
Cardiac-Specific Suppression of Valosin-Containing Protein Induces Progressive Heart Failure and Premature Mortality Correlating with Temporal Dysregulations in mTOR Complex 2 and Protein Phosphatase 1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sufficient VCP in Cardiomyocytes Is Fundamental for Mouse Development and Survival
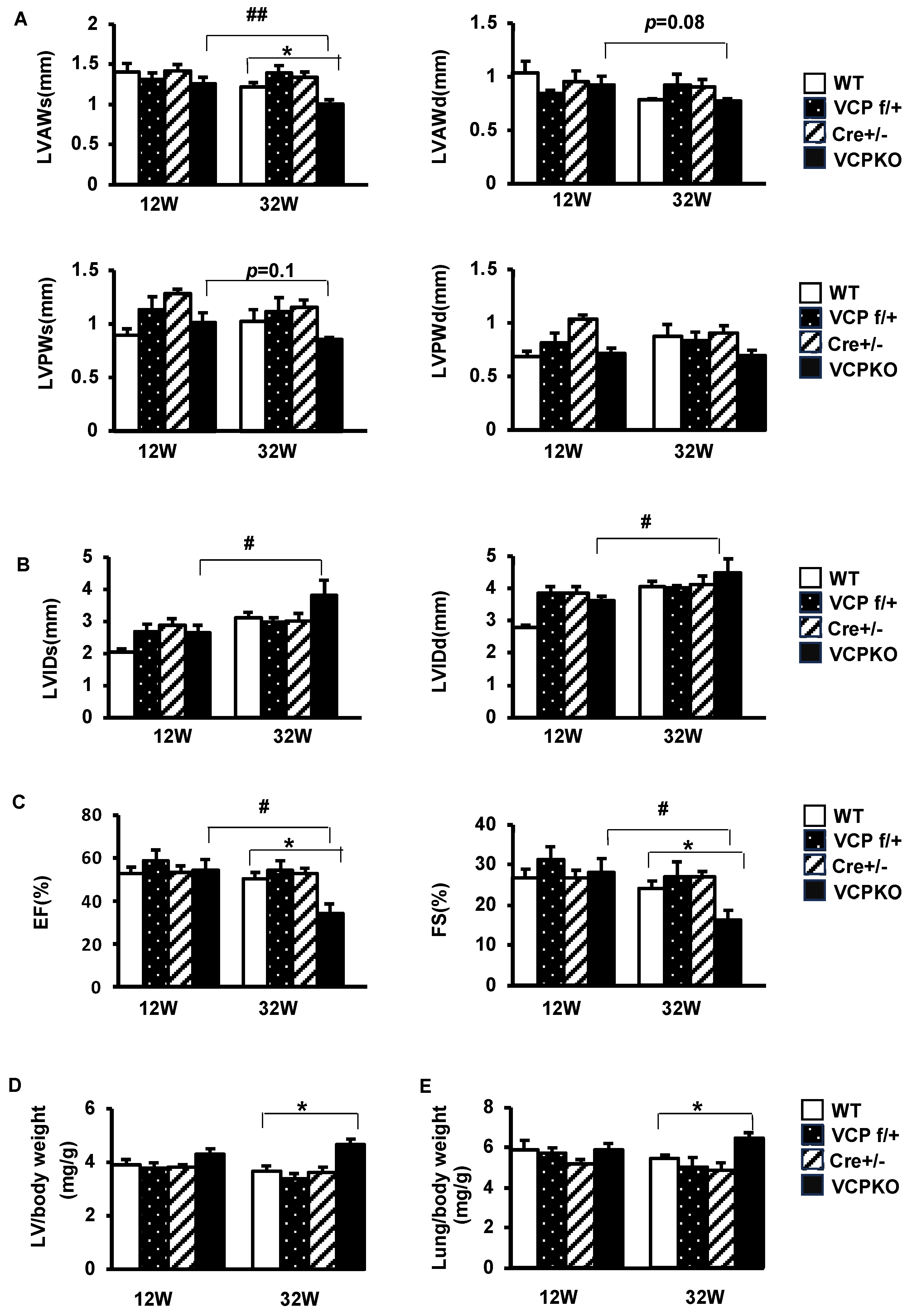
2.2. Suppression of Cardiomyocyte VCP Results in Progressive Cardiomyopathy and Declining Contractile Function
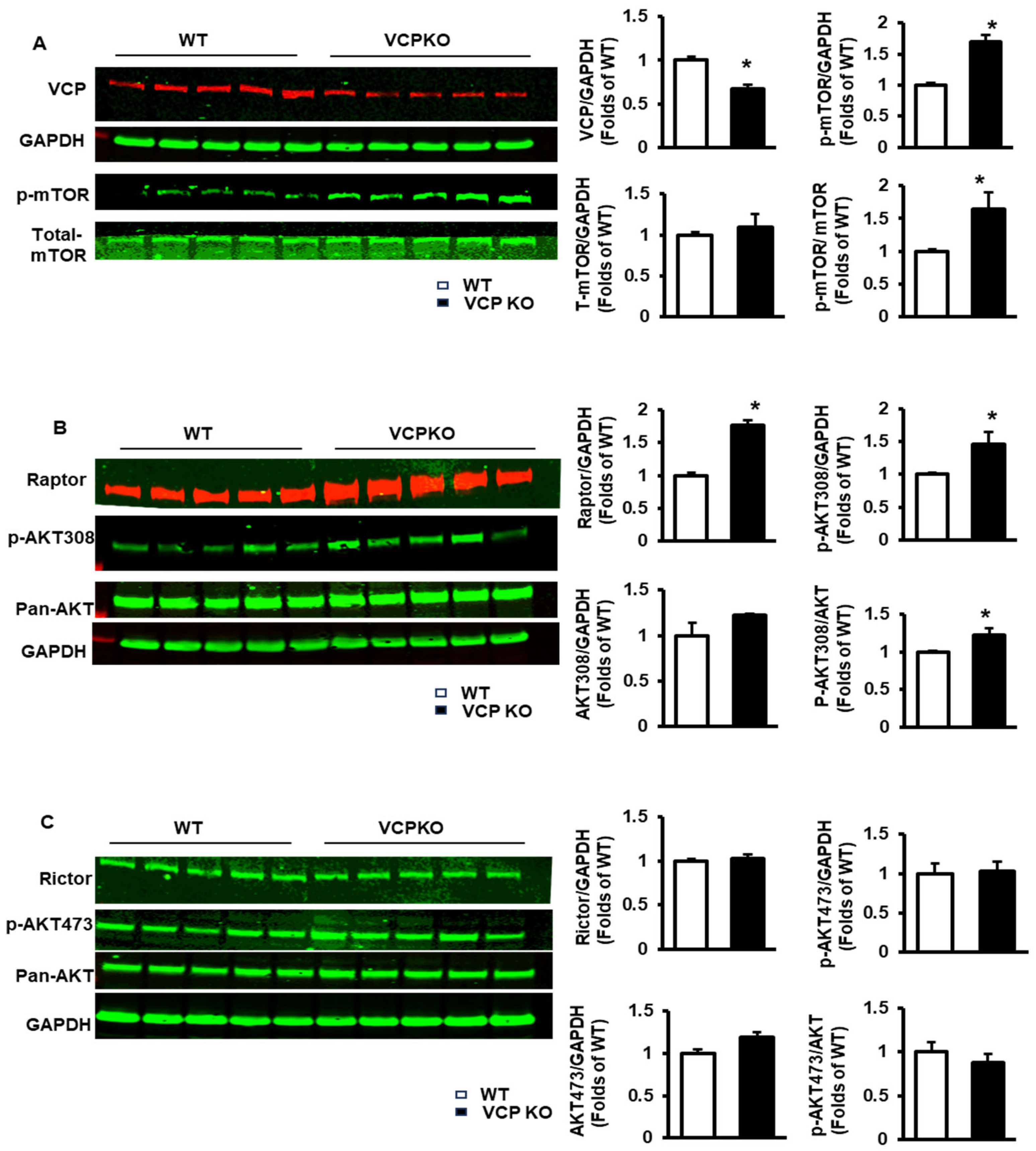
2.3. Reducing VCP Selectively Activates mTORC1 but Not mTORC2 at an Early Stage with Compensatory Cardiac Function
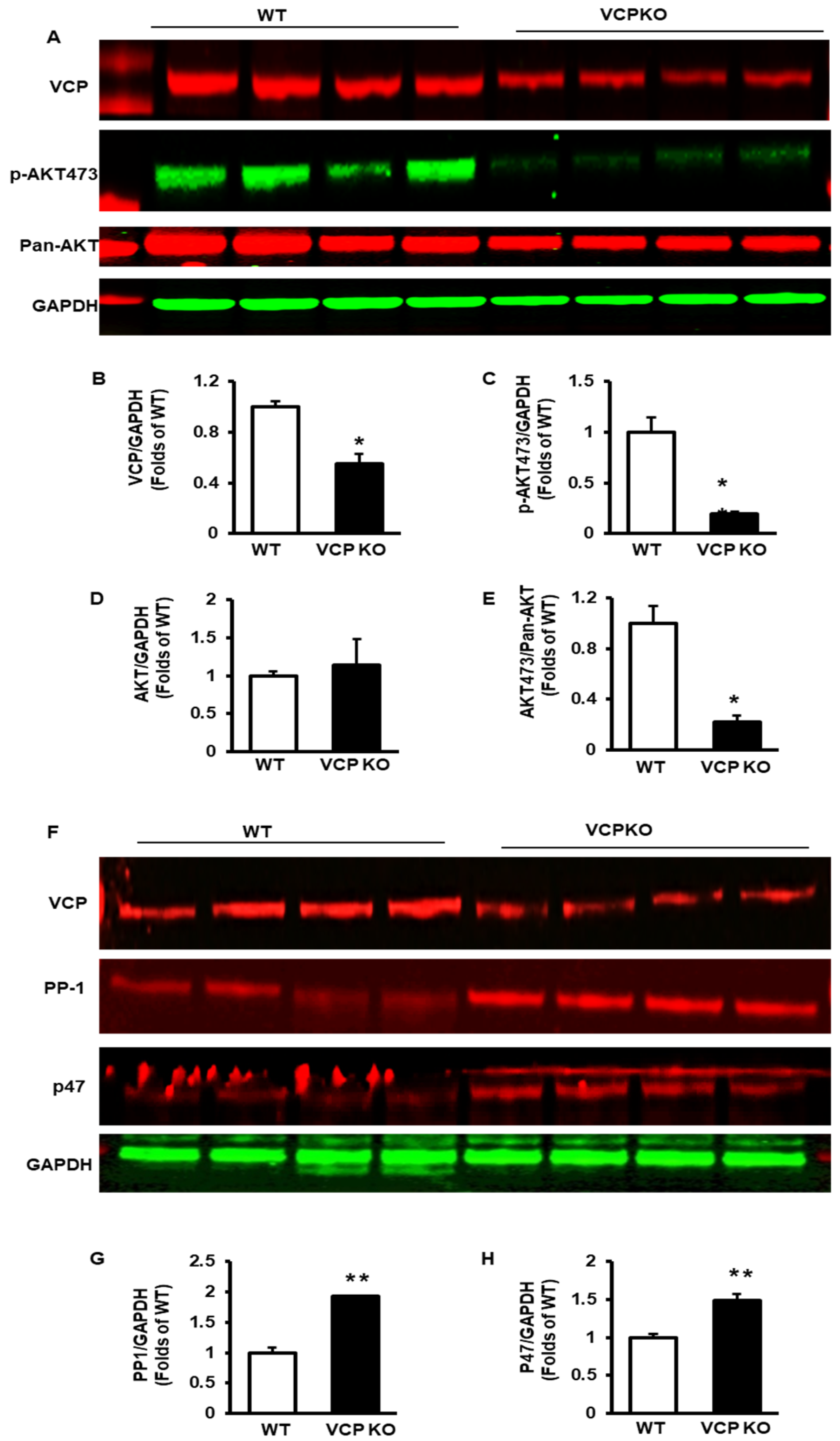
2.4. Prolonged Suppression of VCP Inhibits p-AKT473 and Increases PP1 and p47
2.5. Deficient VCP Expression in Cardiomyocytes Results in Temporal Alterations in the Subcellular Distribution of VCP and PP1
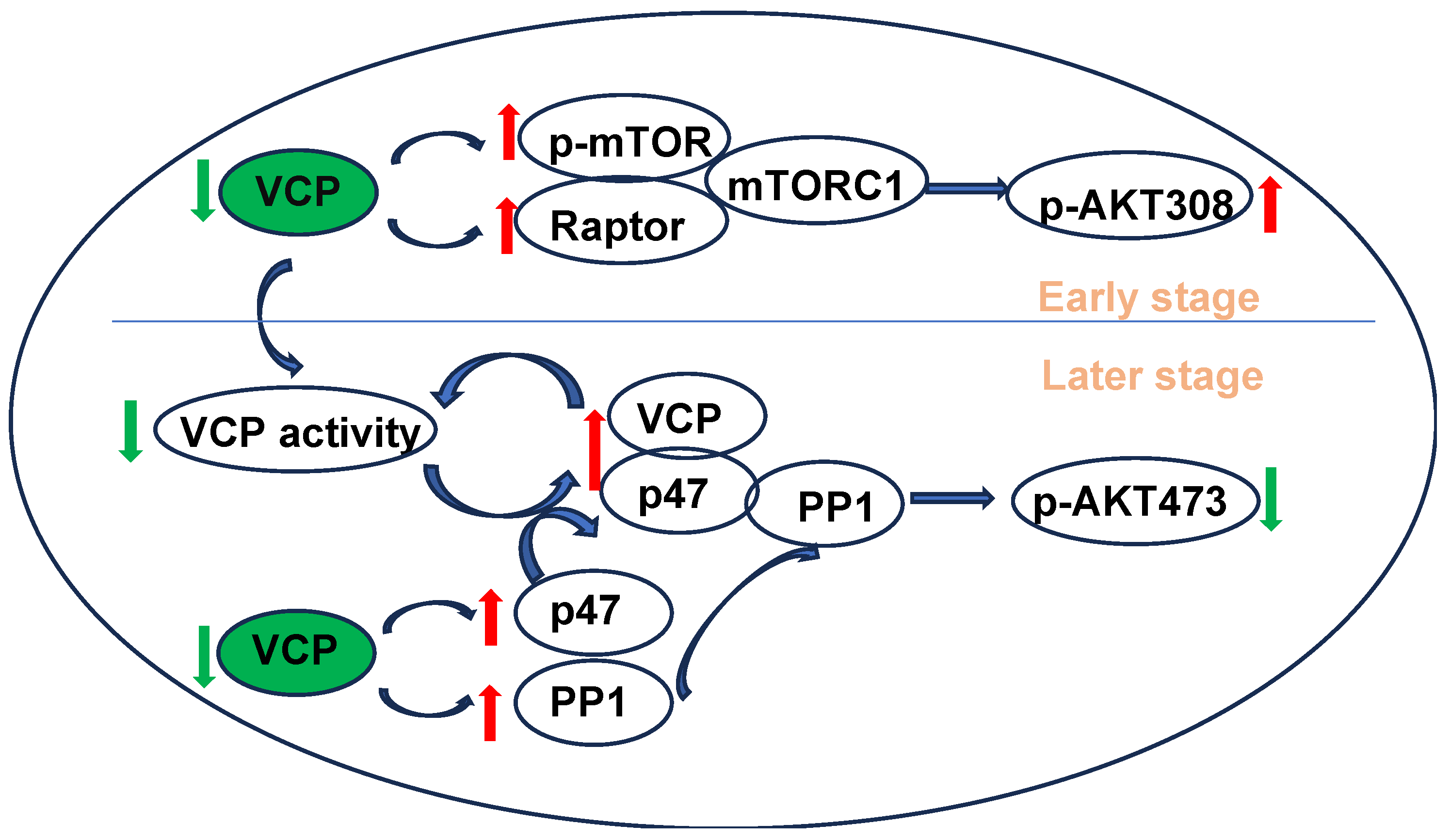
3. Discussion
4. Materials and Methods
4.1. Generation of Cardiac-Specific VCP KO Mice
4.2. Echocardiography
4.3. Histology
4.4. Protein Extraction and Subcellular Fraction
4.5. Western Blotting
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saffert, P.; Enenkel, C.; Wendler, P. Structure and Function of p97 and Pex1/6 Type II AAA+ Complexes. Front. Mol. Biosci. 2017, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Jiang, Y.; Zhang, D.; Liu, X.; Guo, J.; Zhao, J. Valosin-containing protein (VCP) promotes the growth, invasion, and metastasis of colorectal cancer through activation of STAT3 signaling. Mol. Cell. Biochem. 2016, 418, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Yeo, B.K.; Yu, S.W. Valosin-containing protein (VCP): Structure, functions, and implications in neurodegenerative diseases. Anim. Cells Syst. 2016, 20, 303–309. [Google Scholar] [CrossRef]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Barthelme, D.; Sauer, R.T. Origin and Functional Evolution of the Cdc48/p97/VCP AAA+ Protein Unfolding and Remodeling Machine. J. Mol. Biol. 2016, 428, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, K.; Halder, S.; Wiseman, K.; Vaz, B. Strategic role of the ubiquitin-dependent segregase p97 (VCP or Cdc48) in DNA replication. Chromosoma 2017, 126, 17–32. [Google Scholar] [CrossRef]
- Hanzelmann, P.; Schindelin, H. The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97. Front. Mol. Biosci. 2017, 4, 21. [Google Scholar] [CrossRef]
- Frohlich, K.U.; Fries, H.W.; Rudiger, M.; Erdmann, R.; Botstein, D.; Mecke, D. Yeast cell cycle protein CDC48p shows full-length homology to the mammalian protein VCP and is a member of a protein family involved in secretion, peroxisome formation, and gene expression. J. Cell Biol. 1991, 114, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Egerton, M.; Ashe, O.R.; Chen, D.; Druker, B.J.; Burgess, W.H.; Samelson, L.E. VCP, the mammalian homolog of cdc48, is tyrosine phosphorylated in response to T cell antigen receptor activation. EMBO J. 1992, 11, 3533–3540. [Google Scholar] [CrossRef]
- Schulte, R.J.; Campbell, M.A.; Fischer, W.H.; Sefton, B.M. Tyrosine phosphorylation of VCP, the mammalian homologue of the Saccharomyces cerevisiae CDC48 protein, is unusually sensitive to stimulation by sodium vanadate and hydrogen peroxide. J. Immunol. 1994, 153, 5465–5472. [Google Scholar] [CrossRef]
- Pleasure, I.T.; Black, M.M.; Keen, J.H. Valosin-containing protein, VCP, is a ubiquitous clathrin-binding protein. Nature 1993, 365, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, E.; Kerem, A.; Frohlich, K.U.; Diamant, N.; Bar-Nun, S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 2002, 22, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Tomita, Y.; Nakatsuka, S.; Hoshida, Y.; Myoui, A.; Yoshikawa, H.; Aozasa, K. VCP (p97) regulates NFkappaB signaling pathway, which is important for metastasis of osteosarcoma cell line. Jpn J. Cancer Res. 2002, 93, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.M.; Chen, E.; Longo, D.L.; Gorbea, C.M.; Li, C.C. Involvement of valosin-containing protein, an ATPase Co-purified with IkappaBalpha and 26 S proteasome, in ubiquitin-proteasome-mediated degradation of IkappaBalpha. J. Biol. Chem. 1998, 273, 3562–3573. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Latterich, M. The AAA team: Related ATPases with diverse functions. Trends Cell Biol. 1998, 8, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Qiu, H. Valosin-Containing Protein, a Calcium-Associated ATPase Protein, in Endoplasmic Reticulum and Mitochondrial Function and Its Implications for Diseases. Int. J. Mol. Sci. 2020, 21, 3842. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.H.; Shorter, J.G.; Seemann, J.; Pappin, D.; Warren, G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000, 19, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Alford, J.; Qiu, H. Structural and Functional Remodeling of Mitochondria in Cardiac Diseases. Int. J. Mol. Sci. 2021, 22, 4167. [Google Scholar] [CrossRef]
- Ju, J.S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef]
- Dai, R.M.; Li, C.C. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol. 2001, 3, 740–744. [Google Scholar] [CrossRef]
- Choy, N.; Wang, S.; Abbona, P.; Leffler, D.; Kimonis, V. Severe cardiomyopathy associated with the VCP p.R155C and c.177_187del MYBPC3 gene variants. Eur. J. Med. Genet. 2022, 65, 104480. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.K.; Xia, D. Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front. Mol. Biosci. 2016, 3, 79. [Google Scholar] [CrossRef] [PubMed]
- Malpass, K. Neurodegenerative disease: VCP mutations lead to defects in mitochondrial dynamics. Nat. Rev. Neurol. 2013, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.S.; Weihl, C.C. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: A disorder of autophagy. Hum. Mol. Genet. 2010, 19, R38–R45. [Google Scholar] [CrossRef]
- Weihl, C.C.; Pestronk, A.; Kimonis, V.E. Valosin-containing protein disease: Inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul. Disord. NMD 2009, 19, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, V.; Cristofani, R.; Cicardi, M.E.; Tedesco, B.; Crippa, V.; Chierichetti, M.; Casarotto, E.; Cozzi, M.; Mina, F.; Galbiati, M.; et al. Pathogenic variants of Valosin-containing protein induce lysosomal damage and transcriptional activation of autophagy regulators in neuronal cells. Neuropathol. Appl. Neurobiol. 2022, 48, e12818. [Google Scholar] [CrossRef] [PubMed]
- Lizano, P.; Rashed, E.; Kang, H.; Dai, H.; Sui, X.; Yan, L.; Qiu, H.; Depre, C. The valosin-containing protein promotes cardiac survival through the inducible isoform of nitric oxide synthase. Cardiovasc. Res. 2013, 99, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Lizano, P.; Rashed, E.; Stoll, S.; Zhou, N.; Wen, H.; Hays, T.T.; Qin, G.; Xie, L.H.; Depre, C.; Qiu, H. The valosin-containing protein is a novel mediator of mitochondrial respiration and cell survival in the heart in vivo. Sci. Rep. 2017, 7, 46324. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Xi, J.; Ma, B.; Leimena, C.; Behringer, E.J.; Qin, G.; Qiu, H. The Valosin-Containing Protein Protects the Heart Against Pathological Ca2+ Overload by Modulating Ca2+ Uptake Proteins. Toxicol. Sci. 2019, 171, 473–484. [Google Scholar] [CrossRef]
- Zhou, N.; Chen, X.; Xi, J.; Ma, B.; Leimena, C.; Stoll, S.; Qin, G.; Wang, C.; Qiu, H. Novel genomic targets of valosin-containing protein in protecting pathological cardiac hypertrophy. Sci. Rep. 2020, 10, 18098. [Google Scholar] [CrossRef]
- Zhou, N.; Stoll, S.; Qiu, H. VCP represses pathological cardiac hypertrophy. Aging 2017, 9, 2469–2470. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Chen, X.; Xi, J.; Ma, B.; Leimena, C.; Stoll, S.; Qin, G.; Wang, C.; Qiu, H. Genomic characterization reveals novel mechanisms underlying the valosin-containing protein-mediated cardiac protection against heart failure. Redox Biol. 2020, 36, 101662. [Google Scholar] [CrossRef]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Brink, M. mTOR, cardiomyocytes and inflammation in cardiac hypertrophy. Biochim. Et Biophys. Acta 2016, 1863, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, E.; Cannella, G. Regression of left ventricular hypertrophy in kidney transplant recipients: The potential role for inhibition of mammalian target of rapamycin. Transplant. Proc. 2010, 42, S41–S43. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Banjac, K.; Obradovic, M.; Zafirovic, S.; Essack, M.; Gluvic, Z.; Sunderic, M.; Nedic, O.; Isenovic, E.R. The involvement of Akt, mTOR, and S6K in the in vivo effect of IGF-1 on the regulation of rat cardiac Na(+)/K(+)-ATPase. Mol. Biol. Rep. 2024, 51, 517. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Gillick, J.L.; Neil, J.; Tobias, M.; Thwing, Z.E.; Murali, R. Distinct signaling mechanisms of mTORC1 and mTORC2 in glioblastoma multiforme: A tale of two complexes. Adv. Biol. Regul. 2015, 57, 64–74. [Google Scholar] [CrossRef]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef]
- Zhou, N.; Ma, B.; Stoll, S.; Hays, T.T.; Qiu, H. The valosin-containing protein is a novel repressor of cardiomyocyte hypertrophy induced by pressure overload. Aging Cell 2017, 16, 1168–1179. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, N.; Ma, B.; Wu, W.; Stoll, S.; Lai, L.; Qin, G.; Qiu, H. Functional Inhibition of Valosin-Containing Protein Induces Cardiac Dilation and Dysfunction in a New Dominant-Negative Transgenic Mouse Model. Cells 2021, 10, 2891. [Google Scholar] [CrossRef]
- MacDougall, L.K.; Jones, L.R.; Cohen, P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur. J. Biochem. 1991, 196, 725–734. [Google Scholar] [CrossRef]
- Weber, S.; Meyer-Roxlau, S.; Wagner, M.; Dobrev, D.; El-Armouche, A. Counteracting Protein Kinase Activity in the Heart: The Multiple Roles of Protein Phosphatases. Front. Pharmacol. 2015, 6, 270. [Google Scholar] [CrossRef]
- Ferreira, M.; Beullens, M.; Bollen, M.; Van Eynde, A. Functions and therapeutic potential of protein phosphatase 1: Insights from mouse genetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 16–30. [Google Scholar] [CrossRef]
- Klapproth, E.; Kammerer, S.; El-Armouche, A. Function and regulation of phosphatase 1 in healthy and diseased heart. Cell Signal 2022, 90, 110203. [Google Scholar] [CrossRef]
- Nicolaou, P.; Hajjar, R.J.; Kranias, E.G. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J. Mol. Cell Cardiol. 2009, 47, 365–371. [Google Scholar] [CrossRef]
- Meyer-Roxlau, S.; Lammle, S.; Opitz, A.; Kunzel, S.; Joos, J.P.; Neef, S.; Sekeres, K.; Sossalla, S.; Schondube, F.; Alexiou, K.; et al. Differential regulation of protein phosphatase 1 (PP1) isoforms in human heart failure and atrial fibrillation. Basic Res. Cardiol. 2017, 112, 43. [Google Scholar] [CrossRef]
- Mishra, S.; Gupta, R.C.; Tiwari, N.; Sharov, V.G.; Sabbah, H.N. Molecular mechanisms of reduced sarcoplasmic reticulum Ca(2+) uptake in human failing left ventricular myocardium. J. Heart Lung. Transplant. 2002, 21, 366–373. [Google Scholar] [CrossRef]
- Huang, B.; Wang, S.; Qin, D.; Boutjdir, M.; El-Sherif, N. Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: Role of beta-adrenergic pathway, G(i) protein, phosphodiesterase, and phosphatases. Circ. Res. 1999, 85, 848–855. [Google Scholar] [CrossRef]
- Sande, J.B.; Sjaastad, I.; Hoen, I.B.; Bokenes, J.; Tonnessen, T.; Holt, E.; Lunde, P.K.; Christensen, G. Reduced level of serine(16) phosphorylated phospholamban in the failing rat myocardium: A major contributor to reduced SERCA2 activity. Cardiovasc. Res. 2002, 53, 382–391. [Google Scholar] [CrossRef]
- Boknik, P.; Fockenbrock, M.; Herzig, S.; Knapp, J.; Linck, B.; Luss, H.; Muller, F.U.; Muller, T.; Schmitz, W.; Schroder, F.; et al. Protein phosphatase activity is increased in a rat model of long-term beta-adrenergic stimulation. Naunyn Schmiedebergs Arch Pharmacol. 2000, 362, 222–231. [Google Scholar] [CrossRef]
- Gupta, R.C.; Mishra, S.; Yang, X.P.; Sabbah, H.N. Reduced inhibitor 1 and 2 activity is associated with increased protein phosphatase type 1 activity in left ventricular myocardium of one-kidney, one-clip hypertensive rats. Mol. Cell Biochem. 2005, 269, 49–57. [Google Scholar] [CrossRef]
- Gupta, R.C.; Mishra, S.; Rastogi, S.; Imai, M.; Habib, O.; Sabbah, H.N. Cardiac SR-coupled PP1 activity and expression are increased and inhibitor 1 protein expression is decreased in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2373–H2381. [Google Scholar] [CrossRef]
- Carr, A.N.; Schmidt, A.G.; Suzuki, Y.; del Monte, F.; Sato, Y.; Lanner, C.; Breeden, K.; Jing, S.L.; Allen, P.B.; Greengard, P.; et al. Type 1 phosphatase, a negative regulator of cardiac function. Mol. Cell. Biol. 2002, 22, 4124–4135. [Google Scholar] [CrossRef]
- Yan, G.; Ru, Y.; Yan, F.; Xiong, X.; Hu, W.; Pan, T.; Sun, J.; Zhang, C.; Wang, Q.; Li, X. MIIP inhibits the growth of prostate cancer via interaction with PP1alpha and negative modulation of AKT signaling. Cell Commun. Signal 2019, 17, 44. [Google Scholar] [CrossRef]
- Xu, W.; Yuan, X.; Jung, Y.J.; Yang, Y.; Basso, A.; Rosen, N.; Chung, E.J.; Trepel, J.; Neckers, L. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res. 2003, 63, 7777–7784. [Google Scholar]
- Kracht, M.; van den Boom, J.; Seiler, J.; Kroning, A.; Kaschani, F.; Kaiser, M.; Meyer, H. Protein Phosphatase-1 Complex Disassembly by p97 is Initiated through Multivalent Recognition of Catalytic and Regulatory Subunits by the p97 SEP-domain Adapters. J. Mol. Biol. 2020, 432, 6061–6074. [Google Scholar] [CrossRef]
- van den Boom, J.; Kueck, A.F.; Kravic, B.; Muschenborn, H.; Giesing, M.; Pan, D.; Kaschani, F.; Kaiser, M.; Musacchio, A.; Meyer, H. Targeted substrate loop insertion by VCP/p97 during PP1 complex disassembly. Nat. Struct. Mol. Biol. 2021, 28, 964–971. [Google Scholar] [CrossRef]
- Chia, W.S.; Chia, D.X.; Rao, F.; Bar Nun, S.; Geifman Shochat, S. ATP binding to p97/VCP D1 domain regulates selective recruitment of adaptors to its proximal N-domain. PLoS ONE 2012, 7, e50490. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, B.G.; Dumas, C.; Maaroufi, H.; Padmanabhan, P.K.; Papadopoulou, B. The AAA + ATPase valosin-containing protein (VCP)/p97/Cdc48 interaction network in Leishmania. Sci. Rep. 2020, 10, 13135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Li, L.; Wu, J.; Gong, H.; Niu, Y.; Sun, A.; Ge, J.; Zou, Y. Mechanical stress-evoked but angiotensin II-independent activation of angiotensin II type 1 receptor induces cardiac hypertrophy through calcineurin pathway. Biochem. Biophys. Res. Commun. 2010, 397, 263–269. [Google Scholar] [CrossRef]
- Qiu, H.; Lizano, P.; Laure, L.; Sui, X.; Rashed, E.; Park, J.Y.; Hong, C.; Gao, S.; Holle, E.; Morin, D.; et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 2011, 124, 406–415. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Tang, X.; Qiu, H. Cardiac-Specific Suppression of Valosin-Containing Protein Induces Progressive Heart Failure and Premature Mortality Correlating with Temporal Dysregulations in mTOR Complex 2 and Protein Phosphatase 1. Int. J. Mol. Sci. 2024, 25, 6445. https://doi.org/10.3390/ijms25126445
Sun X, Tang X, Qiu H. Cardiac-Specific Suppression of Valosin-Containing Protein Induces Progressive Heart Failure and Premature Mortality Correlating with Temporal Dysregulations in mTOR Complex 2 and Protein Phosphatase 1. International Journal of Molecular Sciences. 2024; 25(12):6445. https://doi.org/10.3390/ijms25126445
Chicago/Turabian StyleSun, Xiaonan, Xicong Tang, and Hongyu Qiu. 2024. "Cardiac-Specific Suppression of Valosin-Containing Protein Induces Progressive Heart Failure and Premature Mortality Correlating with Temporal Dysregulations in mTOR Complex 2 and Protein Phosphatase 1" International Journal of Molecular Sciences 25, no. 12: 6445. https://doi.org/10.3390/ijms25126445