
Diagnostic Algorithm to Subclassify Atypical Spitzoid Tumors in Low and High Risk According to Their Methylation Status

Abstract
:1. Introduction
2. Results
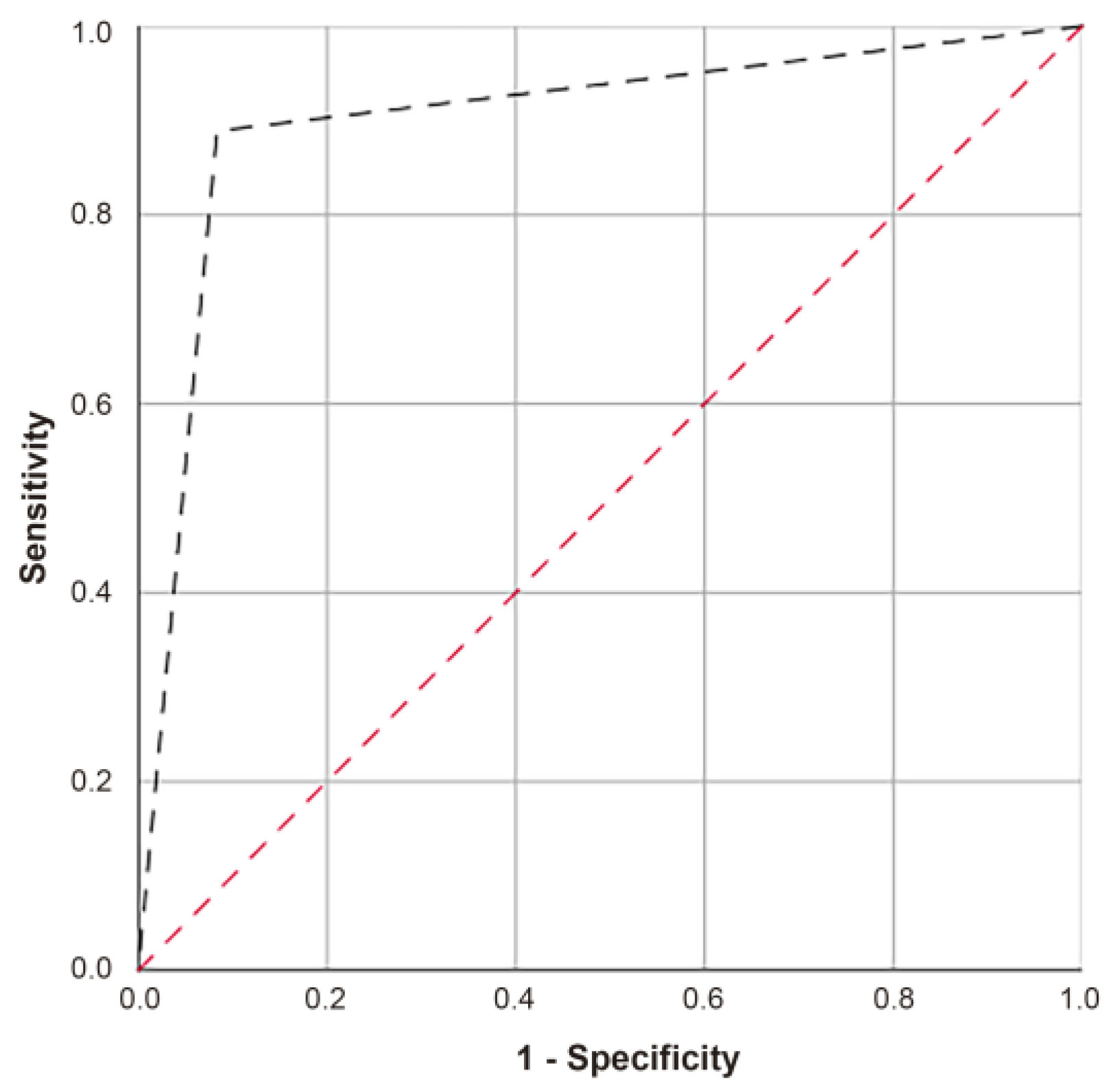
2.1. Differential Methylation Analysis
2.2. Predictive Equations
2.3. Risk Prediction of the Samples
3. Discussion
4. Materials and Methods
4.1. Human Samples
4.2. Nucleic Acid Extraction
4.3. Reduced Representation Bisulfite Sequencing (RRBS)
4.4. Bioinformatics Analysis
4.5. Statistical Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abboud, J.; Stein, M.; Ramien, M.; Malic, C. The diagnosis and management of the Spitz nevus in the pediatric population: A systematic review and meta-analysis protocol. Syst. Rev. 2017, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- WHO. Classification of Tumours Editorial Board, Skin Tumours, 5th ed.; WHO Classification of Tumours Series; WHO: Geneva, Switzerland, 2023; Volume 12.
- Tom, W.L.; Hsu, J.W.; Eichenfield, L.F. Pediatric “STUMP” lesions: Evaluation and management of difficult atypical Spitzoid lesions in children. J. Am. Acad. Dermatol. 2011, 64, 572–599. [Google Scholar] [CrossRef] [PubMed]
- Hillen, L.M.; Van den Oord, J.; Geybels, M.S.; Becker, J.C.; Hausen, A.Z.; Winnepenninckx, V. Genomic landscape of spitzoid neoplasms impacting patient management. Front. Med. 2018, 5, 344. [Google Scholar] [CrossRef] [PubMed]
- Mesbah Ardakani, N.; Thomas, C.; Robinson, C.; Mina, K.; Harvey, N.T.; Amanuel, B.; Wood, B.A. Detection of copy number variations in melanocytic lesions utilising array based comparative genomic hybridisation. Pathology 2017, 49, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Kutzner, H.; Cerroni, L.; Mihm, M.C.; Busam, K.J.; Murali, R. Genomic aberrations in spitzoid melanocytic tumours and their implications for diagnosis, prognosis and therapy. Pathology 2016, 48, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Gerami, P.; Scolyer, R.A.; Xu, X.; Elder, D.E.; Abraham, R.M.; Fullen, D.; Prieto, V.G.; Leboit, P.E.; Barnhill, R.L.; Cooper, C.; et al. Risk assessment for atypical spitzoid melanocytic neoplasms using FISH to identify chromosomal copy number aberrations. Am. J. Surg. Pathol. 2013, 37, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Dimonitsas, E.; Liakea, A.; Sakellariou, S.; Thymara, I.; Giannopoulos, A.; Stratigos, A.; Soura, E.; Saetta, A.; Korkolopoulou, P. An update on molecular alterations in melanocytic tumors with emphasis on Spitzoid lesions. Ann. Transl. Med. 2018, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.S.; Peternel, S.; Mully, T.W.; North, J.P.; Pincus, L.B.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; Yeh, I. Spitz melanoma is a distinct subset of spitzoid melanoma. Mod. Pathol. 2020, 33, 1122–1134. [Google Scholar] [CrossRef]
- van Engen-van Grunsven, A.C.; van Dijk, M.C.; Ruiter, D.J.; Klaasen, A.; Mooi, W.J.; Blokx, W.A. HRAS-mutated spitz tumors: A subtype of spitz tumors with distinct features. Am. J. Surg. Pathol. 2010, 34, 1436–1441. [Google Scholar] [CrossRef]
- Tetzlaff, M.T.; Reuben, A.; Billings, S.D.; Prieto, V.G.; Curry, J.L. Toward a Molecular-Genetic Classification of Spitzoid Neoplasms. Clin. Lab. Med. 2017, 37, 431–448. [Google Scholar] [CrossRef]
- Lee, S.; Barnhill, R.L.; Dummer, R.; Dalton, J.; Wu, J.; Pappo, A.; Bahrami, A. TERT Promoter Mutations Are Predictive of Aggressive Clinical Behavior in Patients with Spitzoid Melanocytic Neoplasms. Sci. Rep. 2015, 5, 11200. [Google Scholar] [CrossRef] [PubMed]
- Requena, C.; Heidenreich, B.; Kumar, R.; Nagore, E. TERT promoter mutations are not always associated with poor prognosis in atypical spitzoid tumors. Pigment Cell Melanoma Res. 2017, 30, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [PubMed]
- Quan, V.L.; Panah, E.; Zhang, B.; Shi, K.; Mohan, L.S.; Gerami, P. The role of gene fusions in melanocytic neoplasms. J. Cutan. Pathol. 2019, 46, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; de la Fouchardiere, A.; Pissaloux, D.; Mully, T.W.; Garrido, M.C.; Vemula, S.S.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2015, 39, 581–591. [Google Scholar] [CrossRef]
- Amin, S.M.; Haugh, A.M.; Lee, C.Y.; Zhang, B.; Bubley, J.A.; Merkel, E.A.; Verzì, A.E.; Gerami, P. A comparison of morphologic and molecular features of BRAF, ALK, and NTRK1 fusion spitzoid neoplasms. Am. J. Surg. Pathol. 2017, 41, 491–498. [Google Scholar] [CrossRef]
- Cho-Vega, J.H. A diagnostic algorithm for atypical spitzoid tumors: Guidelines for immunohistochemical and molecular assessment. Mod. Pathol. 2016, 29, 656–670. [Google Scholar] [CrossRef]
- Lee, C.Y.; Gerami, P. Molecular techniques for predicting behaviour in melanocytic neoplasms. Pathology 2016, 48, 142–146. [Google Scholar] [CrossRef]
- Schinke, C.; Mo, Y.; Yu, Y.; Amiri, K.; Sosman, J.; Greally, J.; Verma, A. Aberrant DNA methylation in malignant melanoma. Melanoma Res. 2010, 20, 253–265. [Google Scholar] [CrossRef]
- Rivera, R.M.; Bennett, L.B. Epigenetics in humans: An overview. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 493–499. [Google Scholar] [CrossRef]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Stockwell, P.A.; Ahn, A.; Rodger, E.J.; Leichter, A.L.; Eccles, M.R. Genome-wide methylation sequencing of paired primary and metastatic cell lines identifies common DNA methylation changes and a role for EBF3 as a candidate epigenetic driver of melanoma metastasis. Oncotarget 2017, 8, 6085–6101. [Google Scholar] [CrossRef] [PubMed]
- Wouters, J.; Vizoso, M.; Martinez-Cardus ACarmona, F.J.; Govaere, O.; Laguna, T.; Joseph, J.; Dynoodt, P.; Aura, C.; Foth, M.; Cloots, R.; et al. Comprehensive DNA methylation study identifies novel progression-related and prognostic markers for cutaneous melanoma. BMC Med. 2017, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ciarpaglini, C.; Gonzalez, J.; Sanchez, B.; Agusti, J.; Navarro, L.; Nieto, G.; Monteagudo, C. The amount of melanin influences p16 Loss in spitzoid melanocytic lesions: Correlation with CDKN2A Status by FISH and MLPA. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Lin, J.; Takayanagi, S.; Suzuki, T.; Ansai, S.; Kimura, T.; Cerroni, L.; Saida, T. Genetic and epigenetic alterations in the differential diagnosis of malignant melanoma and spitzoid lesion. Br. J. Dermatol. 2007, 156, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, A.; Jansen, P.; Murali, R.; Mayakonda, A.; Riedel, A.; Philip, M.; Rose, C.; Schaller, J.; Müller, H.; Kutzner, H.; et al. Genetic and methylation profiles distinguish benign, malignant and spitzoid melanocytic tumors. Int. J. Cancer 2022, 151, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked global DNA hypomethylation is associated with constitutive PD-L1 expression in melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef]
- Xu, S.; Sui, J.; Yang, S.; Liu, Y.; Wang, Y.; Liang, G. Integrative analysis of competing endogenous RNA network focusing on long noncoding RNA associated with progression of cutaneous melanoma. Cancer Med. 2018, 7, 1019–1029. [Google Scholar] [CrossRef]
- Pontén, F.; Jirström, K.; Uhlen, M. The Human Protein Atlas—A tool for pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef]
- Ngollo, M.; Lebert, A.; Daures, M.; Judes, G.; Rifai, K.; Dubois, L.; Kemeny, J.L.; Penault-Llorca, F.; Bignon, Y.J.; Guy, L.; et al. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer 2017, 17, 261. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.S.; Bae, J.A.; Kim, K.Y.; Kim, S.J.; Sun, E.G.; Lee, K.H.; Kim, N.; Kang, H.; Seo, Y.W.; Kim, H.; et al. MYO1D binds with kinase domain of the EGFR family to anchor them to plasma membrane before their activation and contributes carcinogenesis. Oncogene 2019, 38, 7416–7432. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Yuan, P.; Luo, J.; Chen, Y.; Tian, Y.; Ding, L.; Zhao, B.; Wang, X.; Wang, B.; Liu, L. Upregulated SPAG6 promotes acute myeloid leukemia progression through MYO1D that regulates the EGFR family expression. Blood Adv. 2022, 6, 5379–5394. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.G.; Xiong, W.; Wu, X.; Yang, L.; Pfeifer, G.P. The DNA methylation landscape of human melanoma. Genomics 2015, 106, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Rutten-Jacobs LC, A.; Rost, N.S. Emerging Insights from the Genetics of Cerebral Small Vessel Disease. Ann. N. Y. Acad. Sci. 2020, 1471, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Liang, Y.; Hu, J.; Hu, Y.; Liu, Y.; Cheng, Z.; Ou, Y.; Xu, C.; Jiang, H. DNA Methylation Modification Map to Predict Tumor Molecular Subtypes and Efficacy of Immunotherapy in Bladder Cancer. Front. Cell Dev. Biol. 2021, 9, 760369. [Google Scholar] [CrossRef] [PubMed]
- Vizoso, M.; Ferreira, H.J.; Lopez-Serra, P.; Carmona, F.J.; Martínez-Cardús, A.; Girotti, M.R.; Villanueva, A.; Guil, S.; Moutinho, C.; Liz, J.; et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nat. Med. 2015, 21, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Rodger, E.J.; Chatterjee, A.; Stockwell, P.A.; Eccles, M.R. Characterisation of DNA methylation changes in EBF3 and TBC1D16 associated with tumour progression and metastasis in multiple cancer types. Clin Epigenet. 2019, 11, 114. [Google Scholar] [CrossRef]
- Werner, R.J.; Kelly, A.D.; Issa, J.J. Epigenetics and Precision Oncology. Cancer J. 2017, 23, 262–269. [Google Scholar] [CrossRef]
- Broganelli, P.; Ribero, S.; Castagno, I.; Ricceri, F.; Deboli, T.; Marra, E.; Tomasini, C.; Sacerdote, C.; Osella-Abate, S.; Sanlorenzo, M.; et al. The large spectrum of Spitzoid tumors: A retrospective survival study. G. Ital. Dermatol. Venereol. 2019, 154, 315–320. [Google Scholar] [CrossRef]
- Kelley, S.W.; Cockerell, C.J. Sentinel lymph node biopsy as an adjunct to management of histologically difficult to diagnose melanocytic lesions: A proposal. J. Am. Acad. Dermatol. 2000, 42, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Dieng, M.; Cust, A.E.; Kasparian, N.A.; Mann, G.J.; Morton, R.L. Economic evaluations of psychosocial intervention in cancer: A systematic review. Psychooncology 2016, 25, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Bares, C.B.; Trask, P.C.; Schwartz, S.M. An exercise in cost-effectiveness analysis: Treating emotional distress in melanoma patients. J. Clin. Psychol. Med. Settings 2002, 91, 193–200. [Google Scholar] [CrossRef]
- Chapin, N.; Fernandez, J.; Poole, J.; Delatte, B. Anchor-based bisulfite sequencing determines genome-wide DNA methylation. Commun. Biol. 2022, 5, 596. [Google Scholar] [CrossRef] [PubMed]
- Gershman, A.; Sauria ME, G.; Guitart, X.; Vollger, M.R.; Hook, P.W.; Hoyt, S.J.; Jain, M.; Shumate, A.; Razaghi, R.; Koren, S.; et al. Epigenetic patterns in a complete human genome. Science 2022, 376, abj5089. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cao, W.; Zhuang, Y.; Chen, S.; Li, X. Integrative analysis of potential biomarkers and immune cell infiltration in Parkinson’s disease. Brain Res. Bull. 2021, 177, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Latchana, N.; del Campo, S.E.; Grignol, V.P.; Clark, J.R.; Albert, S.P.; Zhang, J.; Wei, L.; Aldrink, J.H.; Nicol, K.K.; Ranalli, M.A.; et al. Classification of Indeterminate Melanocytic Lesions by MicroRNA Profiling. Ann. Surg. Oncol. 2017, 24, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Latchana, N.; Regan, K.; Howard, J.H.; Aldrink, J.H.; Ranalli, M.A.; Peters, S.B.; Zhang, X.; Gru, A.; Payne PR, O.; Sua-rez-Kelly, L.P.; et al. Global microRNA profiling for diagnostic appraisal of melanocytic Spitz tumors. J. Surg. Res. 2016, 205, 350–358. [Google Scholar] [CrossRef]
- Pappo, A.S.; McPherson, V.; Pan, H.; Wang, F.; Wang, L.; Wright, T.; Hussong, M.; Hawkins, D.; Kaste, S.C.; Davidoff, A.M.; et al. A prospective, comprehensive registry that integrates the molecular analysis of pediatric and adolescent mela-nocytic lesions. Cancer 2021, 127, 3825–3831. [Google Scholar] [CrossRef]
- Veillard, A.-C.; Datlinger, P.; Laczik, M.; Squazzo, S.; Bock, C. Diagenode® Premium RRBS technology: Cost-effective DNA methylation mapping with superior coverage. Nat. Methods 2016, 13, i–ii. [Google Scholar] [CrossRef]
- Zhao, S.; Li, J.; Zhang, H.; Qi, L.; Du, Y.; Kogiso, M.; Braun, F.K.; Xiao, S.; Huang, Y.; Li, J.; et al. Epigenetic Alterations of Repeated Relapses in Patient-matched Childhood Ependymomas. Nat. Commun. 2022, 13, 6689. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Calcagno, V.; de Mazancourt, C. glmulti: An R package for easy automated model selection with (generalized) linear models. J. Stat. Softw. 2010, 34, 29. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position (cg) | Chromosome | Gene | Transcript |
|---|---|---|---|
| 31149610 | 17 | MYO1D | NM_015194 |
| 31149858 | 17 | MYO1D | NM_015194 |
| 9826086 | 21 | TEKT4P2 | NR_038327 |
| 9825862 | 21 | TEKT4P2 | NR_038327 |
| 9825882 | 21 | TEKT4P2 | NR_038327 |
| 9826034 | 21 | TEKT4P2 | NR_038327 |
| 156186376 | 1 | PMF1-BGLAP | NM_001199662 |
| Position (cg) | Gene | Specificity | Sensitivity | AUC | Cut-Off |
|---|---|---|---|---|---|
| 31149610 | MYO1D | 0.75 | 0.889 | 0.861 | 17.163 |
| 31149858 | MYO1D | 0.75 | 0.889 | 0.852 | 10.554 |
| 9826086 | TEKT4P2 | 0.75 | 0.889 | 0.861 | 21.219 |
| 9825862 | TEKT4P2 | 0.833 | 0.778 | 0.852 | 11.014 |
| 9825882 | TEKT4P2 | 0.667 | 1 | 0.833 | 10.007 |
| 9826034 | TEKT4P2 | 0.917 | 0.778 | 0.861 | 21.041 |
| 156186376 | PMF1-BGLAP | 0.75 | 0.889 | 0.852 | 15.235 |
| Sample | Diagnosis | Prediction 1 | Prediction 2 |
|---|---|---|---|
| 1 | SM | SM | SM |
| 2 | SM | SM | SM |
| 3 | SM | SM | SN |
| 4 | SM | SM | SM |
| 5 | SM | SM | SM |
| 6 | SM | SM | SM |
| 7 | SM | SM | SM |
| 8 | SM | SM | SM |
| 9 | SN | SN | SN |
| 10 | SN | SN | SN |
| 11 | SN | SN | SN |
| 12 | SN | SN | SN |
| 13 | SN | SN | SN |
| 14 | SN | SN | SN |
| 15 | SN | SN | SN |
| 16 | SN | SN | SM |
| 17 | SN | SN | SN |
| 18 | SM | SM | SM |
| 19 | SN | SN | SN |
| 20 | SN | SN | SN |
| 21 | SN | SN | SN |
| Sample | Diagnosis | Prediction 1 | Prediction 2 |
|---|---|---|---|
| 22 | AST | SN | SN |
| 23 | AST | SN | SN |
| 24 | AST | SN | SM |
| 25 | AST | SM | SM |
| 26 | AST | SN | SN |
| 27 | AST | SM | SM |
| 28 | AST | SN | SN |
| 29 | AST | SM | SM |
| 30 | AST | SM | SM |
| 31 | AST | SM | SM |
| 32 | AST | SN | SN |
| 33 | AST | SM | SM |
| 34 | AST | SM | SM |
| 35 | AST | SM | SM |
| 36 | AST | SM | SM |
| 37 | AST | SN | SN |
| 38 | AST | SN | SM |
| 39 | AST | SN | SM |
| 40 | AST | SN | SN |
| Diagnosis | n | Age (Years) at Diagnosis (Median ± SD #) | Gender | Location | Diameter (mm) (Median ± SD #) | Mitosis/mm2 (Median ± SD #) |
|---|---|---|---|---|---|---|
| SN | 12 | 24.08 ± 18.33 | Male: 25% Female: 75% | Lower limb: 4 Trunk: 2 Upper limb: 4 Head & neck: 1 Not specified: 1 | 6.14 ± 3.18 | 0.33 ± 0.65 |
| AST | 19 | 20.6 ± 14.84 | Male: 37.5% Female: 62.5% | Lower limb: 2 Trunk: 3 Upper limb: 3 Head & neck: 2 Not specified: 9 | 5.22 ± 2.48 | 1.53 ± 1.07 |
| SM | 9 | 47.22 ± 20.31 | Male: 66.67% Female: 33.33% | Lower limb: 1 Trunk: 1 Upper limb: 4 Head & neck: 0 Not specified: 3 | 5.16 ± 1.31 | 5 ± 2.83 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Muñoz, J.F.; Sánchez-Sendra, B.; Monteagudo, C. Diagnostic Algorithm to Subclassify Atypical Spitzoid Tumors in Low and High Risk According to Their Methylation Status. Int. J. Mol. Sci. 2024, 25, 318. https://doi.org/10.3390/ijms25010318
González-Muñoz JF, Sánchez-Sendra B, Monteagudo C. Diagnostic Algorithm to Subclassify Atypical Spitzoid Tumors in Low and High Risk According to Their Methylation Status. International Journal of Molecular Sciences. 2024; 25(1):318. https://doi.org/10.3390/ijms25010318
Chicago/Turabian StyleGonzález-Muñoz, Jose Francisco, Beatriz Sánchez-Sendra, and Carlos Monteagudo. 2024. "Diagnostic Algorithm to Subclassify Atypical Spitzoid Tumors in Low and High Risk According to Their Methylation Status" International Journal of Molecular Sciences 25, no. 1: 318. https://doi.org/10.3390/ijms25010318