

Experimental Models to Study Endothelial to Mesenchymal Transition in Myocardial Fibrosis and Cardiovascular Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
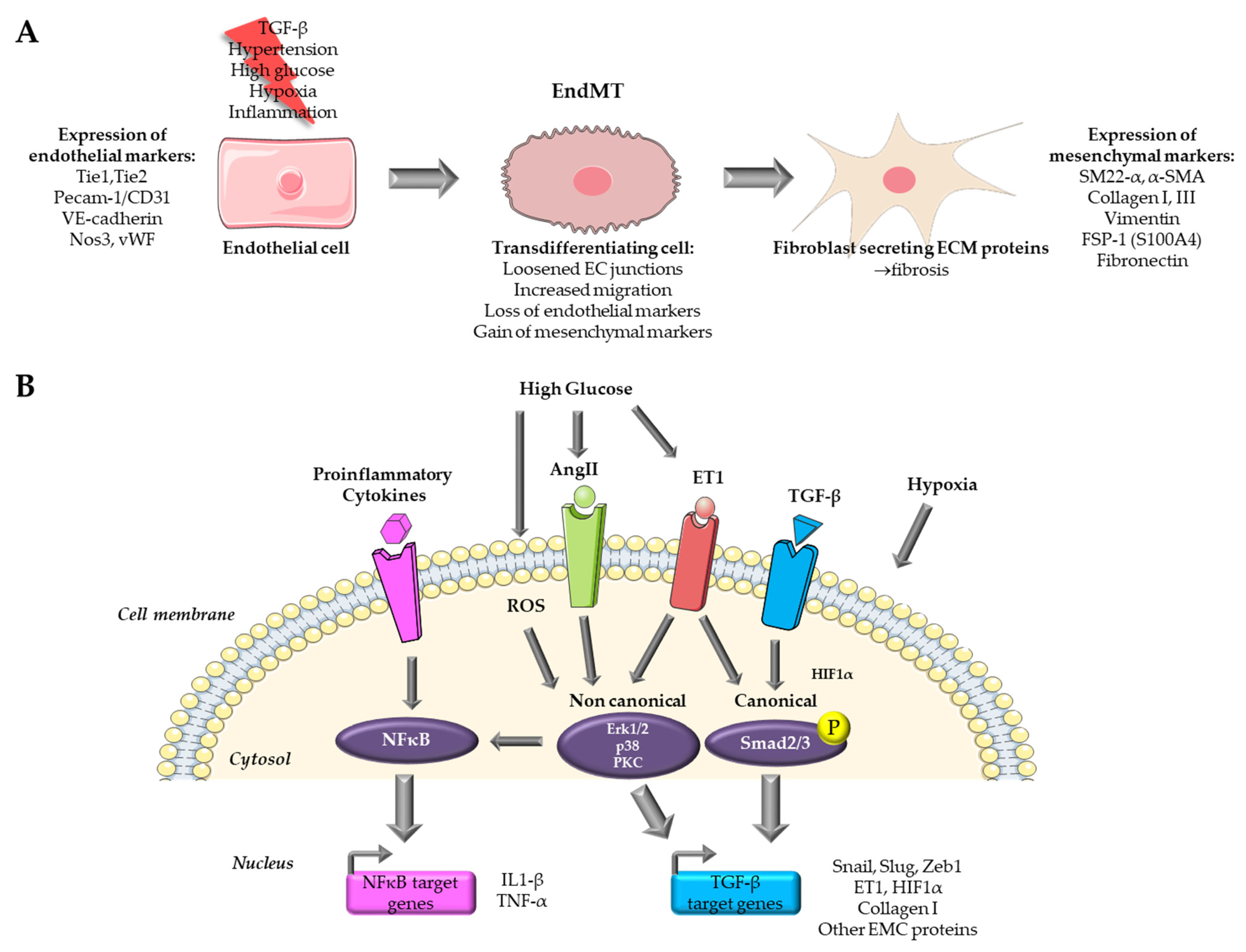
2. Endothelial to Mesenchymal Transition in Cardiovascular Diseases
3. Existing In Vitro Models of Endothelial to Mesenchymal Transition in Cardiovascular Diseases
3.1. Cytokine Based In Vitro Models
3.1.1. TGF-β Signaling
3.1.2. TGF-β Isoform Specific Effects to Induce EndMT
3.1.3. EndMT Induction Level Depend on Endothelial Cell Origin
3.1.4. Induction of EndMT by Co-Treatment with Proinflammatory Signals
3.2. Hypoxia-Based In Vitro Models
3.3. High Glucose Based In Vitro Models of Organ Fibrosis in Diabetes Mellitus
3.4. Important Parameters to Consider
3.4.1. Purity of Cell Preparation
3.4.2. Heterogeneity of Induced EndMT Phenotypes
4. Existing In Vivo Models of EndoMT in Cardiovascular Diseases
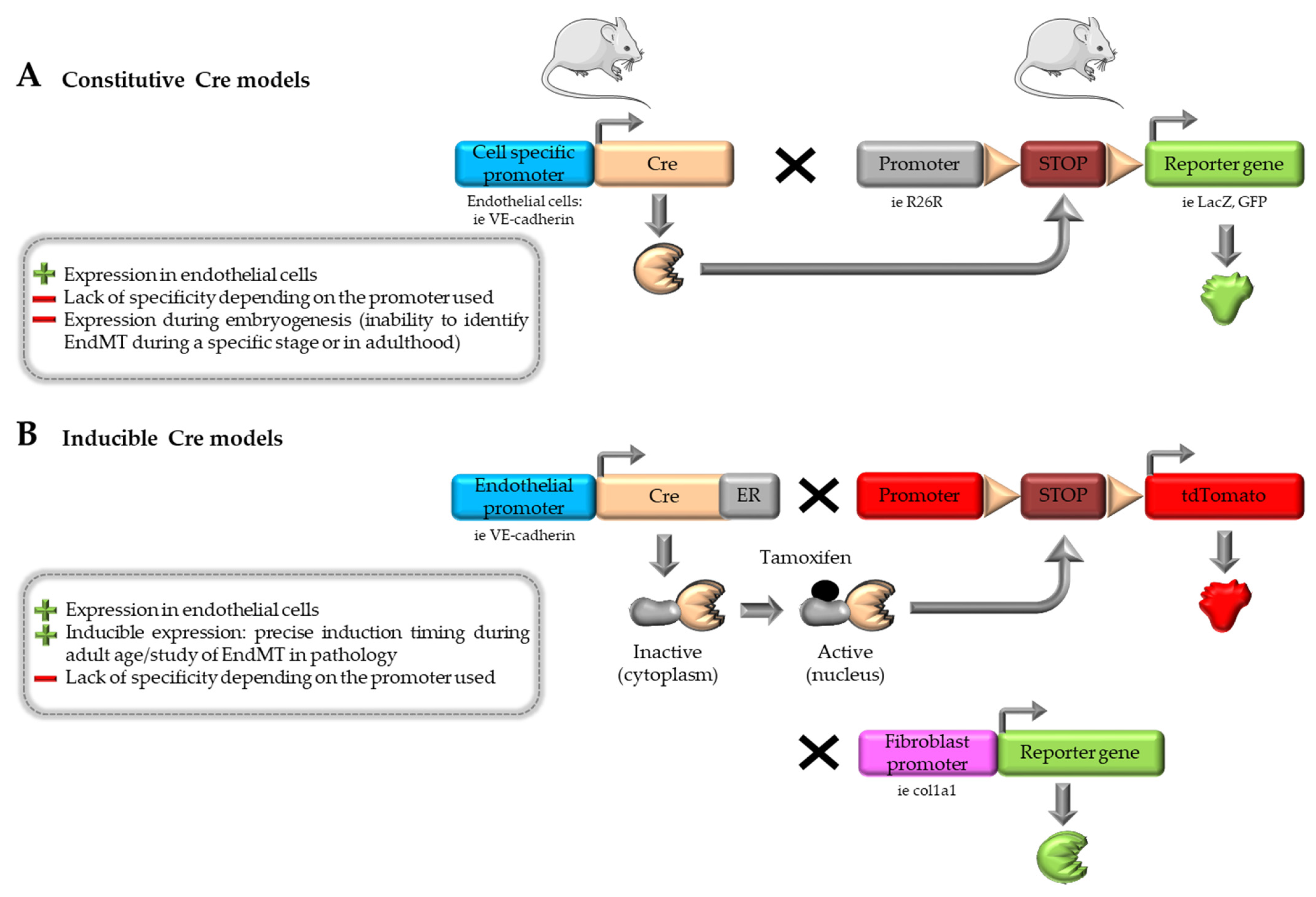
4.1. Cre-loxP System
4.2. Inducible Cre-loxP System and Multiple Reporter Models
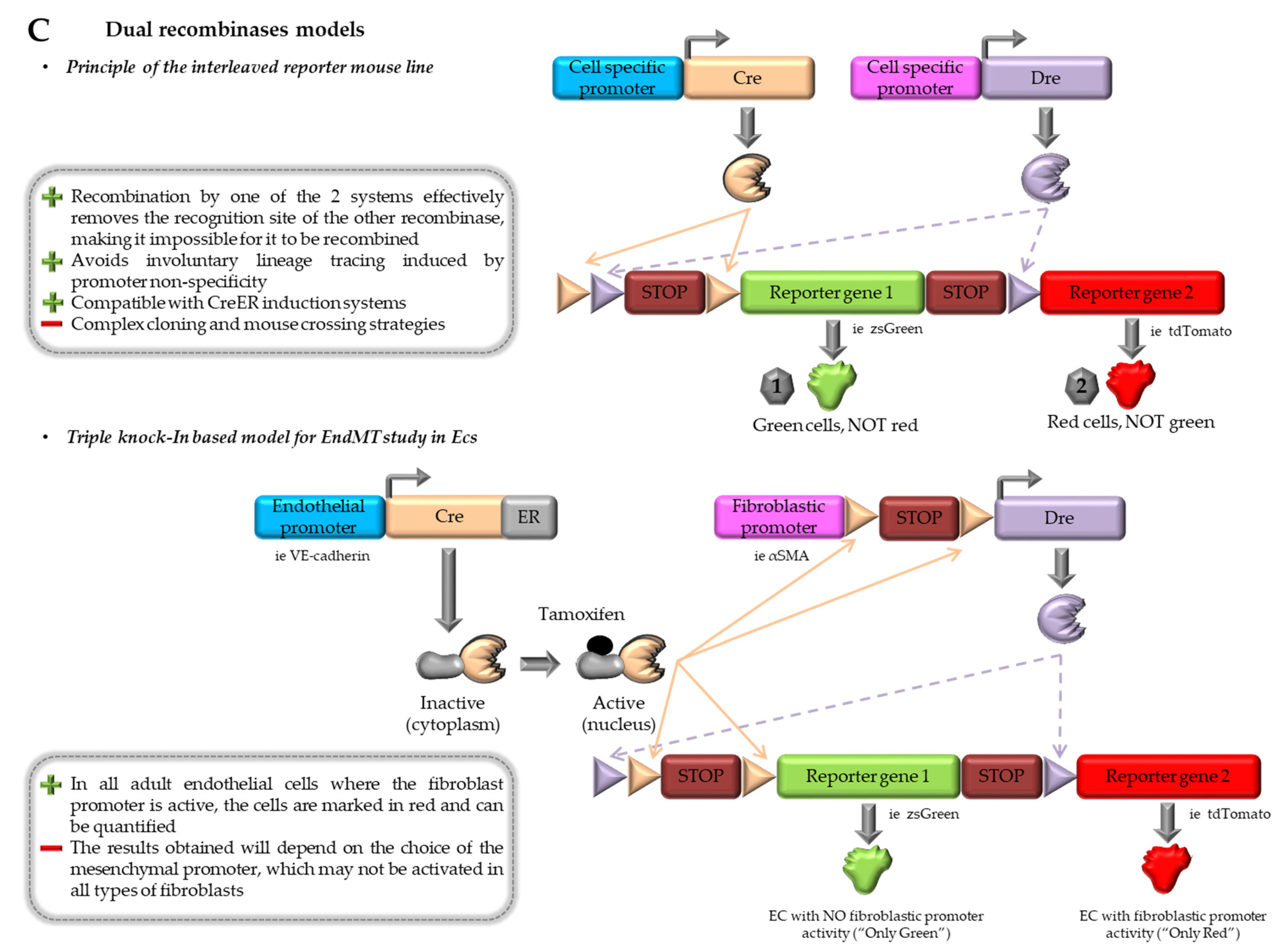
4.3. Combination of Dre-rox and Cre-loxP Models
4.4. Contributions and Limitations of In vivo Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Schimmel, K.; Ichimura, K.; Reddy, S.; Haddad, F.; Spiekerkoetter, E. Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front. Cardiovasc. Med. 2022, 9, 886553. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Imanaka-Yoshida, K. The Pathogenesis of Cardiac Fibrosis: A Review of Recent Progress. Int. J. Mol. Sci. 2022, 23, 2617. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac Fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Kruszewska, J.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Remodeling and Fibrosis of the Cardiac Muscle in the Course of Obesity-Pathogenesis and Involvement of the Extracellular Matrix. Int. J. Mol. Sci. 2022, 23, 4195. [Google Scholar] [CrossRef] [PubMed]
- Valero-Muñoz, M.; Oh, A.; Faudoa, E.; Bretón-Romero, R.; El Adili, F.; Bujor, A.; Sam, F. Endothelial-Mesenchymal Transition in Heart Failure with a Preserved Ejection Fraction: Insights into the Cardiorenal Syndrome. Circ. Heart Fail. 2021, 14, e008372. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Nkennor, B.; Mastikhina, O.; Soon, K.; Nunes, S.S. Endothelium-Mediated Contributions to Fibrosis. Semin. Cell Dev. Biol. 2020, 101, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Anbara, T.; Sharifi, M.; Aboutaleb, N. Endothelial to Mesenchymal Transition in the Cardiogenesis and Cardiovascular Diseases. Curr. Cardiol. Rev. 2020, 16, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Shan, D.; Cui, K.; Li, K.; Zhu, B.; Wu, H.; Wang, B.; Wong, S.; Norton, V.; Dong, Y.; et al. The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells 2022, 11, 1834. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Hong, L.; Du, X.; Li, W.; Mao, Y.; Sun, L.; Li, X. EndMT: A Promising and Controversial Field. Eur. J. Cell Biol. 2018, 97, 493–500. [Google Scholar] [CrossRef]
- Gaikwad, A.V.; Eapen, M.S.; McAlinden, K.D.; Chia, C.; Larby, J.; Myers, S.; Dey, S.; Haug, G.; Markos, J.; Glanville, A.R.; et al. Endothelial to Mesenchymal Transition (EndMT) and Vascular Remodeling in Pulmonary Hypertension and Idiopathic Pulmonary Fibrosis. Expert. Rev. Respir. Med. 2020, 14, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Gorelova, A.; Berman, M.; Al Ghouleh, I. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxid. Redox Signal. 2021, 34, 891–914. [Google Scholar] [CrossRef] [PubMed]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-Mesenchymal Transition in Atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial Cell-Derived Endothelin-1 Promotes Cardiac Fibrosis in Diabetic Hearts through Stimulation of Endothelial-to-Mesenchymal Transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.-N.; Lv, L.-L.; Zhang, J.-D.; Dai, H.-Y.; Li, Q.; Zheng, M.; Ni, J.; Ma, K.-L.; Liu, B.-C. Effects of Angiotensin II Receptor Blocker on Myocardial Endothelial-to-Mesenchymal Transition in Diabetic Rats. Int. J. Cardiol. 2013, 162, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH Oxidase-2 Promotes Interstitial Cardiac Fibrosis and Diastolic Dysfunction through Proinflammatory Effects and Endothelial-Mesenchymal Transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Wang, H.; Chakrabarti, S. Endothelial-to-Mesenchymal Transition: An Underappreciated Mediator of Diabetic Complications. Front. Endocrinol. 2023, 14, 1050540. [Google Scholar] [CrossRef]
- Charytan, D.M.; Padera, R.; Helfand, A.M.; Zeisberg, M.; Xu, X.; Liu, X.; Himmelfarb, J.; Cinelli, A.; Kalluri, R.; Zeisberg, E.M. Increased Concentration of Circulating Angiogenesis and Nitric Oxide Inhibitors Induces Endothelial to Mesenchymal Transition and Myocardial Fibrosis in Patients with Chronic Kidney Disease. Int. J. Cardiol. 2014, 176, 99–109. [Google Scholar] [CrossRef]
- Massagué, J.; Sheppard, D. TGF-β Signaling in Health and Disease. Cell 2023, 186, 4007–4037. [Google Scholar] [CrossRef]
- Goumans, M.-J.; Ten Dijke, P. TGF-β Signaling in Control of Cardiovascular Function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β Signaling in Fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Azhar, M.; Runyan, R.B.; Gard, C.; Sanford, L.P.; Miller, M.L.; Andringa, A.; Pawlowski, S.; Rajan, S.; Doetschman, T. Ligand-Specific Function of Transforming Growth Factor Beta in Epithelial-Mesenchymal Transition in Heart Development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2009, 238, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Sabbineni, H.; Verma, A.; Somanath, P.R. Isoform-Specific Effects of Transforming Growth Factor β on Endothelial-to-Mesenchymal Transition. J. Cell. Physiol. 2018, 233, 8418–8428. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming Growth Factor-Β2 Promotes Snail-Mediated Endothelial-Mesenchymal Transition through Convergence of Smad-Dependent and Smad-Independent Signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef]
- Ursoli Ferreira, F.; Eduardo Botelho Souza, L.; Hassibe Thomé, C.; Tomazini Pinto, M.; Origassa, C.; Salustiano, S.; Marcel Faça, V.; Olsen Câmara, N.; Kashima, S.; Tadeu Covas, D. Endothelial Cells Tissue-Specific Origins Affects Their Responsiveness to TGF-Β2 during Endothelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 458. [Google Scholar] [CrossRef]
- Pinto, M.T.; Covas, D.T.; Kashima, S.; Rodrigues, C.O. Endothelial Mesenchymal Transition: Comparative Analysis of Different Induction Methods. Biol. Proced. Online 2016, 18, 10. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Liu, Y.; Zhang, Y.; Wang, Y.; Peng, Y.; Ding, Y. Diosmetin-7-O-β-D-Glucopyranoside Suppresses Endothelial-Mesenchymal Transformation through Endoplasmic Reticulum Stress in Cardiac Fibrosis. Clin. Exp. Pharmacol. Physiol. 2023, 50, 789–805. [Google Scholar] [CrossRef]
- Wang, B.; Ge, Z.; Wu, Y.; Zha, Y.; Zhang, X.; Yan, Y.; Xie, Y. MFGE8 Is Down-Regulated in Cardiac Fibrosis and Attenuates Endothelial-Mesenchymal Transition through Smad2/3-Snail Signalling Pathway. J. Cell. Mol. Med. 2020, 24, 12799–12812. [Google Scholar] [CrossRef]
- Lai, Y.-J.; Tsai, F.-C.; Chang, G.-J.; Chang, S.-H.; Huang, C.-C.; Chen, W.-J.; Yeh, Y.-H. miR-181b Targets Semaphorin 3A to Mediate TGF-β-Induced Endothelial-Mesenchymal Transition Related to Atrial Fibrillation. J. Clin. Investig. 2022, 132, e142548. [Google Scholar] [CrossRef]
- Wilhelmi, T.; Xu, X.; Tan, X.; Hulshoff, M.S.; Maamari, S.; Sossalla, S.; Zeisberg, M.; Zeisberg, E.M. Serelaxin Alleviates Cardiac Fibrosis through Inhibiting Endothelial-to-Mesenchymal Transition via RXFP1. Theranostics 2020, 10, 3905–3924. [Google Scholar] [CrossRef]
- Chen, H.; Liu, Y.; Gui, Q.; Zhu, X.; Zeng, L.; Meng, J.; Qing, J.; Gao, L.; Jackson, A.O.; Feng, J.; et al. Ghrelin Attenuates Myocardial Fibrosis after Acute Myocardial Infarction via Inhibiting Endothelial-to Mesenchymal Transition in Rat Model. Peptides 2019, 111, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Rinastiti, P.; Ikeda, K.; Rahardini, E.P.; Miyagawa, K.; Tamada, N.; Kuribayashi, Y.; Hirata, K.-I.; Emoto, N. Loss of Family with Sequence Similarity 13, Member A Exacerbates Pulmonary Hypertension through Accelerating Endothelial-to-Mesenchymal Transition. PLoS ONE 2020, 15, e0226049. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Liu, L.; Yu, Y.; Zhang, R.; Li, Y.; Cao, W.; Xiao, Y.; Fang, G.; Li, Z.; Wang, X.; et al. Inhibition of BRD4 Attenuates Transverse Aortic Constriction- and TGF-β-Induced Endothelial-Mesenchymal Transition and Cardiac Fibrosis. J. Mol. Cell. Cardiol. 2019, 127, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hu, J.; Liu, L. MiR-200a Modulates TGF-Β1-Induced Endothelial-to-Mesenchymal Shift via Suppression of GRB2 in HAECs. Biomed. Pharmacother. 2017, 95, 215–222. [Google Scholar] [CrossRef]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; van de Pol, V.; Geerts, M.E.; de Vries, M.R.; Janson, S.G.; van Dam, H.; Lindeman, J.H.; Goumans, M.-J.; Ten Dijke, P. Inflammation Induces Endothelial-to-Mesenchymal Transition and Promotes Vascular Calcification through Downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef]
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2357–2369. [Google Scholar] [CrossRef]
- Pinto, M.T.; Ferreira Melo, F.U.; Malta, T.M.; Rodrigues, E.S.; Plaça, J.R.; Silva, W.A.; Panepucci, R.A.; Covas, D.T.; de Oliveira Rodrigues, C.; Kashima, S. Endothelial Cells from Different Anatomical Origin Have Distinct Responses during SNAIL/TGF-Β2-Mediated Endothelial-Mesenchymal Transition. Am. J. Transl. Res. 2018, 10, 4065–4081. [Google Scholar]
- Monteiro, J.P.; Rodor, J.; Caudrillier, A.; Scanlon, J.P.; Spiroski, A.-M.; Dudnakova, T.; Pflüger-Müller, B.; Shmakova, A.; von Kriegsheim, A.; Deng, L.; et al. MIR503HG Loss Promotes Endothelial-to-Mesenchymal Transition in Vascular Disease. Circ. Res. 2021, 128, 1173–1190. [Google Scholar] [CrossRef]
- Yoshimatsu, Y.; Wakabayashi, I.; Kimuro, S.; Takahashi, N.; Takahashi, K.; Kobayashi, M.; Maishi, N.; Podyma-Inoue, K.A.; Hida, K.; Miyazono, K.; et al. TNF-α Enhances TGF-β-Induced Endothelial-to-Mesenchymal Transition via TGF-β Signal Augmentation. Cancer Sci. 2020, 111, 2385–2399. [Google Scholar] [CrossRef]
- Maleszewska, M.; Moonen, J.-R.A.J.; Huijkman, N.; van de Sluis, B.; Krenning, G.; Harmsen, M.C. IL-1β and TGFβ2 Synergistically Induce Endothelial to Mesenchymal Transition in an NFκB-Dependent Manner. Immunobiology 2013, 218, 443–454. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Moon, H.; Kim, R.; Kim, M.; Jeong, S.; Kim, H.; Kim, S.H.; Hwang, S.S.; Lee, M.Y.; et al. Epigallocatechin-3-Gallate Attenuates Myocardial Dysfunction via Inhibition of Endothelial-to-Mesenchymal Transition. Antioxidants 2023, 12, 1059. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, C.; Chen, J.; Yang, M.; Afzal, T.A.; An, W.; Maguire, E.M.; He, S.; Luo, J.; Wang, X.; et al. miRNA-200c-3p Promotes Endothelial to Mesenchymal Transition and Neointimal Hyperplasia in Artery Bypass Grafts. J. Pathol. 2021, 253, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Miscianinov, V.; Martello, A.; Rose, L.; Parish, E.; Cathcart, B.; Mitić, T.; Gray, G.A.; Meloni, M.; Al Haj Zen, A.; Caporali, A. MicroRNA-148b Targets the TGF-β Pathway to Regulate Angiogenesis and Endothelial-to-Mesenchymal Transition during Skin Wound Healing. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1996–2007. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Kessler, S.P.; West, G.A.; Bhilocha, S.; de la Motte, C.; Sadler, T.M.; Gopalan, B.; Stylianou, E.; Fiocchi, C. Inflammation-Induced Endothelial-to-Mesenchymal Transition: A Novel Mechanism of Intestinal Fibrosis. Am. J. Pathol. 2011, 179, 2660–2673. [Google Scholar] [CrossRef]
- Lin, K.; Luo, W.; Yan, J.; Shen, S.; Shen, Q.; Wang, J.; Guan, X.; Wu, G.; Huang, W.; Liang, G. TLR2 Regulates Angiotensin II-Induced Vascular Remodeling and EndMT through NF-κB Signaling. Aging 2020, 13, 2553–2574. [Google Scholar] [CrossRef]
- Jordan, N.P.; Tingle, S.J.; Shuttleworth, V.G.; Cooke, K.; Redgrave, R.E.; Singh, E.; Glover, E.K.; Ahmad Tajuddin, H.B.; Kirby, J.A.; Arthur, H.M.; et al. MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis. Int. J. Mol. Sci. 2021, 22, 8629. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Zhai, C.; Yang, X.-Y.; Li, H.-B.; Wu, L.-L.; Li, L. Knockdown of CD146 Promotes Endothelial-to-Mesenchymal Transition via Wnt/β-Catenin Pathway. PLoS ONE 2022, 17, e0273542. [Google Scholar] [CrossRef]
- Krishnamoorthi, M.K.; Thandavarayan, R.A.; Youker, K.A.; Bhimaraj, A. An In Vitro Platform to Study Reversible Endothelial-to-Mesenchymal Transition. Front. Pharmacol. 2022, 13, 912660. [Google Scholar] [CrossRef]
- Li, Z.; Kong, X.; Zhang, Y.; Zhang, Y.; Yu, L.; Guo, J.; Xu, Y. Dual Roles of Chromatin Remodeling Protein BRG1 in Angiotensin II-Induced Endothelial-Mesenchymal Transition. Cell Death Dis. 2020, 11, 549. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Lv, Z.; Luo, X.; Deng, W.; Zou, T.; Zhang, Y.; Sang, W.; Wang, X. The miR-214-3p/c-Ski Axis Modulates Endothelial-Mesenchymal Transition in Human Coronary Artery Endothelial Cells in Vitro and in Mice Model in Vivo. Hum. Cell 2022, 35, 486–497. [Google Scholar] [CrossRef]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-Induced Epigenetic Modifications Are Associated with Cardiac Tissue Fibrosis and the Development of a Myofibroblast-like Phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. Snail Is a Direct Target of Hypoxia-Inducible Factor 1α (HIF1α) in Hypoxia-Induced Endothelial to Mesenchymal Transition of Human Coronary Endothelial Cells. J. Biol. Chem. 2015, 290, 16653–16664. [Google Scholar] [CrossRef] [PubMed]
- Sniegon, I.; Prieß, M.; Heger, J.; Schulz, R.; Euler, G. Endothelial Mesenchymal Transition in Hypoxic Microvascular Endothelial Cells and Paracrine Induction of Cardiomyocyte Apoptosis Are Mediated via TGFβ1/SMAD Signaling. Int. J. Mol. Sci. 2017, 18, 2290. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Yang, K.; Wang, X.; Chen, L.; Gill, P.S.; Ha, T.; Liu, L.; Lewis, N.H.; Williams, D.L.; Li, C. Lactate Promotes Endothelial-to-Mesenchymal Transition via Snail1 Lactylation after Myocardial Infarction. Sci. Adv. 2023, 9, eadc9465. [Google Scholar] [CrossRef] [PubMed]
- Helmke, A.; Casper, J.; Nordlohne, J.; David, S.; Haller, H.; Zeisberg, E.M.; von Vietinghoff, S. Endothelial-to-Mesenchymal Transition Shapes the Atherosclerotic Plaque and Modulates Macrophage Function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 2278–2289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-H.; Yu, F.-C.; Li, Y.; Wei, Q.; Song, S.-S.; Zhou, F.-P.; Tong, J.-Y. Prolyl 4-Hydroxylase Domain Protein 3 Overexpression Improved Obstructive Sleep Apnea-Induced Cardiac Perivascular Fibrosis Partially by Suppressing Endothelial-to-Mesenchymal Transition. J. Am. Heart Assoc. 2017, 6, e006680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Yan, L.; Du, W.; Zhang, X.; Zhang, M.; Chen, H.; Zhang, Y.; Zhou, J.; Sun, H.; et al. Bone Morphogenetic Protein-7 Inhibits Endothelial-Mesenchymal Transition in Pulmonary Artery Endothelial Cell under Hypoxia. J. Cell. Physiol. 2018, 233, 4077–4090. [Google Scholar] [CrossRef]
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C.; et al. The Role of PDGF-B/TGF-Β1/Neprilysin Network in Regulating Endothelial-to-Mesenchymal Transition in Pulmonary Artery Remodeling. Cell. Signal. 2016, 28, 1489–1501. [Google Scholar] [CrossRef]
- Woo, K.V.; Shen, I.Y.; Weinheimer, C.J.; Kovacs, A.; Nigro, J.; Lin, C.-Y.; Chakinala, M.; Byers, D.E.; Ornitz, D.M. Endothelial FGF Signaling Is Protective in Hypoxia-Induced Pulmonary Hypertension. J. Clin. Investig. 2021, 131, e141467. [Google Scholar] [CrossRef]
- Zhang, B.; Niu, W.; Dong, H.-Y.; Liu, M.-L.; Luo, Y.; Li, Z.-C. Hypoxia Induces Endothelial-mesenchymal Transition in Pulmonary Vascular Remodeling. Int. J. Mol. Med. 2018, 42, 270–278. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Q.; Zhan, Y.; Ke, J.; Lv, P.; Huang, J. The Role of miR-328 in High Glucose-Induced Endothelial-to-Mesenchymal Transition in Human Umbilical Vein Endothelial Cells. Life Sci. 2018, 207, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Y.; Lv, R.-J.; Zhang, W.; Yan, Y.-G.; Li, P.; Dong, W.-Q.; Liu, X.; Liang, E.-S.; Tian, H.-L.; Lu, Q.-H.; et al. Inhibition of Myocyte-Specific Enhancer Factor 2A Improved Diabetic Cardiac Fibrosis Partially by Regulating Endothelial-to-Mesenchymal Transition. Oncotarget 2016, 7, 31053–31066. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Suriguga; Gong, M.; Liu, W.-J.; Cui, N.-X.; Wang, Y.; Du, X.; Yi, Z.-C. High Glucose Induced Endothelial to Mesenchymal Transition in Human Umbilical Vein Endothelial Cell. Exp. Mol. Pathol. 2017, 102, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.-H.; Lee, C.-H.; Cheng, C.-I.; Fang, Y.-N.; Chung, S.-Y.; Chen, S.-M.; Lin, C.-J.; Wu, C.-J.; Hang, C.-L.; Chen, W.-Y. Liraglutide Inhibits Endothelial-to-Mesenchymal Transition and Attenuates Neointima Formation after Endovascular Injury in Streptozotocin-Induced Diabetic Mice. Cells 2019, 8, 589. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yao, Y.; Shi, S.; Zhou, M.; Zhou, Y.; Wang, M.; Chiu, J.-J.; Huang, Z.; Zhang, W.; Liu, M.; et al. Inhibition of miR-21 Alleviated Cardiac Perivascular Fibrosis via Repressing EndMT in T1DM. J. Cell. Mol. Med. 2020, 24, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin Alleviates Cardiac Fibrosis through Suppressing EndMT and Fibroblast Activation via AMPKα/TGF-β/Smad Signalling in Type 2 Diabetic Rats. J. Cell. Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhong, Z.; Gu, J.; Nan, K.; Zhu, M.; Miao, C. Ets1 Associates with KMT5A to Participate in High Glucose-Mediated EndMT via Upregulation of PFN2 Expression in Diabetic Nephropathy. Mol. Med. 2021, 27, 74. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Yao, J.; Xie, H.; Wang, C.; Chen, J.; Wei, K.; Ji, Y.; Liu, L. MicroRNA-195-5p Downregulation Inhibits Endothelial Mesenchymal Transition and Myocardial Fibrosis in Diabetic Cardiomyopathy by Targeting Smad7 and Inhibiting Transforming Growth Factor Beta 1-Smads-Snail Pathway. Front. Physiol. 2021, 12, 709123. [Google Scholar] [CrossRef]
- Meng, Z.; Shen, W.; Yu, L.; Tong, F.; He, H.; Hu, Y.; Wu, W.; Liu, J. Bach1 Modulates AKT3 Transcription to Participate in Hyperglycaemia-Mediated EndMT in Vascular Endothelial Cells. Clin. Exp. Pharmacol. Physiol. 2023, 50, 443–452. [Google Scholar] [CrossRef]
- Hulshoff, M.S.; Schellinger, I.N.; Xu, X.; Fledderus, J.; Rath, S.K.; Wong, F.C.; Maamari, S.; Haunschild, J.; Krenning, G.; Raaz, U.; et al. miR-132-3p and KLF7 as Novel Regulators of Aortic Stiffening-Associated EndMT in Type 2 Diabetes Mellitus. Diabetol. Metab. Syndr. 2023, 15, 11. [Google Scholar] [CrossRef]
- Zhu, G.-H.; Li, R.; Zeng, Y.; Zhou, T.; Xiong, F.; Zhu, M. MicroRNA-142-3p Inhibits High-Glucose-Induced Endothelial-to-Mesenchymal Transition through Targeting TGF-Β1/Smad Pathway in Primary Human Aortic Endothelial Cells. Int. J. Clin. Exp. Pathol. 2018, 11, 1208–1217. [Google Scholar] [PubMed]
- Geng, H.; Guan, J. MiR-18a-5p Inhibits Endothelial-Mesenchymal Transition and Cardiac Fibrosis through the Notch2 Pathway. Biochem. Biophys. Res. Commun. 2017, 491, 329–336. [Google Scholar] [CrossRef]
- Yuan, C.; Ni, L.; Yang, X.; Zhang, C.; Wu, X. Calcium-Sensing Receptor Participates in High Glucose-Induced EndMT in Primary Human Aortic Endothelial Cells. Front. Physiol. 2020, 11, 629542. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, Y.; Xiong, Z.; Zhu, Z.; Gao, F.; Wang, T.; Man, W.; Sun, D.; Lin, J.; Li, T.; et al. Sirt6-Mediated Endothelial-to-Mesenchymal Transition Contributes Toward Diabetic Cardiomyopathy via the Notch1 Signaling Pathway. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 4801–4808. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-K.; Yu, Q.; Liu, Y.-J.; Du, S.-F.; Huang, L.-Y.; Xu, D.-H.; Ni, X.; Zhu, X.-Y. A Novel Role of Kallikrein-Related Peptidase 8 in the Pathogenesis of Diabetic Cardiac Fibrosis. Theranostics 2021, 11, 4207–4231. [Google Scholar] [CrossRef]
- Peng, H.; Li, Y.; Wang, C.; Zhang, J.; Chen, Y.; Chen, W.; Cao, J.; Wang, Y.; Hu, Z.; Lou, T. ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Sci. Rep. 2016, 6, 20304. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhao, J.; Wang, X.; Chen, Z.; Peng, K.; Lu, X.; Meng, L.; Liu, G.; Guan, G.; Wang, F. Serum Response Factor Induces Endothelial-Mesenchymal Transition in Glomerular Endothelial Cells to Aggravate Proteinuria in Diabetic Nephropathy. Physiol. Genom. 2016, 48, 711–718. [Google Scholar] [CrossRef]
- Yao, Y.; Li, Y.; Zeng, X.; Ye, Z.; Li, X.; Zhang, L. Losartan Alleviates Renal Fibrosis and Inhibits Endothelial-to-Mesenchymal Transition (EMT) Under High-Fat Diet-Induced Hyperglycemia. Front. Pharmacol. 2018, 9, 1213. [Google Scholar] [CrossRef]
- Liu, W.; Wu, Y.; Yu, F.; Hu, W.; Fang, X.; Hao, W. The Implication of Numb-Induced Notch Signaling in Endothelial-Mesenchymal Transition of Diabetic Nephropathy. J. Diabetes Complicat. 2018, 32, 889–899. [Google Scholar] [CrossRef]
- Chang, K.; Xie, Q.; Niu, J.; Gu, Y.; Zhao, Z.; Li, F.; Qin, Q.; Liu, X. Heparanase Promotes Endothelial-to-Mesenchymal Transition in Diabetic Glomerular Endothelial Cells through Mediating ERK Signaling. Cell Death Discov. 2022, 8, 67. [Google Scholar] [CrossRef]
- Ma, Z.; Zhu, L.; Liu, Y.; Wang, Z.; Yang, Y.; Chen, L.; Lu, Q. Lovastatin Alleviates Endothelial-to-Mesenchymal Transition in Glomeruli via Suppression of Oxidative Stress and TGF-Β1 Signaling. Front. Pharmacol. 2017, 8, 473. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhang, X.; Liu, Y.; Zhao, T.; Sun, Z.; Liu, P.; Xiang, Q.; Xiong, J.; Du, X.; Yang, X.; et al. Rutin Alleviates EndMT by Restoring Autophagy through Inhibiting HDAC1 via PI3K/AKT/mTOR Pathway in Diabetic Kidney Disease. Phytomed. Int. J. Phytother. Phytopharm. 2023, 112, 154700. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Xie, J.; Dai, Y.; Han, H. TFPI2 Suppresses the Interaction of TGF-Β2 Pathway Regulators to Promote Endothelial-Mesenchymal Transition in Diabetic Nephropathy. J. Biol. Chem. 2022, 298, 101725. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, J.; Lin, C. Sprouty-Related Proteins with EVH1 Domain (SPRED2) Prevents High-Glucose Induced Endothelial-Mesenchymal Transition and Endothelial Injury by Suppressing MAPK Activation. Bioengineered 2022, 13, 13882–13892. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Song, Y.; Huang, L.-L.; Tian, Y.-J.; Wang, X.-L.; Hua, L.-Y. m6A Transferase METTL3 Regulates Endothelial-Mesenchymal Transition in Diabetic Retinopathy via lncRNA SNHG7/KHSRP/MKL1 Axis. Genomics 2022, 114, 110498. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Xue, L.-D.; Di, Y.; Li, T.; Tian, Y.-J.; Song, Y. MSC-Derived Exosomal lncRNA SNHG7 Suppresses Endothelial-Mesenchymal Transition and Tube Formation in Diabetic Retinopathy via miR-34a-5p/XBP1 Axis. Life Sci. 2021, 272, 119232. [Google Scholar] [CrossRef]
- Wang, H.; Feng, Z.; Han, X.; Xing, Y.; Zhang, X. Downregulation of Acylglycerol Kinase Suppresses High-Glucose-Induced Endothelial-Mesenchymal Transition in Human Retinal Microvascular Endothelial Cells through Regulating the LPAR1/TGF-β/Notch Signaling Pathway. Can. J. Physiol. Pharmacol. 2022, 100, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zeng, Y.; Chen, J.; Cai, D.; Chen, C.; Zhang, S.; Chen, Z. miR-29a/b Cluster Suppresses High Glucose-Induced Endothelial-Mesenchymal Transition in Human Retinal Microvascular Endothelial Cells by Targeting Notch2. Exp. Ther. Med. 2019, 17, 3108–3116. [Google Scholar] [CrossRef]
- Cecoltan, S.; Ciortan, L.; Macarie, R.D.; Vadana, M.; Mihaila, A.C.; Tucureanu, M.; Vlad, M.-L.; Droc, I.; Gherghiceanu, M.; Simionescu, A.; et al. High Glucose Induced Changes in Human VEC Phenotype in a 3D Hydrogel Derived From Cell-Free Native Aortic Root. Front. Cardiovasc. Med. 2021, 8, 714573. [Google Scholar] [CrossRef]
- Li, Y.; Lui, K.O.; Zhou, B. Reassessing Endothelial-to-Mesenchymal Transition in Cardiovascular Diseases. Nat. Rev. Cardiol. 2018, 15, 445–456. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 Counteracts TGF-Beta1-Induced Epithelial-to-Mesenchymal Transition and Reverses Chronic Renal Injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-Mesenchymal Transition Contributes to Cardiac Fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Saxena, A.; Su, Y.; Frangogiannis, N.G. Lack of Specificity of Fibroblast-Specific Protein 1 in Cardiac Remodeling and Fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1363–H1372. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, Y.; Li, Y.; Pu, W.; Huang, X.; Tian, X.; Wang, Y.; Zhang, H.; Liu, Q.; Zhang, L.; et al. Enhancing the Precision of Genetic Lineage Tracing Using Dual Recombinases. Nat. Med. 2017, 23, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident Fibroblast Lineages Mediate Pressure Overload-Induced Cardiac Fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic Lineage Tracing Defines Myofibroblast Origin and Function in the Injured Heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-β Signaling Mediates Endothelial-to-Mesenchymal Transition (EndMT) during Vein Graft Remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.-R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to Mesenchymal Transition Is Common in Atherosclerotic Lesions and Is Associated with Plaque Instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.; Huang, X.; Liu, K.; Wang, Q.-D.; Chen, A.F.; Sun, K.; Lui, K.O.; Zhou, B. Seamless Genetic Recording of Transiently Activated Mesenchymal Gene Expression in Endothelial Cells During Cardiac Fibrosis. Circulation 2021, 144, 2004–2020. [Google Scholar] [CrossRef]
- Kurose, H. Cardiac Fibrosis and Fibroblasts. Cells 2021, 10, 1716. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mimouni, M.; Lajoix, A.-D.; Desmetz, C. Experimental Models to Study Endothelial to Mesenchymal Transition in Myocardial Fibrosis and Cardiovascular Diseases. Int. J. Mol. Sci. 2024, 25, 382. https://doi.org/10.3390/ijms25010382
Mimouni M, Lajoix A-D, Desmetz C. Experimental Models to Study Endothelial to Mesenchymal Transition in Myocardial Fibrosis and Cardiovascular Diseases. International Journal of Molecular Sciences. 2024; 25(1):382. https://doi.org/10.3390/ijms25010382
Chicago/Turabian StyleMimouni, Mohammed, Anne-Dominique Lajoix, and Caroline Desmetz. 2024. "Experimental Models to Study Endothelial to Mesenchymal Transition in Myocardial Fibrosis and Cardiovascular Diseases" International Journal of Molecular Sciences 25, no. 1: 382. https://doi.org/10.3390/ijms25010382
APA StyleMimouni, M., Lajoix, A.-D., & Desmetz, C. (2024). Experimental Models to Study Endothelial to Mesenchymal Transition in Myocardial Fibrosis and Cardiovascular Diseases. International Journal of Molecular Sciences, 25(1), 382. https://doi.org/10.3390/ijms25010382