
The Role of the Toll-like Receptor 2 and the cGAS-STING Pathways in Breast Cancer: Friends or Foes?
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of TLR2 in Anticancer Immune Responses
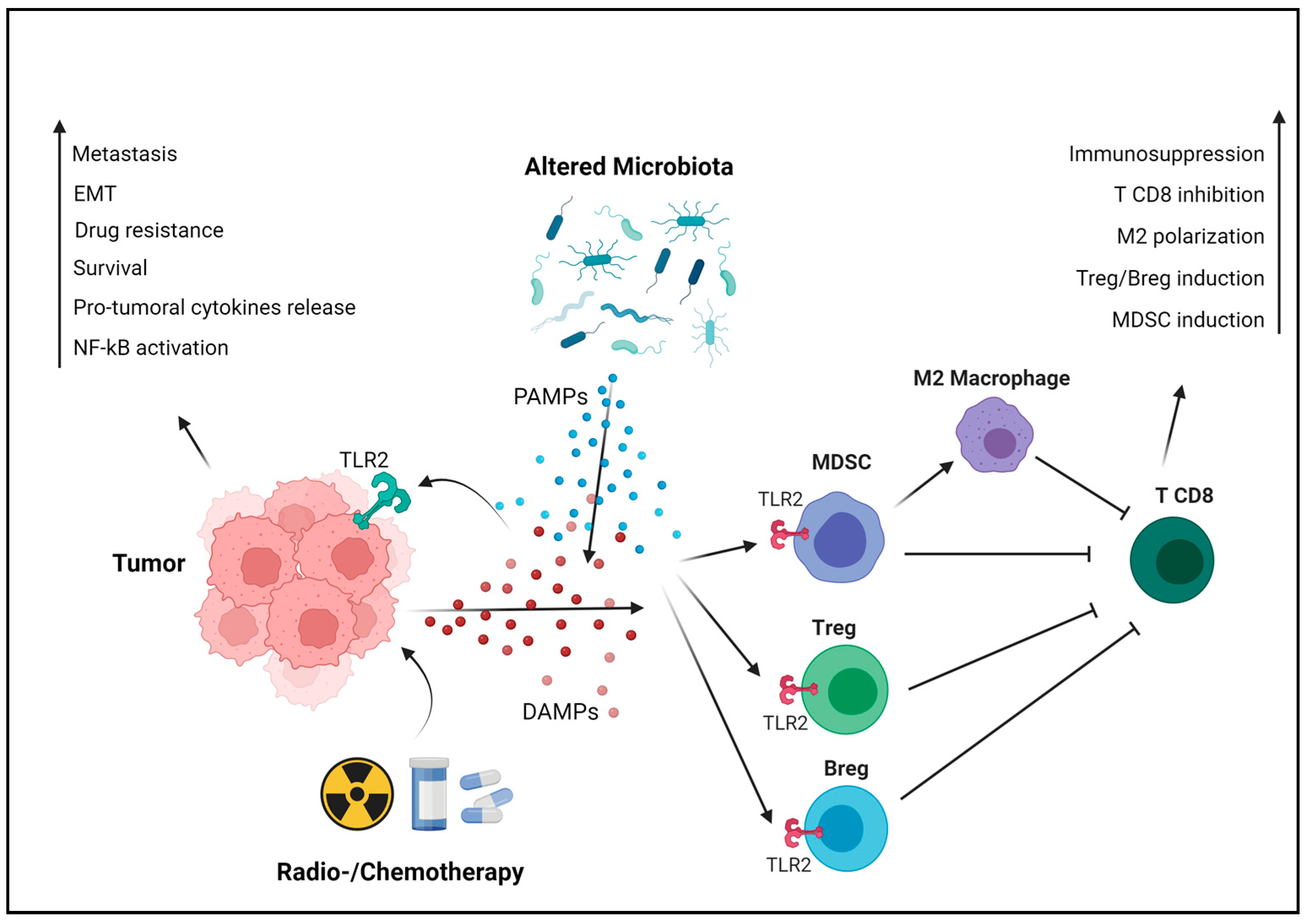
3. The Protumoral Role of TLR2 in Breast Cancer
3.1. The Cancer Cell-Intrinsic Protumoral Effects of TLR2
3.2. The Cancer Cell-Extrinsic Protumoral Effects of TLR2
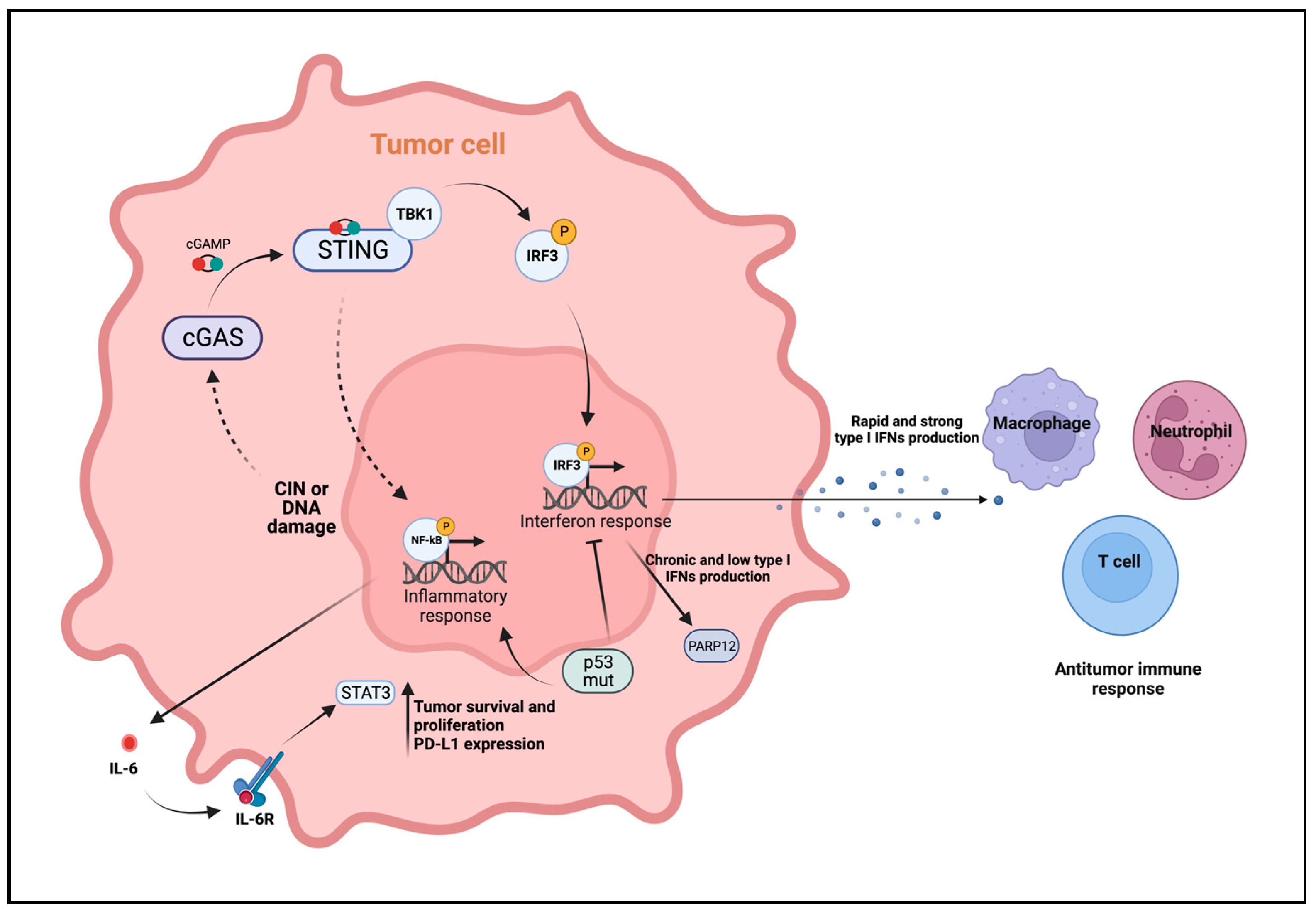
4. Role of cGAS-STING in Antitumor Immunity
5. The Protumoral Role of cGAS-STING in Breast Cancer
6. Therapeutic Potential of Innate Immune Molecules Targeting for Breast Cancer Treatment
6.1. Activation
6.2. Inhibition
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L.; Albini, A.; Ferrone, S.; Bruno, A.; Lollini, P.-L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Jenkins, B.J. Context-Dependent Functions of Pattern Recognition Receptors in Cancer. Nat. Rev. Cancer 2022, 22, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Xu, C.; Fang, X.; Zhang, Y.; Li, H.; Wen, W.; Yang, G. Expression Profile of Toll-like Receptors in Human Breast Cancer. Mol. Med. Rep. 2020, 21, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, S.A.; Palaniyandi, T.; Parthasarathy, U.; Surendran, H.; Viswanathan, S.; Wahab, M.R.A.; Baskar, G.; Natarajan, S.; Ranjan, K. Implications of Toll-like Receptors (TLRs) and Their Signaling Mechanisms in Human Cancers. Pathol. Res. Pr. 2023, 248, 154673. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Wang, J.; Hu, X.; Ouyang, L.; Wang, Y. Small-Molecule Modulators Targeting Toll-like Receptors for Potential Anticancer Therapeutics. J. Med. Chem. 2023, 66, 6437–6462. [Google Scholar] [CrossRef] [PubMed]
- Dajon, M.; Iribarren, K.; Cremer, I. Toll-like Receptor Stimulation in Cancer: A pro-and Anti-Tumor Double-Edged Sword. Immunobiology 2017, 222, 89–100. [Google Scholar] [CrossRef]
- Turnbull, A.; Gerratana, L.; Matteo Luca Battisti, N.; Marsden, R.; Yang, J.; Wang, Y.; Liu, S.; Zhang, Y. Dysregulation of TLR2 Serves as a Prognostic Biomarker in Breast Cancer and Predicts Resistance to Endocrine Therapy in the Luminal B Subtype. Front. Oncol. 2020, 10, 547. [Google Scholar] [CrossRef]
- West, A.C.; Tang, K.; Tye, H.; Yu, L.; Deng, N.; Najdovska, M.; Lin, S.J.; Balic, J.J.; Okochi-Takada, E.; Mcguirk, P.; et al. Identification of a TLR2-Regulated Gene Signature Associated with Tumor Cell Growth in Gastric Cancer. Oncogene 2017, 36, 5134–5144. [Google Scholar] [CrossRef]
- Lundy, J.; Gearing, L.J.; Gao, H.; West, A.C.; Mcleod, L.; Deswaerte, V.; Yu, L.; Porazinski, S.; Pajic, M.; Hertzog, P.J.; et al. TLR2 Activation Promotes Tumour Growth and Associates with Patient Survival and Chemotherapy Response in Pancreatic Ductal Adenocarcinoma. Oncogene 1992, 40, 6007–6022. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Leading Edge Review Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; Mcphillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through Trans-Activation of LRP on the Phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Leggatt, G.R.; Land, W.G.; Bianchi, M.E.; Vénéreau, E.; Ceriotti, C. DAMPs from Cell Death to New Life. Article 2015, 6, 422. [Google Scholar] [CrossRef]
- Chen, C.; Xu, P. Cellular Functions of CGAS-STING Signaling Cell Biology. Trends Cell Biol. 2023, 33, 630–648. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.-P.; Hornung, V. Cell Intrinsic Recognition and Defence Systems against Foreign Genetic Material Encompass. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Samson, N.; Ablasser, A. The CGAS–STING Pathway and Cancer. Nat. Cancer 2022, 3, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Mann, C.C.; Orzalli, M.H.; King, D.S.; Kagan, J.C.; Lee, A.S.Y.; Kranzusch, P.J. Modular Architecture of the STING C-Terminal Tail Allows Interferon and NF-ΚB Signaling Adaptation. Cell Rep. 2019, 27, 1165–1175.e5. [Google Scholar] [CrossRef]
- Huang, L.; Li, L.; Lemos, H.; Chandler, P.R.; Pacholczyk, G.; Baban, B.; Barber, G.N.; Hayakawa, Y.; McGaha, T.L.; Ravishankar, B.; et al. Cutting Edge: DNA Sensing via the STING Adaptor in Myeloid Dendritic Cells Induces Potent Tolerogenic Responses. An Alternative Splicing Isoform of MITA Antagonizes MITA-Mediated Induction of Type I IFNs. J. Immunol. 2013, 191, 3509–3513. [Google Scholar] [CrossRef]
- Larkin, B.; Ilyukha, V.; Sorokin, M.; Buzdin, A.; Vannier, E.; Poltorak, A. Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J. Immunol. 2017, 199, 397–402. [Google Scholar] [CrossRef]
- Kabelitz, D.; Lathia, J.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R.; Khan, M.M.; Allen Hamilton, B.; Urban-Wojciuk, Z.; Oyler, B.L.; Fåhraeus, R.; et al. The Role of TLRs in Anti-Cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Lu, H.; Yang, Y.; Gad, E.; Inatsuka, C.; Wenner, C.A.; Disis, M.L.; Standish, L.J. TLR2 Agonist PSK Activates Human NK Cells and Enhances the Antitumor Effect of HER2-Targeted Monoclonal Antibody Therapy. Clin. Cancer Res. 2011, 17, 6742–6753. [Google Scholar] [CrossRef] [PubMed]
- Ke, M.; Wang, H.; Zhang, M.; Tian, Y.; Wang, Y.; Li, B.; Yu, J.; Dou, J.; Xi, T.; Zhou, C. The Anti-Lung Cancer Activity of SEP Is Mediated by the Activation and Cytotoxicity of NK Cells via TLR2/4 in Vivo. Biochem. Pharmacol. 2014, 89, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Yang, J.; Qian, J.; Liu, R.; Huang, E.; Wang, Y.; Luo, F.; Chu, Y. TLR1/TLR2 Signaling Blocks the Suppression of Monocytic Myeloid-Derived Suppressor Cell by Promoting Its Differentiation into M1-Type Macrophage. Mol. Immunol. 2019, 112, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Su, L.; Morin, M.D.; Jones, B.T.; Mifune, Y.; Shi, H.; Wang, K.-W.; Zhan, X.; Liu, A.; Wang, J.; et al. Adjuvant Effect of the Novel TLR1/TLR2 Agonist Diprovocim Synergizes with Anti-PD-L1 to Eliminate Melanoma in Mice. Proc. Natl. Acad. Sci. USA 2018, 115, E8698–E8706. [Google Scholar] [CrossRef] [PubMed]
- Kiura, K.; Hasebe, A.; Saeki, A.; Segawa, T.; Okada, F.; Shamsul, H.M.; Ohtani, M.; Into, T.; Inoue, N.; Wakita, M.; et al. In Vivo Anti- and pro-Tumour Activities of the TLR2 Ligand FSL-1. Immunobiology 2011, 216, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Singh, S.; Anang, V.; Bhatt, A.N.; Natarajan, K.; Dwarakanath, B.S. Pattern Recognition Receptors in Cancer Progression and Metastasis. Cancer Growth Metastasis 2015, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- El-Kharashy, G.; Gowily, A.; Okda, T.; Houssen, M. Association between Serum Soluble Toll-like Receptor 2 and 4 and the Risk of Breast Cancer. Mol. Clin. Oncol. 2021, 14, 38. [Google Scholar] [CrossRef]
- Blazquez, R.; Chuang, H.-N.; Wenske, B.; Trigueros, L.; Wlochowitz, D.; Liguori, R.; Ferrazzi, F.; Regen, T.; Proescholdt, M.A.; Rohde, V.; et al. ARTICLE Intralesional TLR4 Agonist Treatment Strengthens the Organ Defense against Colonizing Cancer Cells in the Brain. Oncogene 2022, 41, 5008–5019. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Bolli, E.; Ruiu, R.; Ferrauto, G.; Di Gregorio, E.; Avalle, L.; Savino, A.; Poggio, P.; Merighi, I.F.; Riccardo, F.; et al. Toll-like Receptor 2 Promotes Breast Cancer Progression and Resistance to Chemotherapy. Oncoimmunology 2022, 11, 2086752. [Google Scholar] [CrossRef]
- De Angelis, M.L.; Francescangeli, F.; Zeuner, A. Cancers Breast Cancer Stem Cells as Drivers of Tumor Chemoresistance, Dormancy and Relapse: New Challenges and Therapeutic Opportunities. Cancers 2019, 11, 1569. [Google Scholar] [CrossRef]
- Conti, L.; Lanzardo, S.; Arigoni, M.; Antonazzo, R.; Radaelli, E.; Cantarella, D.; Calogero, R.A.; Cavallo, F. The Noninflammatory Role of High Mobility Group Box 1/Toll-like Receptor 2 Axis in the Self-Renewal of Mammary Cancer Stem Cells. FASEB J. Res. Commun. 2013, 27, 4731–4744. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The Mechanism of HMGB1 Secretion and Release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, C.; Li, L.; Jin, X.; Zhang, Z.; Zheng, H.; Pan, J.; Shi, L.; Jiang, Z.; Su, K.; et al. Oncogenesis Tumor-Derived HMGB1 Induces CD62L Dim Neutrophil Polarization and Promotes Lung Metastasis in Triple-Negative Breast Cancer. Oncogenesis 2020, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Seclì, L.; Avalle, L.; Poggio, P.; Fragale, G.; Cannata, C.; Conti, L.; Iannucci, A.; Carr, G.; Rubinetto, C.; Miniscalco, B.; et al. Targeting the Extracellular HSP90 Co-Chaperone Morgana Inhibits Cancer Cell Migration and Promotes Anticancer Immunity. Cancer Res. 2021, 81, 4794–4807. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Takahashi, H.; Lin, W.-W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.-L.; Karin, M. LETTERS Carcinoma-Produced Factors Activate Myeloid Cells through TLR2 to Stimulate Metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Bolli, E.; Tarone, L.; Cavallo, F.; Conti, L. Toll-like Receptor 2 at the Crossroad between Cancer Cells, the Immune System, and the Microbiota. Int. J. Mol. Sci. 2020, 21, 9418. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Wang, Y.; Huang, Y.; Yang, H.; Wang, J.; Hu, Z. Toll-like Receptor 2 Mediates Invasion via Activating NF-ΚB in MDA-MB-231 Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2009, 379, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Scheeren, F.A.; Kuo, A.H.; Van Weele, L.J.; Cai, S.; Glykofridis, I.; Sikandar, S.S.; Zabala, M.; Qian, D.; Lam, J.S.; Johnston, D.; et al. A Cell-Intrinsic Role for TLR2-MYD88 in Intestinal and Breast Epithelia and Oncogenesis. Nat. Cell Biol. 2014, 16, 1238–1248. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Gö Ktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Tye, H.; Kennedy, C.L.; Najdovska, M.; Mcleod, L.; Mccormack, W.; Hughes, N.; Dev, A.; Sievert, W.; Ooi, C.H.; Ishikawa, T.-O.; et al. Article STAT3-Driven Upregulation of TLR2 Promotes Gastric Tumorigenesis Independent of Tumor Inflammation. Cancer Cell 2012, 22, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Y.; Fu, Y. The Critical Role of Toll-like Receptor-Mediated Signaling in Cancer Immunotherapy. Med. Drug Discov. 2022, 14, 100122. [Google Scholar] [CrossRef]
- Parida, S.; Sharma, D. The Power of Small Changes: Comprehensive Analyses of Microbial Dysbiosis in Breast Cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 392. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhao, J.; Shen, S.; Li, H.; He, K.-L.; Shen, G.-X.; Mayer, L.; Unkeless, J.; Li, D.; Yuan, Y.; et al. Listeria Monocytogenes Promotes Tumor Growth via Tumor Cell Toll-Like Receptor 2 Signaling. Cancer Res. 2007, 67, 4346–4352. [Google Scholar] [CrossRef] [PubMed]
- Krzysiek-Maczka, G.; Targosz, A.; Szczyrk, U.; Strzalka, M.; Brzozowski, T.; Ptak-Belowska, A. Involvement of Epithelial-Mesenchymal Transition-Inducing Transcription Factors in the Mechanism of Helicobacter Pylori-Induced Fibroblasts Activation. J. Physiol. Pharmacol. 2019, 70, 727–736. [Google Scholar] [CrossRef]
- Parida, S.; Wu, S.; Siddharth, S.; Wang, G.; Muniraj, N.; Nagalingam, A.; Hum, C.; Mistriotis, P.; Hao, H.; Conover, C.; et al. A Procarcinogenic Colon Microbe Promotes Breast Tumorigenesis and Metastatic Progression and Concomitantly Activates Notch and B-Catenin Axes. Cancer Discov. 2021, 11, 1138–1157. [Google Scholar] [CrossRef] [PubMed]
- Kahraman Gürsoy, U.; Kantarci, A.; Enersen, M.; Guncu, G.N.; Bachrach, G.; Abed, J.; Maalouf, N.; Parhi, L.; Chaushu, S.; Mandelboim, O. Tumor Targeting by Fusobacterium Nucleatum: A Pilot Study and Future Perspectives. Front. Cell. Infect. Microbiol. 2017, 1, 295. [Google Scholar] [CrossRef]
- Jia, Y.-P.; Wang, K.; Zhang, Z.-J.; Tong, Y.-N.; Han, D.; Hu, C.-Y.; Li, Q.; Xiang, Y.; Mao, X.-H.; Tang, B. TLR2/TLR4 Activation Induces Tregs and Suppresses Intestinal Inflammation Caused by Fusobacterium Nucleatum in vivo. PLoS ONE 2017, 12, e0186179. [Google Scholar] [CrossRef]
- Chiba, A.; Bawaneh, A.; Velazquez, C.; Clear, K.Y.J.; Wilson, A.S.; Howard-Mcnatt, M.; Levine, E.A.; Levi-Polyachenko, N.; Yates-Alston, S.A.; Diggle, S.P.; et al. Neoadjuvant Chemotherapy Shifts Breast Tumor Microbiota Populations to Regulate Drug Responsiveness and the Development of Metastasis. Mol. Cancer Res. 2019, 18, 130–139. [Google Scholar] [CrossRef]
- Montassier, E.; Gastinne, T.; Vangay, P.; Al-Ghalith, G.A.; Bruley Des Varannes, S.; Massart, S.; Moreau, P.; Potel, G.; De La Cochetière, M.F.; Batard, E.; et al. Chemotherapy-Driven Dysbiosis in the Intestinal Microbiome. Aliment. Pharmacol. Ther. 2015, 42, 515–528. [Google Scholar] [CrossRef]
- Bernardo, G.; Le Noci, V.; Ottaviano, E.; De Cecco, L.; Camisaschi, C.; Guglielmetti, S.; Di Modica, M.; Gargari, G.; Bianchi, F.; Indino, S.; et al. Reduction of Staphylococcus Epidermidis in the Mammary Tumor Microbiota Induces Antitumor Immunity and Decreases Breast Cancer Aggressiveness. Cancer Lett. 2023, 555, 216041. [Google Scholar] [CrossRef] [PubMed]
- Sutmuller, R.P.M.; Den Brok, M.H.M.G.M.; Kramer, M.; Bennink, E.J.; Toonen, L.W.J.; Kullberg, B.-J.; Joosten, L.A.; Akira, S.; Netea, M.G.; Adema, G.J. Toll-like Receptor 2 Controls Expansion and Function of Regulatory T Cells. J Clin Invest. 2006, 116, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Mcbride, A.; Konowich, J.; Salgame, P. Host Defense and Recruitment of Foxp3 + T Regulatory Cells to the Lungs in Chronic Mycobacterium Tuberculosis Infection Requires Toll-like Receptor 2. PLoS Pathog. 2013, 9, e1003397. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, Q.; Cheng, Y.; Chen, X.; Wang, G.; Shi, M.; Zhang, T.; Cao, Y.; Pan, H.; Zhang, L.; et al. Tumor-Derived Exosomal HMGB1 Fosters Hepatocellular Carcinoma Immune Evasion by Promoting TIM-1+ Regulatory B Cell Expansion. J. Immunother. Cancer 2018, 6, 145. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Q.; Li, P.-C.; Pan, N.; Gao, R.; Wen, Z.-F.; Zhang, T.-Y.; Huang, F.; Wu, F.-Y.; Ou, X.-L.; Zhang, J.-P.; et al. Tumor-Released Autophagosomes Induces CD4 + T Cell-Mediated Immunosuppression via a TLR2-IL-6 Cascade. J. Immunother. Cancer 2019, 7, 178. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Yin, L.; Yang, X.; Yang, Y.; Gu, Y.; Sun, Y.; Yang, M.; Wang, Y.; Zhang, Q.; Ji, H. Serum Amyloid A 1 Induces Suppressive Neutrophils through the Toll-like Receptor 2–Mediated Signaling Pathway to Promote Progression of Breast Cancer. Cancer Sci. 2022, 113, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Spanier, J.; Kasnitz, N.; Kröger, A.; Jin, L.; Brinkmann, M.M.; Kalinke, U.; Weiss, S.; Jablonska, J.; Lienenklaus, S. Growing Tumors Induce a Local STING Dependent Type I IFN Response in Dendritic Cells. Int. J. Cancer 2016, 139, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef]
- Vargas-Rondón, N.; Villegas, V.E.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2018, 10, 4. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I Interferon Is Selectively Required by Dendritic Cells for Immune Rejection of Tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, X.; Wang, Z.; Liu, P.; Hou, Y.; Xu, Y.; Su, H.; Koci, M.D.; Yin, H.; Zhang, C. NF-ΚB Activation Enhances STING Signaling by Altering Microtubule-Mediated STING Trafficking. Cell Rep. 2023, 42, 112185. [Google Scholar] [CrossRef] [PubMed]
- Kitai, Y.; Kawasaki, T.; Sueyoshi, T.; Kobiyama, K.; Ishii, K.J.; Zou, J.; Akira, S.; Matsuda, T.; Kawai, T. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J. Immunol. 2017, 198, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.; Cordova, A.F.; Hess, G.T.; Bassik, M.C.; Li, L. SLC19A1 Is an Importer of the Immunotransmitter CGAMP. Mol. Cell 2019, 75, 372–381.e5. [Google Scholar] [CrossRef] [PubMed]
- Maltbaek, J.H.; Cambier, S.; Snyder, J.M.; Stetson, D.B. ABCC1 Transporter Exports the Immunostimulatory Cyclic Dinucleotide CGAMP. Immunity 2022, 55, 1799–1812.e4. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.A.; Vance, R.E.; Raulet, D.H. Tumor-Derived CGAMP Triggers a STING-Mediated Interferon Response in Non-Tumor Cells to Activate the NK Cell Response. Immunity 2018, 49, 754–763.e4. [Google Scholar] [CrossRef]
- Lu, L.; Yang, C.; Zhou, X.; Wu, L.; Hong, X.; Li, W.; Wang, X.; Yang, Y.; Cao, D.; Zhang, A.; et al. STING Signaling Promotes NK Cell Antitumor Immunity and Maintains a Reservoir of TCF-1+ NK Cells. Cell Rep. 2023, 42, 113108. [Google Scholar] [CrossRef]
- Cordova, A.F.; Ritchie, C.; Böhnert, V.; Li, L. Human SLC46A2 Is the Dominant CGAMP Importer in Extracellular CGAMP-Sensing Macrophages and Monocytes. ACS Cent. Sci. 2021, 7, 1073–1088. [Google Scholar] [CrossRef]
- Wang, Q.; Bergholz, J.S.; Ding, L.; Lin, Z.; Kabraji, S.K.; Hughes, M.E.; He, X.; Xie, S.; Jiang, T.; Wang, W.; et al. STING Agonism Reprograms Tumor-Associated Macrophages and Overcomes Resistance to PARP Inhibition in BRCA1-Deficient Models of Breast Cancer. Nat. Commun. 2022, 13, 3022. [Google Scholar] [CrossRef]
- An, X.; Zhu, Y.; Zheng, T.; Wang, G.; Zhang, M.; Li, J.; Ji, H.; Li, S.; Yang, S.; Xu, D.; et al. An Analysis of the Expression and Association with Immune Cell Infiltration of the CGAS/STING Pathway in Pan-Cancer. Mol. Ther. Nucleic Acids 2019, 14, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates with Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Falahat, R.; Berglund, A.; Putney, R.M.; Perez-Villarroel, P.; Aoyama, S.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. Epigenetic Reprogramming of Tumor Cell-Intrinsic STING Function Sculpts Antigenicity and T Cell Recognition of Melanoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2013598118. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Wu, L.; Xiao, Q.; Meng, X.; Hafiz, A.; Yan, Q.; Lu, R.; Cao, J. Epigenetically Suppressed Tumor Cell Intrinsic STING Promotes Tumor Immune Escape. Biomed. Pharmacother. 2023, 157, 114033. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. The Landscape of Chromosome Instability in Breast Cancers and Associations with the Tumor Mutation Burden: An Analysis of Data from TCGA. Cancer Investig. 2021, 39, 25–38. [Google Scholar] [CrossRef]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. CGAS–STING Drives the IL-6-Dependent Survival of Chromosomally Instable Cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef]
- Vasiyani, H.; Mane, M.; Rana, K.; Shinde, A.; Roy, M.; Singh, J.; Gohel, D.; Currim, F.; Srivastava, R.; Singh, R. DNA Damage Induces STING Mediated IL-6-STAT3 Survival Pathway in Triple-Negative Breast Cancer Cells and Decreased Survival of Breast Cancer Patients. Apoptosis 2022, 27, 961–978. [Google Scholar] [CrossRef]
- Cheon, H.; Holvey-Bates, E.G.; Mcgrail, D.J.; Stark, G.R.; Diaz, L.A.; Hertzog, P.J. PD-L1 Sustains Chronic, Cancer Cell-Intrinsic Responses to Type I Interferon, Enhancing Resistance to DNA Damage. Proc. Natl. Acad. Sci. USA 2021, 118, e2112258118. [Google Scholar] [CrossRef]
- Gaston, J.; Cheradame, L.; Yvonnet, V.; Deas, O.; Poupon, M.-F.; Judde, J.-G.; Cairo, S.; Goffin, V. Intracellular STING Inactivation Sensitizes Breast Cancer Cells to Genotoxic Agents. Oncotarget 2016, 7, 77205–77224. [Google Scholar] [CrossRef]
- Wang, J.; Yi, S.; Zhou, J.; Zhang, Y.; Guo, F. The NF-B Subunit RelB Regulates the Migration and Invasion Abilities and the Radio-Sensitivity of Prostate Cancer Cells. Int. J. Oncol. 2016, 49, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.M.; Martinez, L.A. Mutant P53 Suppresses Innate Immune Signaling to Promote Tumorigenesis. Cancer Cell 2021, 39, 494–508.e5. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Bijker, M.S.; Weterings, J.J.; Tanke, H.J.; Adema, G.J.; Van Hall, T.; Drijfhout, J.W.; Melief, C.J.M.; Overkleeft, H.S.; Van Der Marel, G.A.; et al. Distinct Uptake Mechanisms but Similar Intracellular Processing of Two Different Toll-like Receptor Ligand-Peptide Conjugates in Dendritic Cells. J. Biol. Chem. 2007, 282, 21145–21159. [Google Scholar] [CrossRef] [PubMed]
- Zom, G.G.; Khan, S.; Britten, C.M.; Sommandas, V.; Camps, M.G.M.; Loof, N.M.; Budden, C.F.; Meeuwenoord, N.J.; Filippov, D.V.; van der Marel, G.A.; et al. Efficient Induction of Antitumor Immunity by Synthetic Toll-like Receptor Ligand-Peptide Conjugates. Cancer Immunol. Res. 2014, 2, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aal, A.B.M.; Lakshminarayanan, V.; Thompson, P.; Supekar, N.; Bradley, J.M.; Wolfert, M.A.; Cohen, P.A.; Gendler, S.J.; Boons, G.J. Immune and Anticancer Responses Elicited by Fully Synthetic Aberrantly Glycosylated MUC1 Tripartite Vaccines Modified by a TLR2 or TLR9 Agonist. ChemBioChem 2014, 15, 1508–1513. [Google Scholar] [CrossRef]
- Shi, W.; Tong, Z.; Chen, S.; Qiu, Q.; Zhou, J.; Qian, H. Development of Novel Self-Assembled Vaccines Based on Tumour-Specific Antigenic Peptide and TLR2 Agonist for Effective Breast Cancer Immunotherapy via Activating CD8+ T Cells and Enhancing Their Function. Immunology 2023, 169, 454–466. [Google Scholar] [CrossRef]
- Liu, B.; Huang, J.; Xiao, J.; Xu, W.; Zhang, H.; Yuan, Y.; Yin, Y.; Zhang, X. RESEARCH Open Access the Streptococcus Virulence Protein PepO Triggers Anti-Tumor Immune Responses by Reprograming Tumor-Associated Macrophages in a Mouse Triple Negative Breast Cancer Model. Cell Biosci. 2023, 13, 198. [Google Scholar] [CrossRef]
- Lu, H.; Yang, Y.; Gad, E.; Wenner, C.A.; Chang, A.; Larson, E.R.; Dang, Y.; Martzen, M.; Standish, L.J.; Disis, M.L. Cancer Therapy: Preclinical Polysaccharide Krestin Is a Novel TLR2 Agonist That Mediates Inhibition of Tumor Growth via Stimulation of CD8 T Cells and NK Cells. Clin. Cancer Res. 2011, 17, 67–76. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; Dubensky, T.W.; Gajewski, T.F. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef]
- Baird, J.R.; Friedman, D.; Cottam, B.; Dubensky, T.W.; Kanne, D.B.; Bambina, S.; Bahjat, K.; Crittenden, M.R.; Gough, M.J. Microenvironment and Immunology Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res 2016, 76, 50–61. [Google Scholar] [CrossRef]
- Lu, X.; Wang, X.; Cheng, H.; Wang, X.; Liu, C.; Tan, X. Anti-Triple-Negative Breast Cancer Metastasis Efficacy and Molecular Mechanism of the STING Agonist for Innate Immune Pathway. Ann. Med. 2023, 55, 2210845. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Quispe-Tintaya, W.; Jahangir, A.; Asafu-Adjei, D.; Ramos, I.; Sintim, H.O.; Zhou, J.; Hayakawa, Y.; Karaolis, D.K.R.; Gravekamp, C. STING Ligand C-Di-GMP Improves Cancer Vaccination against Metastatic Breast Cancer. Cancer Immunol. Res. 2014, 2, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Hu, J.; Yuan, Z.; Luo, G.; Yao, J.; Wang, R.; Liu, D.; Cao, B.; Wu, W.; Hu, Z. STING Agonist Enhances the Efficacy of Programmed Death-Ligand 1 Monoclonal Antibody in Breast Cancer Immunotherapy by Activating the Interferon-β Signalling Pathway. Cell Cycle 2022, 21, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Watkins-Schulz, R.; Batty, C.J.; Stiepel, R.T.; Schmidt, M.E.; Sandor, A.M.; Chou, W.C.; Ainslie, K.M.; Bachelder, E.M.; Ting, J.P.Y. Microparticle Delivery of a STING Agonist Enables Indirect Activation of NK Cells by Antigen-Presenting Cells. Mol. Pharm. 2022, 19, 3125–3138. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.; Burdette, D.L.; Sharma, S.; Bhat, N.; Thompson, M.; Jiang, Z.; Rathinam, V.A.K.; Monks, B.; Jin, T.; Xiao, T.S.; et al. Mouse, but Not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. J. Immunol. 2013, 190, 5216–5225. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ascano, M.; Zillinger, T.; Wang, W.; Dai, P.; Serganov, A.A.; Gaffney, B.L.; Shuman, S.; Jones, R.A.; Deng, L.; et al. Structure-Function Analysis of STING Activation by c[G(2′,5′)pA(3′,5′)p] and Targeting by Antiviral DMXAA. Cell 2013, 154, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.B.; Kacew, A.J.; Sweis, R.F. The Development of STING Agonists and Emerging Results as a Cancer Immunotherapy. Curr. Oncol. Rep. 2023, 25, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-S.; Huang, J.-H.; Hung, L.-Y.; Wu, S.-Y.; Wu-Hsieh, B.A. Distinct Roles of Complement Receptor 3, Dectin-1, and Sialic Acids in Murine Macrophage Interaction with Histoplasma Yeast. J. Leukoc. Biol. 2010, 88, 95–106. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Jabbour, E.J.; Konopleva, M.Y.; Daver, N.G.; Borthakur, G.; DiNardo, C.D.; Bose, P.; Patel, P.; Komrokji, R.S.; Shastri, A.; et al. A Clinical Study of Tomaralimab (OPN-305), a Toll-like Receptor 2 (TLR-2) Antibody, in Heavily Pre-Treated Transfusion Dependent Patients with Lower Risk Myelodysplastic Syndromes (MDS) That Have Received and Failed on Prior Hypomethylating Agent (HMA) Therapy. Blood 2018, 132, 798. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, W.; Hu, X. Robinin Inhibits Pancreatic Cancer Cell Proliferation, EMT and Inflammation via Regulating TLR2-PI3k-AKT Signaling Pathway. Cancer Cell Int. 2023, 23, 328. [Google Scholar] [CrossRef]
- Farnebo, L.; Shahangian, A.; Lee, Y.; Shin, J.H.; Scheeren, F.A.; Sunwoo, J.B.; Farnebo, L.; Shahangian, A.; Lee, Y.; Shin, J.H.; et al. Targeting Toll-like Receptor 2 Inhibits Growth of Head and Neck Squamous Cell Carcinoma. Oncotarget 2015, 6, 9897–9907. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cossu, C.; Di Lorenzo, A.; Fiorilla, I.; Todesco, A.M.; Audrito, V.; Conti, L. The Role of the Toll-like Receptor 2 and the cGAS-STING Pathways in Breast Cancer: Friends or Foes? Int. J. Mol. Sci. 2024, 25, 456. https://doi.org/10.3390/ijms25010456
Cossu C, Di Lorenzo A, Fiorilla I, Todesco AM, Audrito V, Conti L. The Role of the Toll-like Receptor 2 and the cGAS-STING Pathways in Breast Cancer: Friends or Foes? International Journal of Molecular Sciences. 2024; 25(1):456. https://doi.org/10.3390/ijms25010456
Chicago/Turabian StyleCossu, Chiara, Antonino Di Lorenzo, Irene Fiorilla, Alberto Maria Todesco, Valentina Audrito, and Laura Conti. 2024. "The Role of the Toll-like Receptor 2 and the cGAS-STING Pathways in Breast Cancer: Friends or Foes?" International Journal of Molecular Sciences 25, no. 1: 456. https://doi.org/10.3390/ijms25010456