian, U.; Nagarajan, D. Radiation-induced H3K9 methylation on E-cadherin promoter mediated by ROS/Snail axis: Role of G9a signalling during lung epithelial-mesenchymal transition. Toxicol. Vitro 2021, 70, 105037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, G.; Zhang, Y.; Zhang, M.; Zhou, J.; Gao, W.; Xuan, X.; Yang, X.; Yang, D.; Tian, Z.; et al. Critical effects of long non-coding RNA on fibrosis diseases. Exp. Mol. Med. 2018, 50, e428. [Google Scholar] [CrossRef]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef]
- Wu, C.; Bao, S.; Sun, H.; Chen, X.; Yang, L.; Li, R.; Peng, Y. Noncoding RNAs regulating ferroptosis in cardiovascular diseases: Novel roles and therapeutic strategies. Mol. Cell. Biochem. 2023, in press. [CrossRef]
- Li, H.; Zhao, X.; Shan, H.; Liang, H. MicroRNAs in idiopathic pulmonary fibrosis: Involvement in pathogenesis and potential use in diagnosis and therapeutics. Acta Pharm. Sin. B 2016, 6, 531–539. [Google Scholar] [CrossRef]
- Liang, H.; Xu, C.; Pan, Z.; Zhang, Y.; Xu, Z.; Chen, Y.; Li, T.; Li, X.; Liu, Y.; Huangfu, L.; et al. The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol. Ther. 2014, 22, 1122–1133. [Google Scholar] [CrossRef]
- Yu, X.; Zhai, R.; Hua, B.; Bao, L.; Wang, D.; Li, Y.; Yao, W.; Fan, H.; Hao, C. miR-let-7d attenuates EMT by targeting HMGA2 in silica-induced pulmonary fibrosis. RSC Adv. 2019, 9, 19355–19364. [Google Scholar] [CrossRef]
- Wang, Y.C.; Liu, J.S.; Tang, H.K.; Nie, J.; Zhu, J.X.; Wen, L.L.; Guo, Q.L. miR-221 targets HMGA2 to inhibit bleomycin-induced pulmonary fibrosis by regulating TGF-β1/Smad3-induced EMT. Int. J. Mol. Med. 2016, 38, 1208–1216. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Z.; Jiang, X.; Wang, Y.; Yang, Z.; Mao, Y.; Wu, Z.; Li, G.; Chen, H. Mouse mesenchymal stem cell-derived exosomal miR-466f-3p reverses EMT process through inhibiting AKT/GSK3β pathway via c-MET in radiation-induced lung injury. J. Exp. Clin. Cancer Res. 2022, 41, 128. [Google Scholar] [CrossRef]
- Wang, D.; Liu, Z.; Yan, Z.; Liang, X.; Liu, X.; Liu, Y.; Wang, P.; Bai, C.; Gu, Y.; Zhou, P.K. MiRNA-155-5p inhibits epithelium-to-mesenchymal transition (EMT) by targeting GSK-3β during radiation-induced pulmonary fibrosis. Arch. Biochem. Biophys. 2021, 697, 108699. [Google Scholar] [CrossRef]
- Liang, X.; Yan, Z.; Wang, P.; Liu, Y.; Ao, X.; Liu, Z.; Wang, D.; Liu, X.; Zhu, M.; Gao, S.; et al. Irradiation Activates MZF1 to Inhibit miR-541-5p Expression and Promote Epithelial-Mesenchymal Transition (EMT) in Radiation-Induced Pulmonary Fibrosis (RIPF) by Upregulating Slug. Int. J. Mol. Sci. 2021, 22, 11309. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Shi, H.; Chen, X.; Li, C.; Shi, B.; Yeo, A.J.; Lavin, M.F.; Jia, Q.; Shao, H.; Zhang, J.; et al. miRNA-34c-5p targets Fra-1 to inhibit pulmonary fibrosis induced by silica through p53 and PTEN/PI3K/Akt signalling pathway. Environ. Toxicol. 2022, 37, 2019–2032. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Song, L.; Zhang, Z.; Bai, X.X.; Cui, M.F.; Ma, L.J. MicroRNA-21 promotes TGF-β1-induced epithelial-mesenchymal transition in gastric cancer through up-regulating PTEN expression. Oncotarget 2016, 7, 66989–67003. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zmijewska, A.; Zhi, D.; Mannon, R.B. Cyclosporine-mediated allograft fibrosis is associated with micro-RNA-21 through AKT signalling. Transpl. Int. 2015, 28, 232–245. [Google Scholar] [CrossRef]
- Bao, H.; Hu, S.; Zhang, C.; Shi, S.; Qin, W.; Zeng, C.; Zen, K.; Liu, Z. Inhibition of miRNA-21 prevents fibrogenic activation in podocytes and tubular cells in IgA nephropathy. Biochem. Biophys. Res. Commun. 2014, 444, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Chung, A.C.; Chen, H.-Y.; Meng, X.-M.; Lan, H.Y. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1668–1681. [Google Scholar] [CrossRef]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 2010, 39, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-J.; Hong, Q.; Wang, Z.; Yu, Y.-y.; Zou, X.; Xu, L.-h. MicroRNA21 promotes interstitial fibrosis via targeting DDAH1: A potential role in renal fibrosis. Mol. Cell. Biochem. 2016, 411, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Nagasu, H.; Morita, Y.; Yamaguchi, T.P.; Kanwar, Y.S.; Kashihara, N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signalling. Am. J. Physiol. Ren. Physiol. 2012, 303, F1641–F1651. [Google Scholar] [CrossRef]
- Koh, N.; Fujimori, T.; Nishiguchi, S.; Tamori, A.; Shiomi, S.; Nakatani, T.; Sugimura, K.; Kishimoto, T.; Kinoshita, S.; Kuroki, T. Severely reduced production of klotho in human chronic renal failure kidney. Biochem. Biophys. Res. Commun. 2001, 280, 1015–1020. [Google Scholar] [CrossRef]
- Gluba-Sagr, A.; Franczyk, B.; Rysz-Górzyńska, M.; Ławiński, J.; Rysz, J. The Role of miRNA in Renal Fibrosis Leading to Chronic Kidney Disease. Biomedicines 2023, 11, 2358. [Google Scholar] [CrossRef]
- Ai, K.; Zhu, X.; Kang, Y.; Li, H.; Zhang, L. miR-130a-3p inhibition protects against renal fibrosis in vitro via the TGF-β1/Smad pathway by targeting SnoN. Exp. Mol. Pathol. 2020, 112, 104358. [Google Scholar] [CrossRef]
- Bai, L.; Lin, Y.; Xie, J.; Zhang, Y.; Wang, H.; Zheng, D. MiR-27b-3p inhibits the progression of renal fibrosis via suppressing STAT1. Hum. Cell 2021, 34, 383–393. [Google Scholar] [CrossRef]
- Zhao, D.; Jia, J.; Shao, H. miR-30e targets GLIPR-2 to modulate diabetic nephropathy: In vitro and in vivo experiments. J. Mol. Endocrinol. 2017, 59, 181–190. [Google Scholar] [CrossRef]
- Patel, V.; Noureddine, L. MicroRNAs and fibrosis. Curr. Opin. Nephrol. Hypertens. 2012, 21, 410. [Google Scholar] [CrossRef] [PubMed]
- Tang, O.; Chen, X.-M.; Shen, S.; Hahn, M.; Pollock, C.A. MiRNA-200b represses transforming growth factor-β1-induced EMT and fibronectin expression in kidney proximal tubular cells. Am. J. Physiol. Ren. Physiol. 2013, 304, F1266–F1273. [Google Scholar] [CrossRef]
- Li, Q.; Li, Z.; Lin, Y.; Che, H.; Hu, Y.; Kang, X.; Zhang, Y.; Wang, L.; Zhang, Y. High glucose promotes hepatic fibrosis via miR-32/MTA3-mediated epithelial-to-mesenchymal transition. Mol. Med. Rep. 2019, 19, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Gao, L.; Liu, Y.; Liu, Y.; Yao, R.; Li, Y.; Xiao, L.; Wu, L.; Du, B.; Huang, Z.; et al. MiR-451 antagonist protects against cardiac fibrosis in streptozotocin-induced diabetic mouse heart. Life Sci. 2019, 224, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.X.; Chen, L.L. Circular RNAs: Characterization, cellular roles, and applications. Cell 2022, 185, 2016–2034. [Google Scholar] [CrossRef]
- Yao, W.; Li, Y.; Han, L.; Ji, X.; Pan, H.; Liu, Y.; Yuan, J.; Yan, W.; Ni, C. The CDR1as/miR-7/TGFBR2 Axis Modulates EMT in Silica-Induced Pulmonary Fibrosis. Toxicol. Sci. 2018, 166, 465–478. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Song, X.; Sun, W.; Zhang, J.; Liu, Y.; Li, H.; Meng, C.; Zhang, J.; Zheng, Q.; et al. Potential regulatory role of circular RNA in idiopathic pulmonary fibrosis. Int. J. Mol. Med. 2018, 42, 3256–3268. [Google Scholar] [CrossRef]
- Qi, F.; Li, Y.; Yang, X.; Wu, Y.; Lin, L.; Liu, X. Hsa_circ_0044226 knockdown attenuates progression of pulmonary fibrosis by inhibiting CDC27. Aging 2020, 12, 14808–14818. [Google Scholar] [CrossRef]
- Yang, X.; Wang, J.; Zhou, Z.; Jiang, R.; Huang, J.; Chen, L.; Cao, Z.; Chu, H.; Han, B.; Cheng, Y.; et al. Silica-induced initiation of circular ZC3H4 RNA/ZC3H4 pathway promotes the pulmonary macrophage activation. FASEB J. 2018, 32, 3264–3277. [Google Scholar] [CrossRef]
- Jiang, R.; Zhou, Z.; Liao, Y.; Yang, F.; Cheng, Y.; Huang, J.; Wang, J.; Chen, H.; Zhu, T.; Chao, J. The emerging roles of a novel CCCH-type zinc finger protein, ZC3H4, in silica-induced epithelial to mesenchymal transition. Toxicol. Lett. 2019, 307, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Sebastian-delaCruz, M.; Gonzalez-Moro, I.; Olazagoitia-Garmendia, A.; Castellanos-Rubio, A.; Santin, I. The Role of lncRNAs in Gene Expression Regulation through mRNA Stabilization. Non-Coding RNA 2021, 7, 3. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Xu, Q.; Yao, W.; Wu, Q.; Yuan, J.; Yan, W.; Xu, T.; Ji, X.; Ni, C. Long non-coding RNA-ATB promotes EMT during silica-induced pulmonary fibrosis by competitively binding miR-200c. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 420–431. [Google Scholar] [CrossRef]
- Xu, Q.; Cheng, D.; Liu, Y.; Pan, H.; Li, G.; Li, P.; Li, Y.; Sun, W.; Ma, D.; Ni, C. LncRNA-ATB regulates epithelial-mesenchymal transition progression in pulmonary fibrosis via sponging miR-29b-2-5p and miR-34c-3p. J. Cell. Mol. Med. 2021, 25, 7294–7306. [Google Scholar] [CrossRef]
- Sun, H.; Chen, J.; Qian, W.; Kang, J.; Wang, J.; Jiang, L.; Qiao, L.; Chen, W.; Zhang, J. Integrated long non-coding RNA analyses identify novel regulators of epithelial-mesenchymal transition in the mouse model of pulmonary fibrosis. J. Cell. Mol. Med. 2016, 20, 1234–1246. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, Y.; Yang, S.X.; Zhang, H.M.; Li, H.; Zong, Q.B.; Li, L.W.; Zhao, L.L.; Xia, R.H.; Li, C.; et al. MIR99AHG inhibits EMT in pulmonary fibrosis via the miR-136-5p/USP4/ACE2 axis. J. Transl. Med. 2022, 20, 426. [Google Scholar] [CrossRef]
- Zhan, H.; Chang, X.; Wang, X.; Yang, M.; Gao, Q.; Liu, H.; Li, C.; Li, S.; Sun, Y. LncRNA MEG3 mediates nickel oxide nanoparticles-induced pulmonary fibrosis via suppressing TGF-β1 expression and epithelial-mesenchymal transition process. Environ. Toxicol. 2021, 36, 1099–1110. [Google Scholar] [CrossRef]
- Qian, W.; Cai, X.; Qian, Q.; Peng, W.; Yu, J.; Zhang, X.; Tian, L.; Wang, C. lncRNA ZEB1-AS1 promotes pulmonary fibrosis through ZEB1-mediated epithelial–mesenchymal transition by competitively binding miR-141-3p. Cell Death Dis. 2019, 10, 129. [Google Scholar] [CrossRef]
- Dong, Z.; Li, S.; Wang, X.; Si, L.; Ma, R.; Bao, L.; Bo, A. lncRNA GAS5 restrains CCl4-induced hepatic fibrosis by targeting miR-23a through the PTEN/PI3K/Akt signalling pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G539–G550. [Google Scholar] [CrossRef]
- Chen, T.; Lin, H.; Chen, X.; Li, G.; Zhao, Y.; Zheng, L.; Shi, Z.; Zhang, K.; Hong, W.; Han, T. LncRNA Meg8 suppresses activation of hepatic stellate cells and epithelial-mesenchymal transition of hepatocytes via the Notch pathway. Biochem. Biophys. Res. Commun. 2020, 521, 921–927. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| ncRNAs | Target | Action Modes | Outcomes | References |
|---|---|---|---|---|
| miR-26a | HMGA2 | Downregulation of miR-26a promotes EMT process | Pulmonary fibrosis | [152] |
| miRNA let-7d | reduces HMGA2 expression | Overexpression of miRNA let-7d inhibits EMT process | Silicone-induced lung fibrosis | [154] |
| miR-221 | reduces HMGA2 expression | Overexpression of miR-221 inhibits EMT process | Bleomycin (BLM)-induced pulmonary fibrosis | [155] |
| miR-466f-3p | mMSCs-exo | Antifibrotic features of miR-466f-3p that prevent radiation-induced EMT | Radiation-induced pulmonary fibrosis | [156] |
| miR-155–5p | GSK-3β | Downregulation of miR-155–5p in radiation-induced pulmonary fibrosis | Radiation-induced pulmonary fibrosis | [157] |
| mir-486–3p | Snail gene | Downregulation of mir-486–3p promotes EMT process | Radiation-induced pulmonary fibrosis | [158] |
| miR-541-5p | MZF1, Slug gene | miR-541-5p repression induces EMT process | Radiation-induced pulmonary fibrosis | [158] |
| miR-34c-5p | Fra-1 | miR-34c-5p/Fra-1 axis represses activation of EMT process | Pulmonary fibrosis | [159] |
| miR-21 | MMPs, activation of TGF-β/Smad3 signaling | Upregulation of miR-21 induces TGF-β1, promoting EMT | Kidney fibrosis | [160,163,164] |
| miR-21 | DDAH1 | Upregulation of miR-21 decreases DDAH1 and increases ADMA, diminishing the production of NO | Kidney fibrosis | [165] |
| miR-34a | 3′ UTR of Klotho mRNA | Increased expression of miR-34a downregulates Kloto | Kidney fibrosis | [154] |
| miR-34a-5p | SnoN | Overexpression of miR-34a-5p determines the downregulation of SnoN and induction of EMT | Kidney fibrosis | [168] |
| miR-130a-3p | Smads | Downregulation of miR-130a-3p modulates Smads, induction of EMT | Kidney fibrosis | [169] |
| miR-27b-3p | α-SMA, fibronectin, collagen III, vimentin | Overexpression of miR-27b-3p reduces EMT process. Overexpression of miR-27b-3p reduces p-STAT1 and STAT1 induces EMT. | Kidney fibrosis | [170] |
| miR-30e | Downregulation of miR-30e expression in diabetic nephropathy, increases EMT | Kidney fibrosis | [171] | |
| miR-200b | miR-200b inhibits TGF-β1-induced EMT, ameliorates renal fibrosis | Kidney fibrosis | [172] | |
| miR200a | miR-200a represses TGF-β2 expression, inhibiting the development of renal fibrosis | Kidney fibrosis | [172,173] | |
| miR-32 | MTA3 | Profibrotic role of miR-32 | Liver fibrosis | [174] |
| miR-451 | Induced by EndMT | Cardiac fibrosis | [175] | |
| CDR1 | sponge for miR-7 | Upregulation of CDR1 promotes EMT process | Silica-induced pulmonary fibrosis | [177] |
| hsa_circ_0044226 | CDC27 | Upregulation of hsa_circ_0044226 promotes EMT | Idiopatic pulmonary fibrosis | [178] |
| circZC3H4 | sponge for miR-212 | Modulation of ZC3H4 expression induces EMT process | Idiopatic pulmonary fibrosis | [181] |
| lncRNA-ATB | sponge for miR-200c | lncRNA-ATB promotes EMT program | Silica-induced pulmonary fibrosis | [184] |
| uc.77 and 2700086A05Rik | Zeb2 and Hoxa3 | Upregulation of uc.77 and 2700086A05Rik induces EMT process | Pulmonary fibrosis | [186] |
| lncRNA miR99AHG | miR-136–5p | miR99AHG regulates the expression of ACE2, a target gene of USP4, and induces EMT process | Pulmonary fibrosis | [187] |
| lncRNA MEG3 | MEG3 induces NiO NPs via activating TGF-β1 and activation of EMT process | Pulmonary fibrosis | [188] | |
| ZEB1-AS1 | Upregulation of EB1-AS1 induces EMT process | Pulmonary fibrosis | [189] | |
| GAS5 | sponge for miR-23a | Activation of PTEN/PI3K/Akt pathway induces EMT process | Hepatic fibrosis | [190] |
| Meg8 | Overepression of Meg8 promotes EMT process | Hepatic fibrosis | [191] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sisto, M.; Lisi, S. Epigenetic Regulation of EMP/EMT-Dependent Fibrosis. Int. J. Mol. Sci. 2024, 25, 2775. https://doi.org/10.3390/ijms25052775
Sisto M, Lisi S. Epigenetic Regulation of EMP/EMT-Dependent Fibrosis. International Journal of Molecular Sciences. 2024; 25(5):2775. https://doi.org/10.3390/ijms25052775
Chicago/Turabian StyleSisto, Margherita, and Sabrina Lisi. 2024. "Epigenetic Regulation of EMP/EMT-Dependent Fibrosis" International Journal of Molecular Sciences 25, no. 5: 2775. https://doi.org/10.3390/ijms25052775
APA StyleSisto, M., & Lisi, S. (2024). Epigenetic Regulation of EMP/EMT-Dependent Fibrosis. International Journal of Molecular Sciences, 25(5), 2775. https://doi.org/10.3390/ijms25052775