
Monitoring the Intracellular pH and Metabolic State of Cancer Cells in Response to Chemotherapy Using a Combination of Phosphorescence Lifetime Imaging Microscopy and Fluorescence Lifetime Imaging Microscopy
 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Results
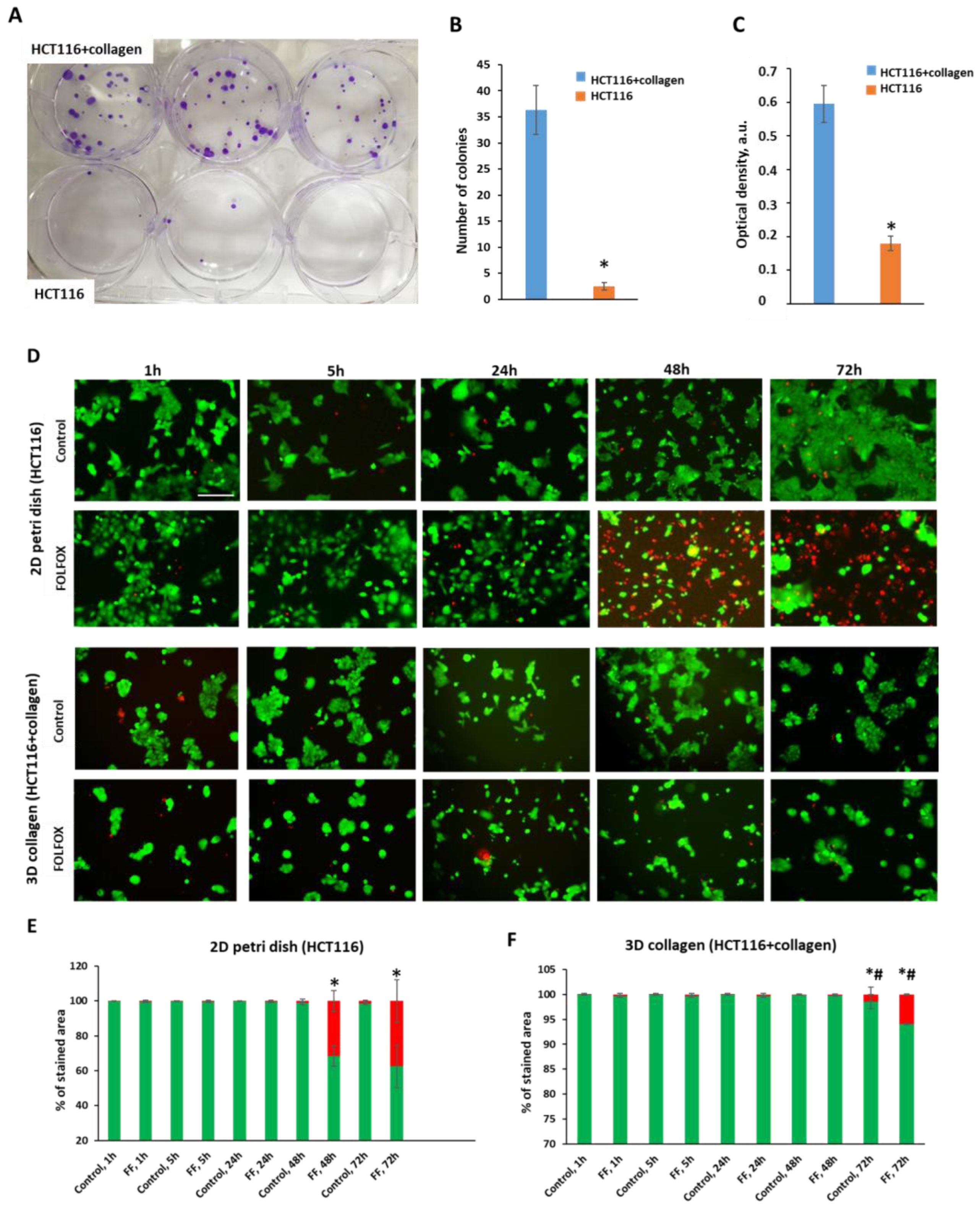
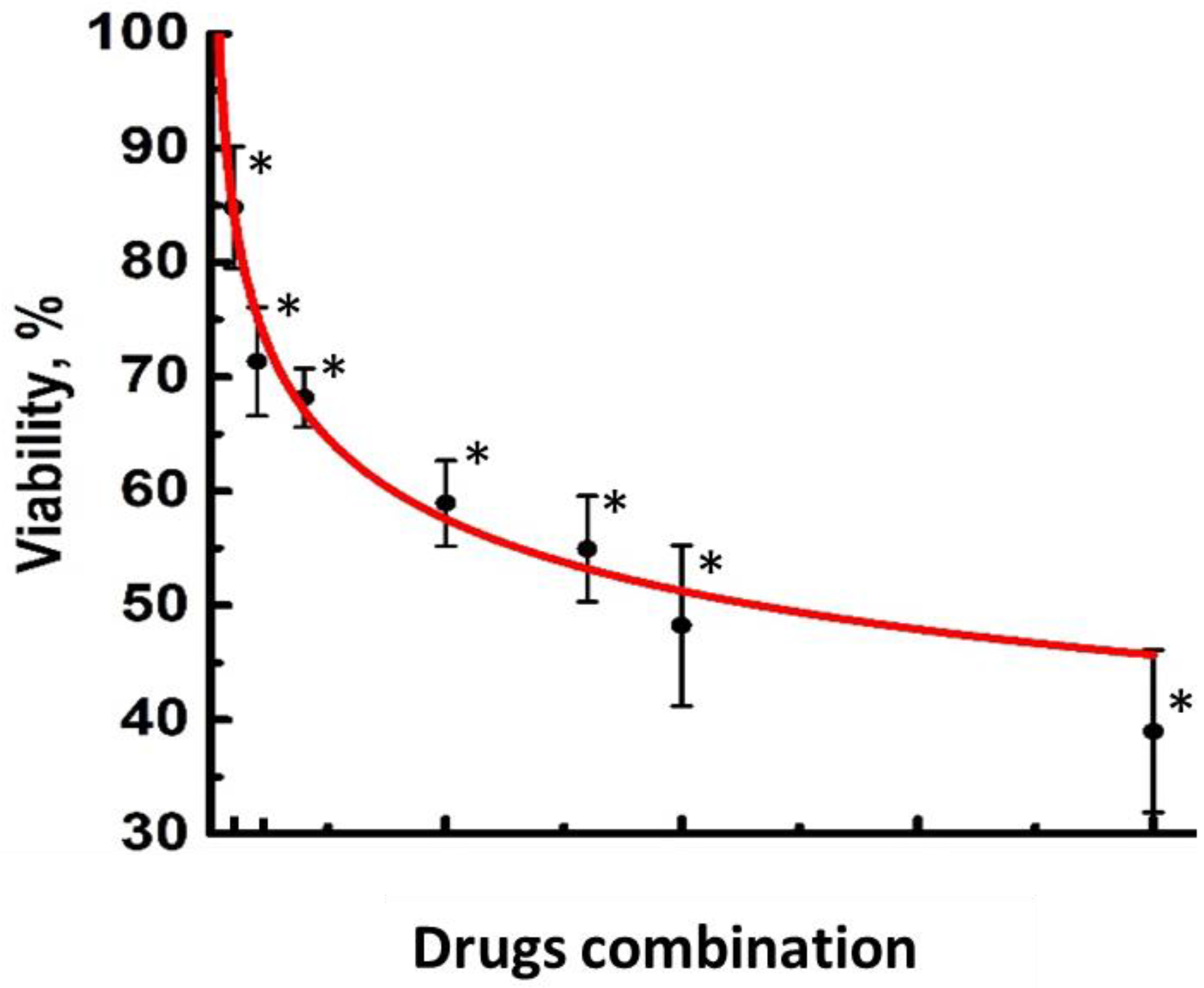
2.1. Effects of Collagen on Cytotoxicity of FOLFOX
2.2. Intracellular pH after Treatment with FOLFOX Regimen
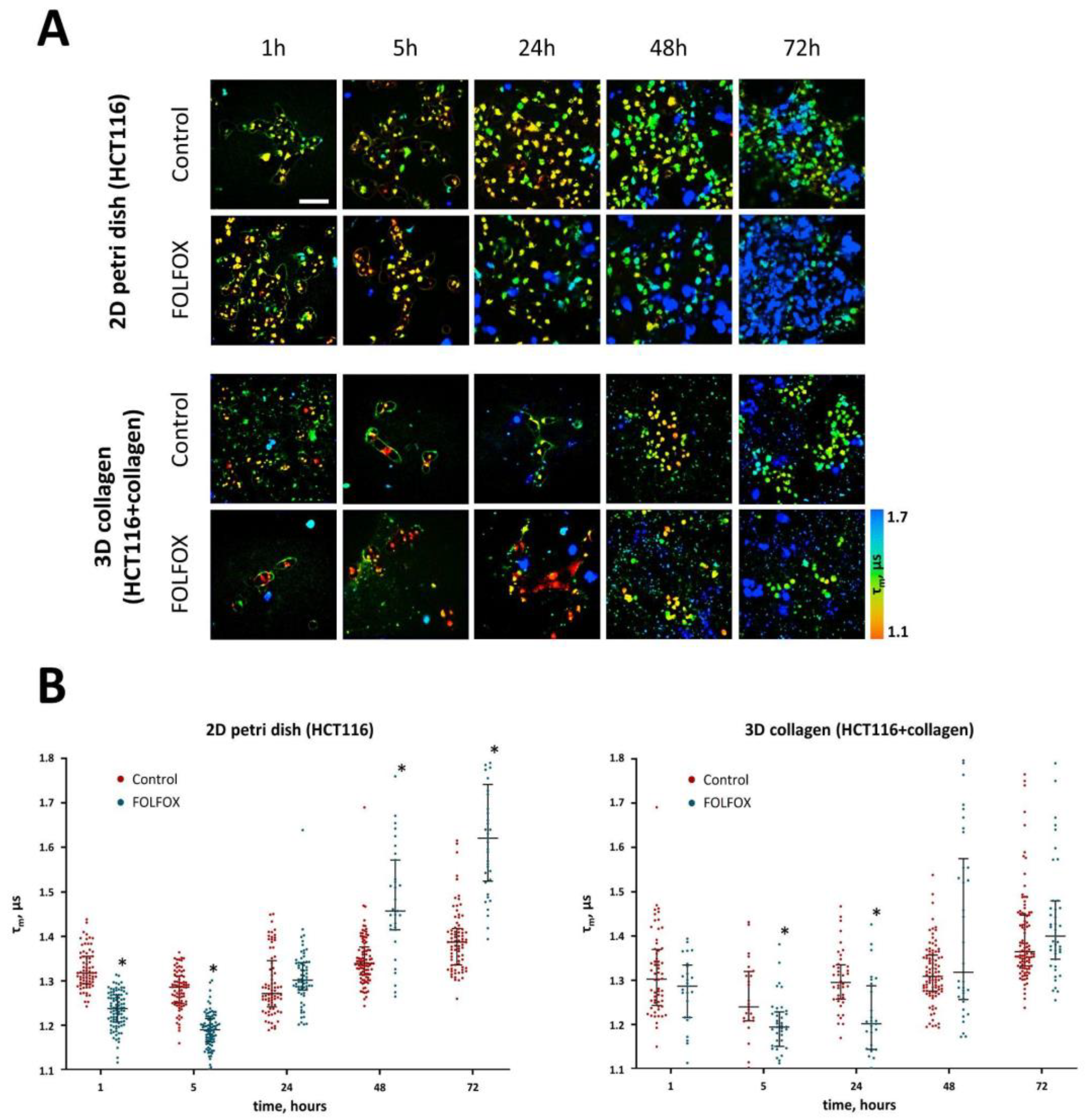
2.3. Phosphorescence Lifetime Imaging Microscopy (PLIM)
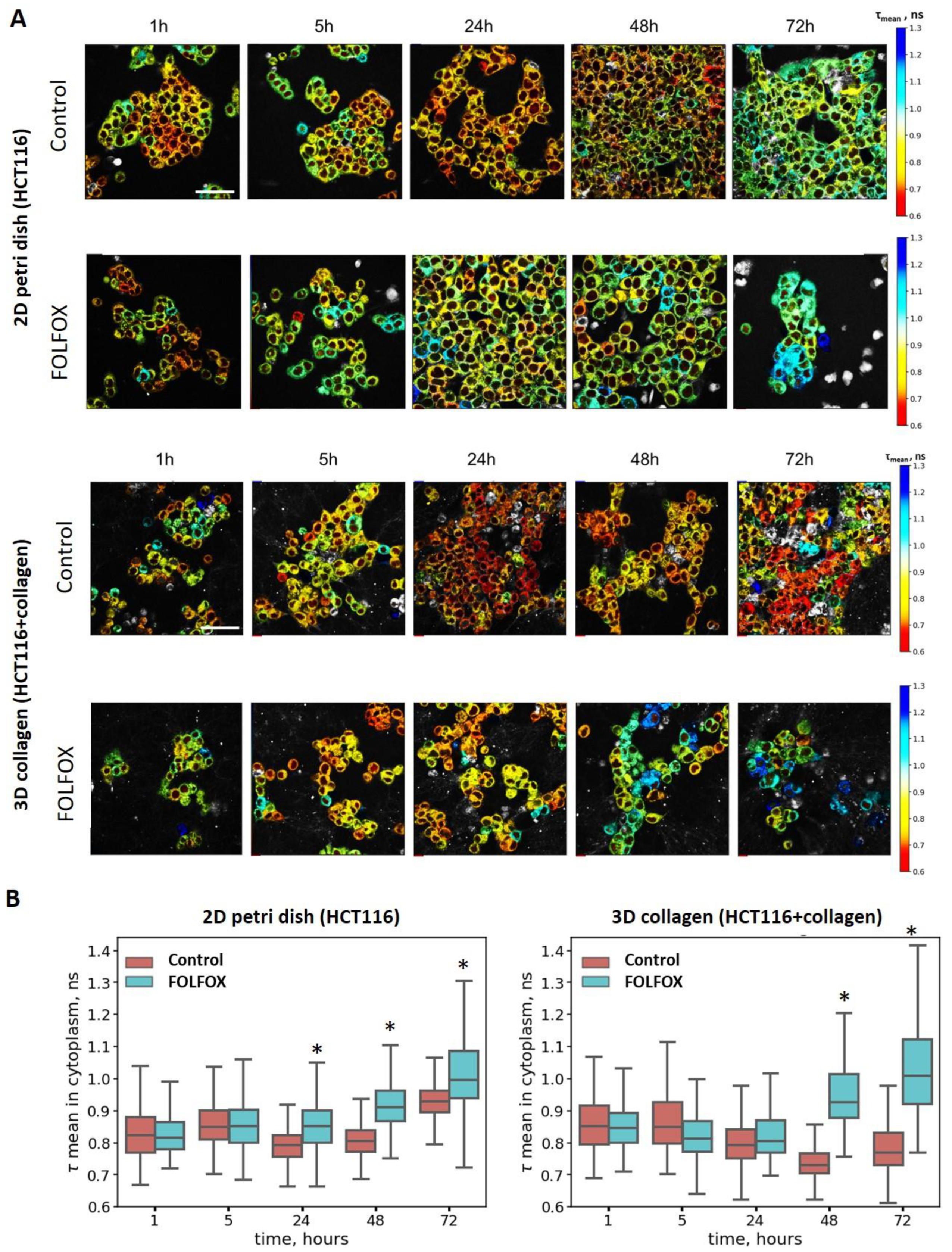
2.4. Metabolic Activity of Cells after Treatment with FOLFOX Regimen
3. Discussion
4. Materials and Methods
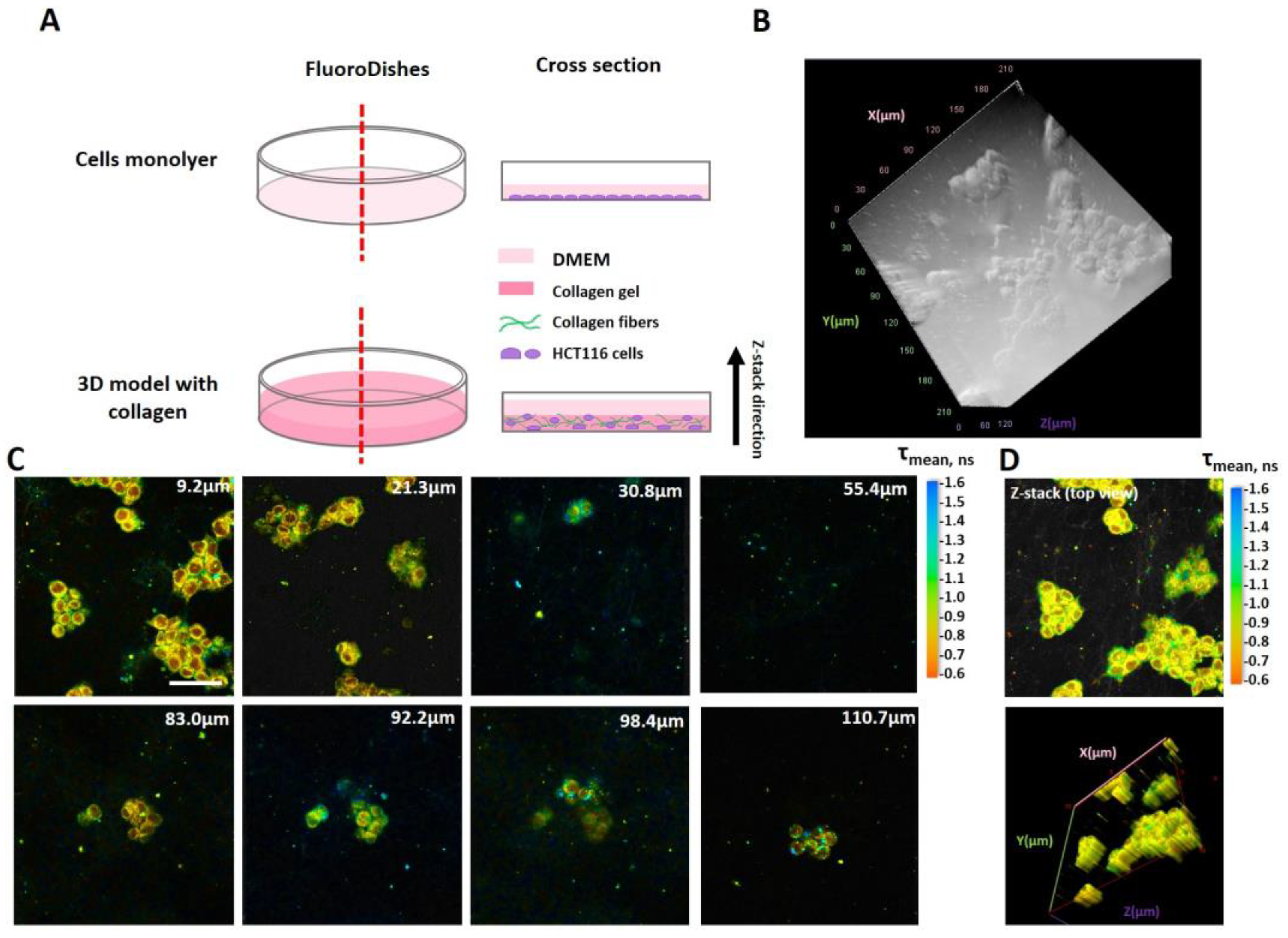
4.1. Cell Cultures and Models
4.2. MTT Assay and In Vitro Chemotherapy
4.3. pH Probe and Staining Protocol
4.4. Phosphorescence Lifetime Imaging Microscopy (PLIM)
4.5. Fluorescence Lifetime Imaging Microscopy (FLIM)
4.6. Second Harmonic Generation (SHG) Microscopy
4.7. Colony-Forming Assay
4.8. Live/Dead Cell Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. CR 2019, 38, 115. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Tian, M.; Pei, Q.; Tan, F.; Pei, H. Extracellular Matrix Stiffness: New Areas Affecting Cell Metabolism. Front. Oncol. 2021, 11, 631991. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e110. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.D.; Yin, B.; Li, Z.; Yuan, W.; Zhang, Q.; Xie, X.; Tan, Y.; Wong, N.; Zhang, K.; Bian, L. Mechanical manipulation of cancer cell tumorigenicity via heat shock protein signaling. Sci. Adv. 2023, 9, eadg9593. [Google Scholar] [CrossRef]
- Swaminathan, V.; Mythreye, K.; O’Brien, E.T.; Berchuck, A.; Blobe, G.C.; Superfine, R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. 2011, 71, 5075–5080. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Eikenes, L.; Tari, M.; Tufto, I.; Bruland, O.S.; de Lange Davies, C. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br. J. Cancer 2005, 93, 81–88. [Google Scholar] [CrossRef]
- Rossow, L.; Veitl, S.; Vorlová, S.; Wax, J.K.; Kuhn, A.E.; Maltzahn, V.; Upcin, B.; Karl, F.; Hoffmann, H.; Gätzner, S.; et al. LOX-catalyzed collagen stabilization is a proximal cause for intrinsic resistance to chemotherapy. Oncogene 2018, 37, 4921–4940. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell. Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef]
- Bonatelli, M.; Silva, E.C.A.; Cárcano, F.M.; Zaia, M.G.; Lopes, L.F.; Scapulatempo-Neto, C.; Pinheiro, C. The Warburg Effect Is Associated with Tumor Aggressiveness in Testicular Germ Cell Tumors. Front. Endocrinol. 2019, 10, 417. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Calderón Montaño, J.M.; Burgos Morón, E.; Pérez Guerrero, M.C.; Salvador Bofill, F.J.; Robles Frías, A.; López Lázaro, M. Role of the Intracellular pH in the Metabolic Switch between Oxidative Phosphorylation and Aerobic Glycolysis-Relevance to Cancer. WebmedCentral 2011, 2, 1–10. [Google Scholar]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH sensing and regulation in cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Talley, K.; Alexov, E. On the pH-optimum of activity and stability of proteins. Proteins 2010, 78, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, A.; Sahara, H. The Metabolic Heterogeneity and Flexibility of Cancer Stem Cells. Cancers 2020, 12, 2780. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Grillo-Hill, B.K.; Choi, C.; Jimenez-Vidal, M.; Barber, D.L. Increased H+ efflux is sufficient to induce dysplasia and necessary for viability with oncogene expression. Elife 2015, 4, e03270. [Google Scholar] [CrossRef]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef]
- Shirmanova, M.V.; Druzhkova, I.N.; Lukina, M.M.; Dudenkova, V.V.; Ignatova, N.I.; Snopova, L.B.; Shcheslavskiy, V.I.; Belousov, V.V.; Zagaynova, E.V. Chemotherapy with cisplatin: Insights into intracellular pH and metabolic landscape of cancer cells in vitro and in vivo. Sci. Rep. 2017, 7, 8911. [Google Scholar] [CrossRef] [PubMed]
- Druzhkova, I.; Lukina, M.; Dudenkova, V.; Ignatova, N.; Snopova, L.; Gavrina, A.; Shimolina, L.; Belousov, V.; Zagaynova, E.; Shirmanova, M. Tracing of intracellular pH in cancer cells in response to Taxol treatment. Cell Cycle 2021, 20, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.T.; Diggins, K.E.; Walsh, A.J.; Irish, J.M.; Skala, M.C. In Vivo Autofluorescence Imaging of Tumor Heterogeneity in Response to Treatment. Neoplasia 2015, 17, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.R.; Wallrabe, H.; Svindrych, Z.; Chaudhary, A.K.; Christopher, K.G.; Chandra, D.; Periasamy, A. Investigation of Mitochondrial Metabolic Response to Doxorubicin in Prostate Cancer Cells: An NADH, FAD and Tryptophan FLIM Assay. Sci. Rep. 2017, 7, 10451. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.J.; Tveit, K.M.; Gibson, F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin. Color. Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Venook, A.P. Flashback Foreword: IFL/FOLFOX/IROX in Advanced Colorectal Cancer and FOLFIRI and FOLFOX6 in Colorectal Cancer. J. Clin. Oncol. 2023, 41, 3459–3460. [Google Scholar] [CrossRef]
- Shakirova, J.R.; Baigildin, V.A.; Solomatina, A.I.; Aghakhanpour, R.B.; Pavlovskiy, V.V.; Porsev, V.V.; Tunik, S.P. Intracellular pH Sensor Based on Heteroleptic Bis-Cyclometalated Iridium (III) Complex Embedded into Block-Copolymer Nanospecies: Application in Phosphorescence Lifetime Imaging Microscopy. Adv. Funct. Mater. 2023, 33, 2212390. [Google Scholar] [CrossRef]
- Papkovsky, D.B.; O’Riordan, T.C. Emerging Applications of Phosphorescent Metalloporphyrins. J. Fluoresc. 2005, 15, 569–584. [Google Scholar] [CrossRef]
- Ji, M.; Ma, X. Recent progress with the application of organic room-temperature phosphorescent materials. Ind. Chem. Mater. 2023, 1, 582–594. [Google Scholar] [CrossRef]
- Shcheslavskiy, V.I.; Neubauer, A.; Bukowiecki, R.; Dinter, F.; Becker, W. Combined fluorescence and phosphorescence lifetime imaging. Appl. Phys. Lett. 2016, 108, 091111. [Google Scholar] [CrossRef]
- Papkovsky, D.B.; Dmitriev, R.I.; Borisov, S. Quenched-Phosphorescence Detection of Molecular Oxygen: Applications in Life Sciences; Papkovsky, D.B., Dmitriev, R.I., Eds.; The Royal Society of Chemistry: London, UK, 2018; ISBN 1788011759. [Google Scholar]
- Esipova, T.V.; Karagodov, A.; Miller, J.; Wilson, D.F.; Busch, T.M.; Vinogradov, S.A. Two new “protected” oxyphors for biological oximetry: Properties and application in tumor imaging. Anal. Chem. 2011, 83, 8756–8765. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Hirakawa, Y.; Hosaka, M.; Nangaku, M.; Tobita, S. Oxygen imaging of living cells and tissues using luminescent molecular probes. J. Photochem. Photobiol. C Photochem. Rev. 2017, 30, 71–95. [Google Scholar] [CrossRef]
- Parshina, Y.P.; Komarova, A.D.; Bochkarev, L.N.; Kovylina, T.A.; Plekhanov, A.A.; Klapshina, L.G.; Konev, A.N.; Mozherov, A.M.; Shchechkin, I.D.; Sirotkina, M.A.; et al. Simultaneous Probing of Metabolism and Oxygenation of Tumors In Vivo Using FLIM of NAD(P)H and PLIM of a New Polymeric Ir(III) Oxygen Sensor. Int. J. Mol. Sci. 2022, 23, 10263. [Google Scholar] [CrossRef] [PubMed]
- Heikal, A.A. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark. Med. 2010, 4, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Georgakoudi, I.; Quinn, K.P. Optical imaging using endogenous contrast to assess metabolic state. Annu. Rev. Biomed. Eng. 2012, 14, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Druzhkova, I.; Nikonova, E.; Ignatova, N.; Koryakina, I.; Zyuzin, M.; Mozherov, A.; Kozlov, D.; Krylov, D.; Kuznetsova, D.; Lisitsa, U.; et al. Effect of Collagen Matrix on Doxorubicin Distribution and Cancer Cells’ Response to Treatment in 3D Tumor Model. Cancers 2022, 14, 5487. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, S.; Breymayer, J.; Schäfer, P.; Calzia, E.; Shcheslavskiy, V.; Becker, W.; Rück, A. Correlative NAD(P)H-FLIM and oxygen sensing-PLIM for metabolic mapping. J. Biophotonics 2016, 9, 800–811. [Google Scholar] [CrossRef]
- Ma, Y.; Liang, H.; Zeng, Y.; Yang, H.; Ho, C.-L.; Xu, W.; Zhao, Q.; Huang, W.; Wong, W.-Y. Phosphorescent soft salt for ratiometric and lifetime imaging of intracellular pH variations. Chem. Sci. 2016, 7, 3338–3346. [Google Scholar] [CrossRef]
- Rebillard, A.; Tekpli, X.; Meurette, O.; Sergent, O.; LeMoigne-Muller, G.; Vernhet, L.; Gorria, M.; Chevanne, M.; Christmann, M.; Kaina, B.; et al. Cisplatin-induced apoptosis involves membrane fluidification via inhibition of NHE1 in human colon cancer cells. Cancer Res. 2007, 67, 7865–7874. [Google Scholar] [CrossRef]
- Nishino, K.; Yamasaki, S.; Nakashima, R.; Zwama, M.; Hayashi-Nishino, M. Function and Inhibitory Mechanisms of Multidrug Efflux Pumps. Front. Microbiol. 2021, 12, 737288. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.; Kumar, G. Cancer multidrug-resistance reversal by ABCB1 inhibition: A recent update. Eur. J. Med. Chem. 2022, 239, 114542. [Google Scholar] [CrossRef] [PubMed]
- Gourdier, I.; Crabbe, L.; Andreau, K.; Pau, B.; Kroemer, G. Oxaliplatin-induced mitochondrial apoptotic response of colon carcinoma cells does not require nuclear DNA. Oncogene 2004, 23, 7449–7457. [Google Scholar] [CrossRef]
- Tafani, M.; Cohn, J.A.; Karpinich, N.O.; Rothman, R.J.; Russo, M.A.; Farber, J.L. Regulation of intracellular pH mediates Bax activation in HeLa cells treated with staurosporine or tumor necrosis factor-alpha. J. Biol. Chem. 2002, 277, 49569–49576. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Avila, A.; Gollahon, L. Paclitaxel induces apoptosis in breast cancer cells through different calcium—Regulating mechanisms depending on external calcium conditions. Int. J. Mol. Sci. 2014, 15, 2672–2694. [Google Scholar] [CrossRef] [PubMed]
- Danthuluri, N.; Kim, D.; Brock, T. Intracellular alkalinization leads to Ca2+ mobilization from agonist-sensitive pools in bovine aortic endothelial cells. J. Biol. Chem. 1990, 265, 19071–19076. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hao, B.; Lu, Y.; Yu, P.; Lee, H.-C.; Yue, J. Intracellular alkalinization induces cytosolic Ca2+ increases by inhibiting sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). PLoS ONE 2012, 7, e31905. [Google Scholar] [CrossRef]
- Shirmanova, M.V.; Druzhkova, I.N.; Lukina, M.M.; Matlashov, M.E.; Belousov, V.V.; Snopova, L.B.; Prodanetz, N.N.; Dudenkova, V.V.; Lukyanov, S.A.; Zagaynova, E.V. Intracellular pH imaging in cancer cells in vitro and tumors in vivo using the new genetically encoded sensor SypHer2. Biochim. Biophys. Acta 2015, 1850, 1905–1911. [Google Scholar] [CrossRef]
- Lukina, M.M.; Dudenkova, V.V.; Shimolina, L.E.; Snopova, L.B.; Zagaynova, E.V.; Shirmanova, M.V. In vivo metabolic and SHG imaging for monitoring of tumor response to chemotherapy. Cytometry. Part A J. Int. Soc. Anal. Cytol. 2019, 95, 47–55. [Google Scholar] [CrossRef]
- Shirshin, E.A.; Shirmanova, M.V.; Gayer, A.V.; Lukina, M.M.; Nikonova, E.E.; Yakimov, B.P.; Budylin, G.S.; Dudenkova, V.V.; Ignatova, N.I.; Komarov, D.V.; et al. Label-free sensing of cells with fluorescence lifetime imaging: The quest for metabolic heterogeneity. Proc. Natl. Acad. Sci. USA 2022, 119, e2118241119. [Google Scholar] [CrossRef]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chen, G.; Jiang, W.; Fu, M.; Liu, W.; Sui, J.; Xu, S.; Liu, Z.; Zheng, X.; Chi, L.; et al. Association of the Collagen Signature in the Tumor Microenvironment with Lymph Node Metastasis in Early Gastric Cancer. JAMA Surg. 2019, 154, e185249. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xue, F.; Li, M.; Zhu, X.; Lu, X.; Wang, C.; Xu, E.; Wang, X.; Zhang, L.; Yu, H.; et al. Extracellular Matrix Characterization in Gastric Cancer Helps to Predict Prognosis and Chemotherapy Response. Front. Oncol. 2021, 11, 753330. [Google Scholar] [CrossRef] [PubMed]
- Devarasetty, M.; Dominijanni, A.; Herberg, S.; Shelkey, E.; Skardal, A.; Soker, S. Simulating the human colorectal cancer microenvironment in 3D tumor-stroma co-cultures in vitro and in vivo. Sci. Rep. 2020, 10, 9832. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Jia, H.; Wu, Q.; Zhang, Y.; Ma, Q.; Yao, D.; Gao, X.; Xie, D.; Xu, Z.; Zhao, Q.; et al. Inhibition of Angiogenesis and Extracellular Matrix Remodeling: Synergistic Effect of Renin-Angiotensin System Inhibitors and Bevacizumab. Front. Oncol. 2022, 12, 829059. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhang, R.; Feng, H. Fibronectin promotes tumor cells growth and drugs resistance through a CDC42-YAP-dependent signaling pathway in colorectal cancer. Cell Biol. Int. 2020, 44, 1840–1849. [Google Scholar] [CrossRef]
- D’Angelo, E.; Natarajan, D.; Sensi, F.; Ajayi, O.; Fassan, M.; Mammano, E.; Pilati, P.; Pavan, P.; Bresolin, S.; Preziosi, M.; et al. Patient-Derived Scaffolds of Colorectal Cancer Metastases as an Organotypic 3D Model of the Liver Metastatic Microenvironment. Cancers 2020, 12, 364. [Google Scholar] [CrossRef]
- Druzhkova, I.; Shirmanova, M.; Ignatova, N.; Dudenkova, V.; Lukina, M.; Zagaynova, E.; Safina, D.; Kostrov, S.; Didych, D.; Kuzmich, A.; et al. Expression of EMT-Related Genes in Hybrid E/M Colorectal Cancer Cells Determines Fibroblast Activation and Collagen Remodeling. Int. J. Mol. Sci. 2020, 21, 8119. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combination | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Oxaliplatin, μM | 0.5 | 1 | 2 | 5 | 8 | 10 | 20 |
| Leucovorin, μM | 1 | 2 | 4 | 8 | 16 | 20 | 40 |
| 5-Fluorouracil, μM | 2 | 4 | 7 | 14 | 25 | 35 | 70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Druzhkova, I.; Komarova, A.; Nikonova, E.; Baigildin, V.; Mozherov, A.; Shakirova, Y.; Lisitsa, U.; Shcheslavskiy, V.; Ignatova, N.; Shirshin, E.; et al. Monitoring the Intracellular pH and Metabolic State of Cancer Cells in Response to Chemotherapy Using a Combination of Phosphorescence Lifetime Imaging Microscopy and Fluorescence Lifetime Imaging Microscopy. Int. J. Mol. Sci. 2024, 25, 49. https://doi.org/10.3390/ijms25010049
Druzhkova I, Komarova A, Nikonova E, Baigildin V, Mozherov A, Shakirova Y, Lisitsa U, Shcheslavskiy V, Ignatova N, Shirshin E, et al. Monitoring the Intracellular pH and Metabolic State of Cancer Cells in Response to Chemotherapy Using a Combination of Phosphorescence Lifetime Imaging Microscopy and Fluorescence Lifetime Imaging Microscopy. International Journal of Molecular Sciences. 2024; 25(1):49. https://doi.org/10.3390/ijms25010049
Chicago/Turabian StyleDruzhkova, Irina, Anastasiya Komarova, Elena Nikonova, Vadim Baigildin, Artem Mozherov, Yuliya Shakirova, Uliana Lisitsa, Vladislav Shcheslavskiy, Nadezhda Ignatova, Evgeny Shirshin, and et al. 2024. "Monitoring the Intracellular pH and Metabolic State of Cancer Cells in Response to Chemotherapy Using a Combination of Phosphorescence Lifetime Imaging Microscopy and Fluorescence Lifetime Imaging Microscopy" International Journal of Molecular Sciences 25, no. 1: 49. https://doi.org/10.3390/ijms25010049