
A Knockout of the IFITM3 Gene Increases the Sensitivity of WI-38 VA13 Cells to the Influenza A Virus

 , , , and
, , , and
Abstract
:1. Introduction
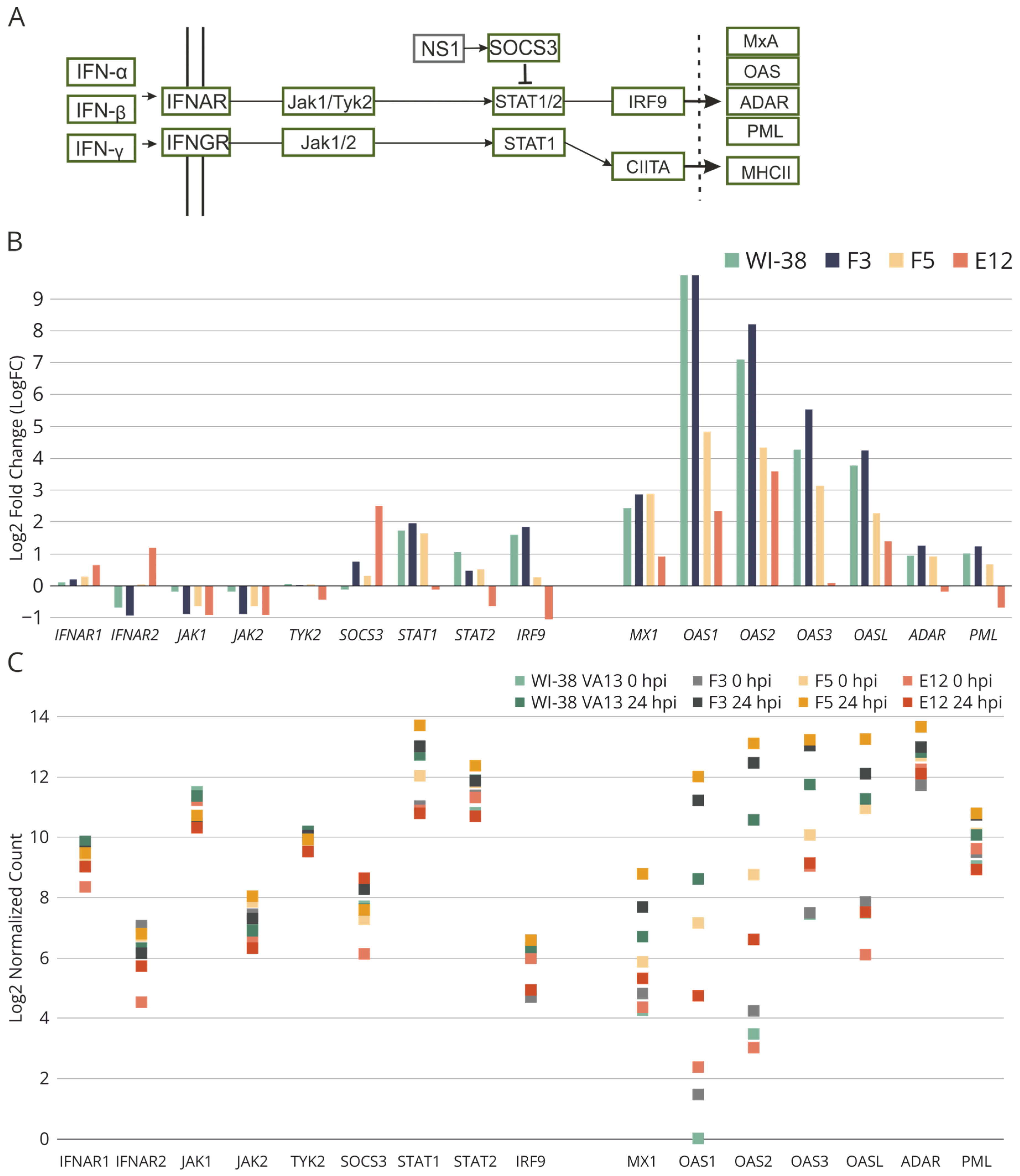
2. Results
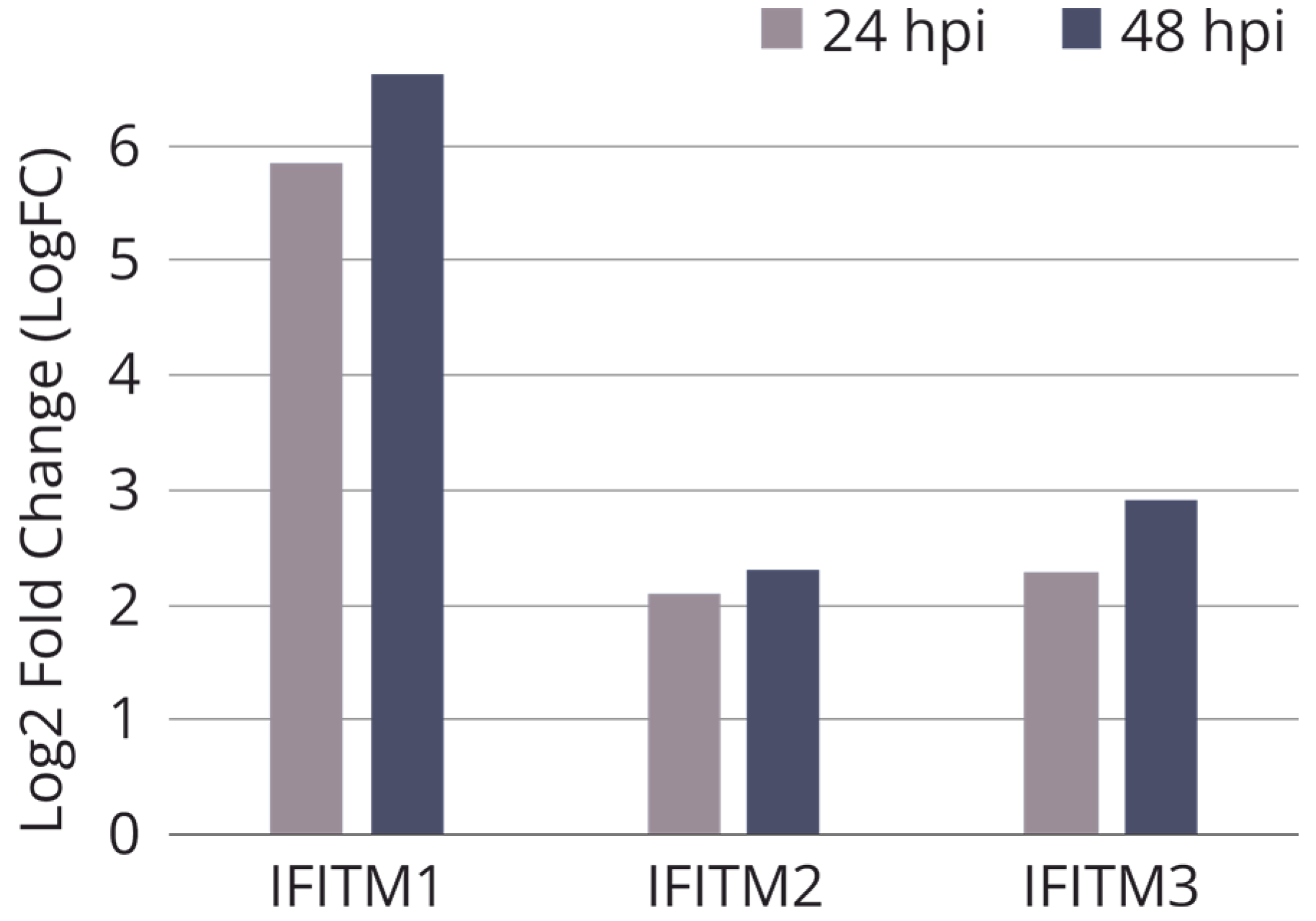
2.1. The Choice of a Target Gene and Cell Line for the CRISPR/Cas9 Editing
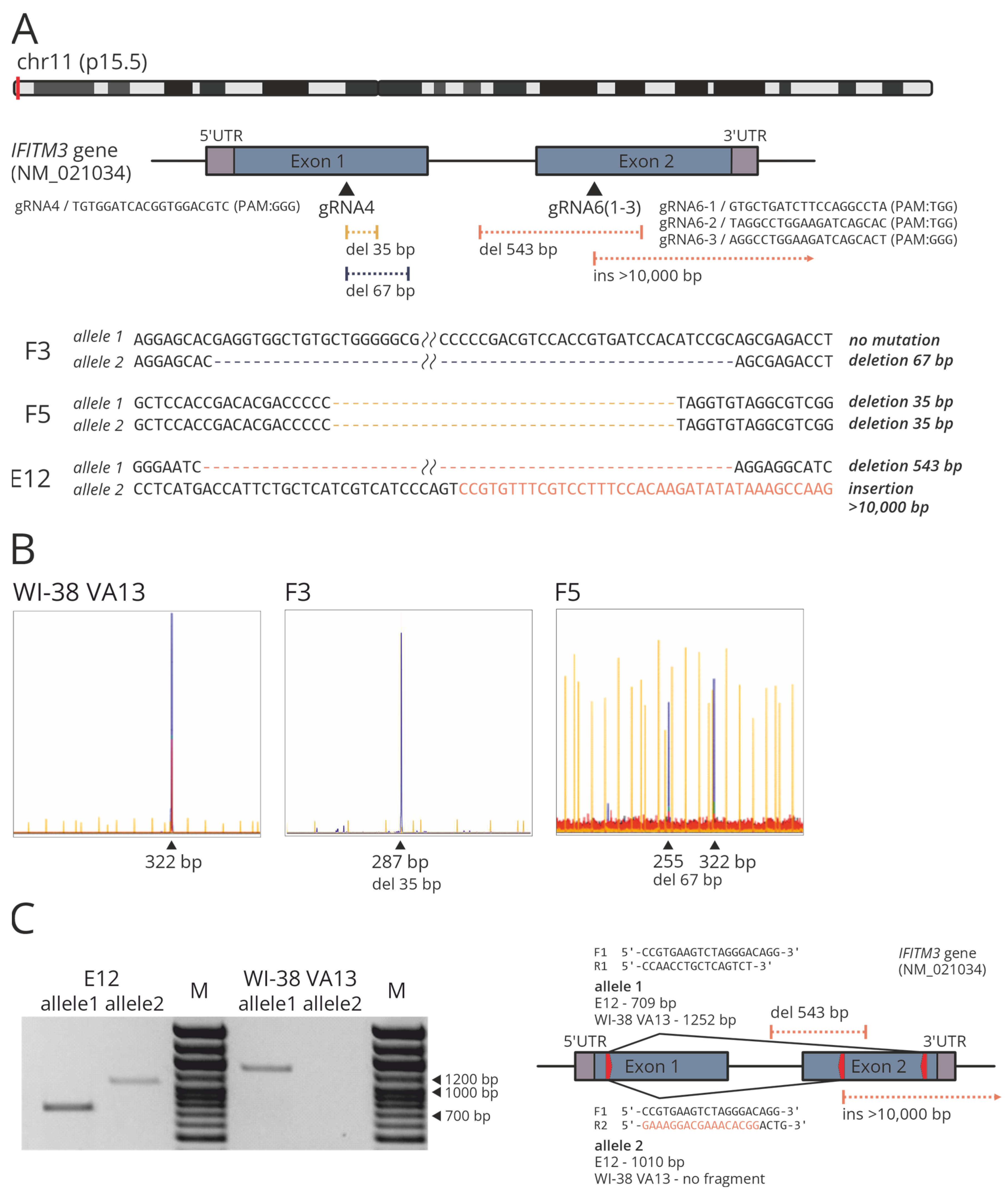
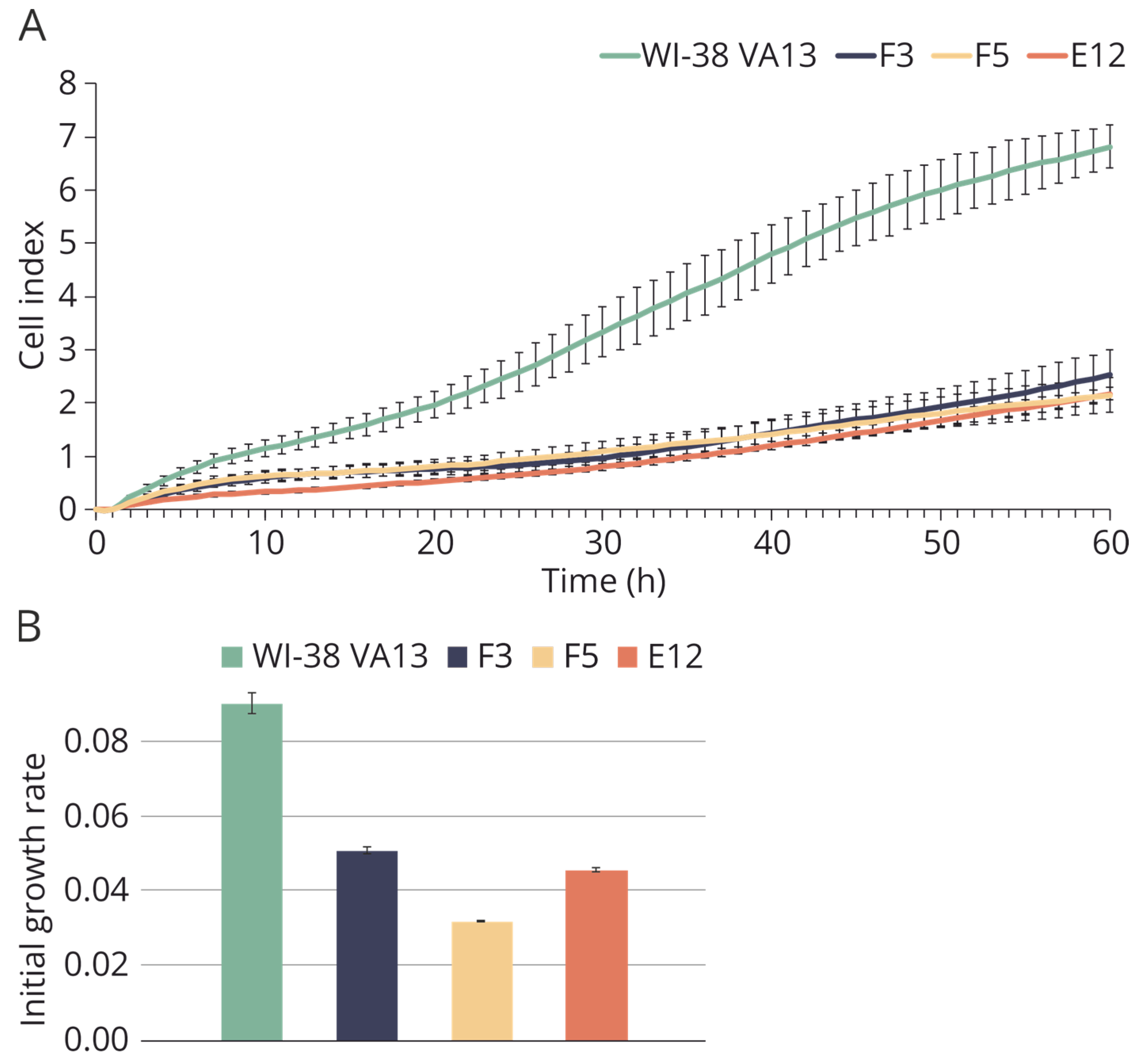
2.2. Construction of Cell Lines with Knocked Out IFITM3
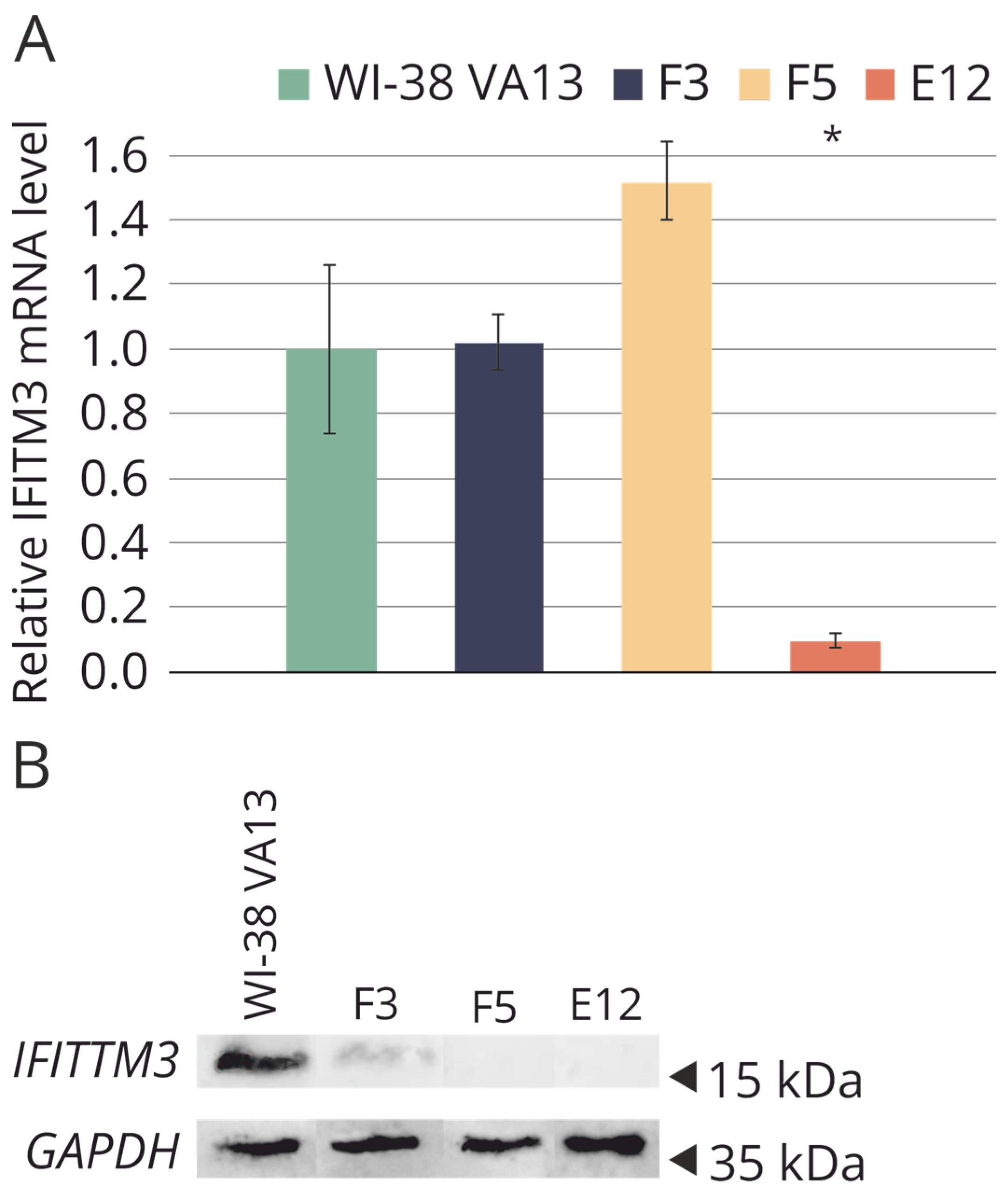
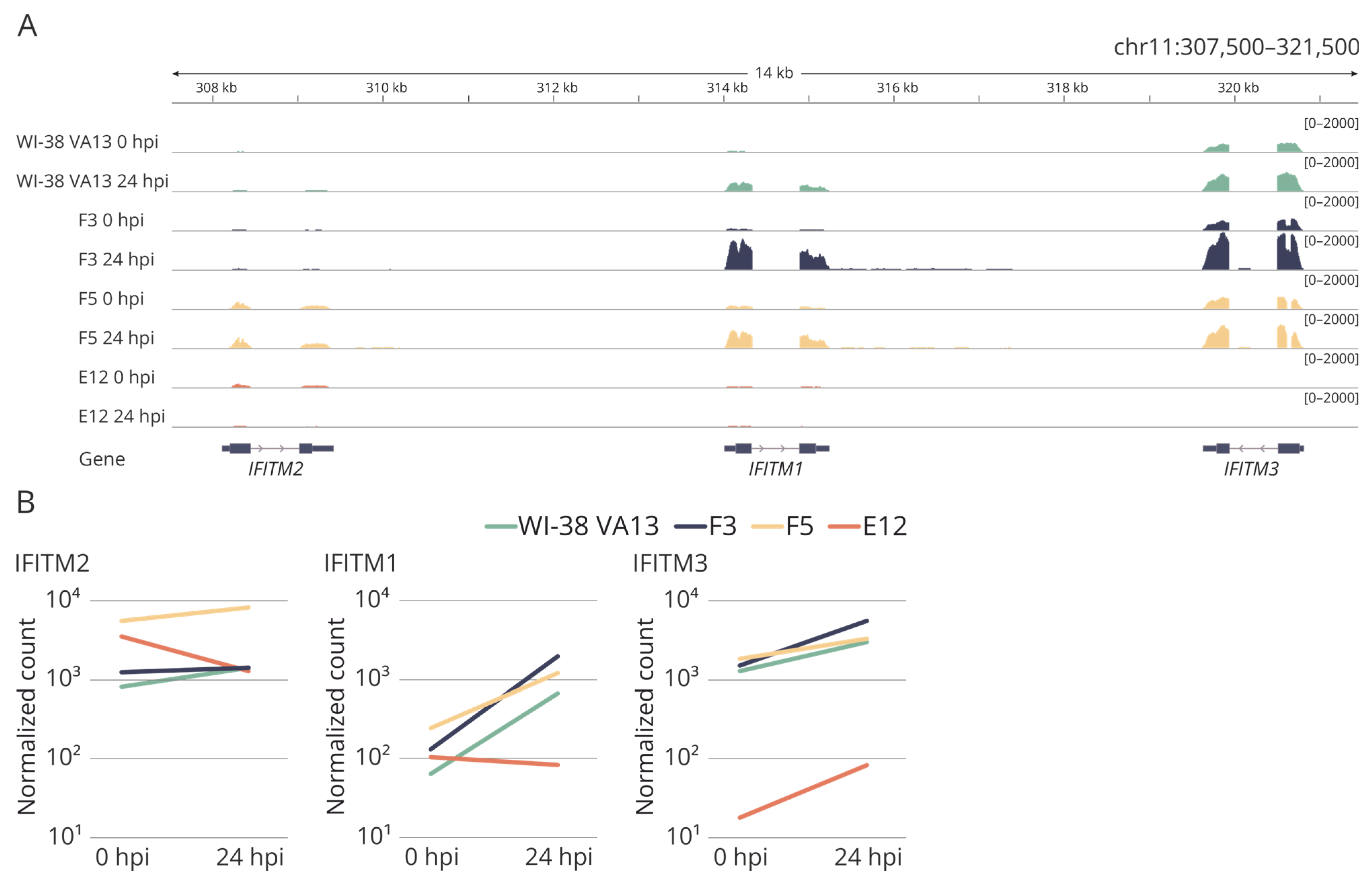
2.3. Evaluation of IFITM3 mRNA Expression
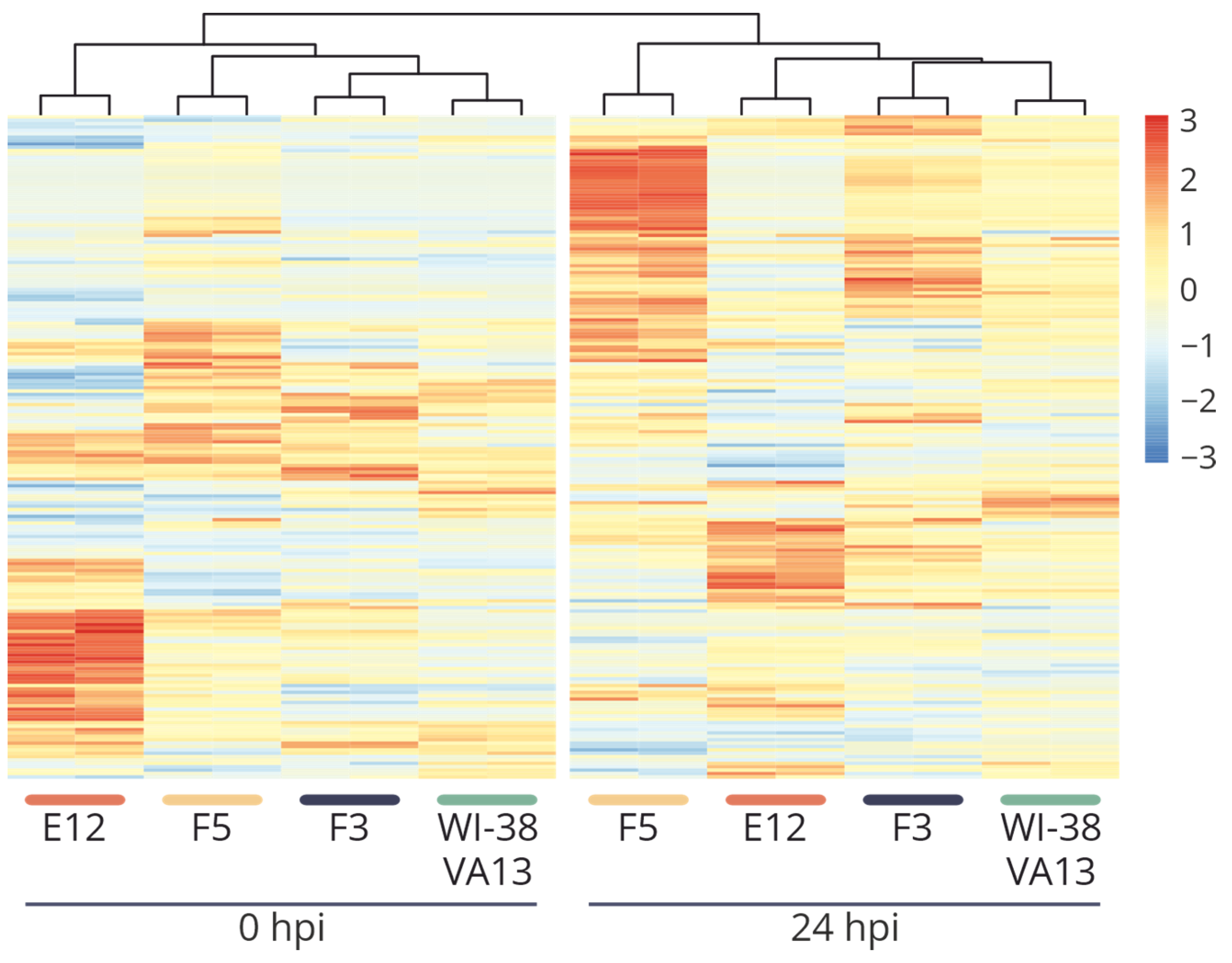
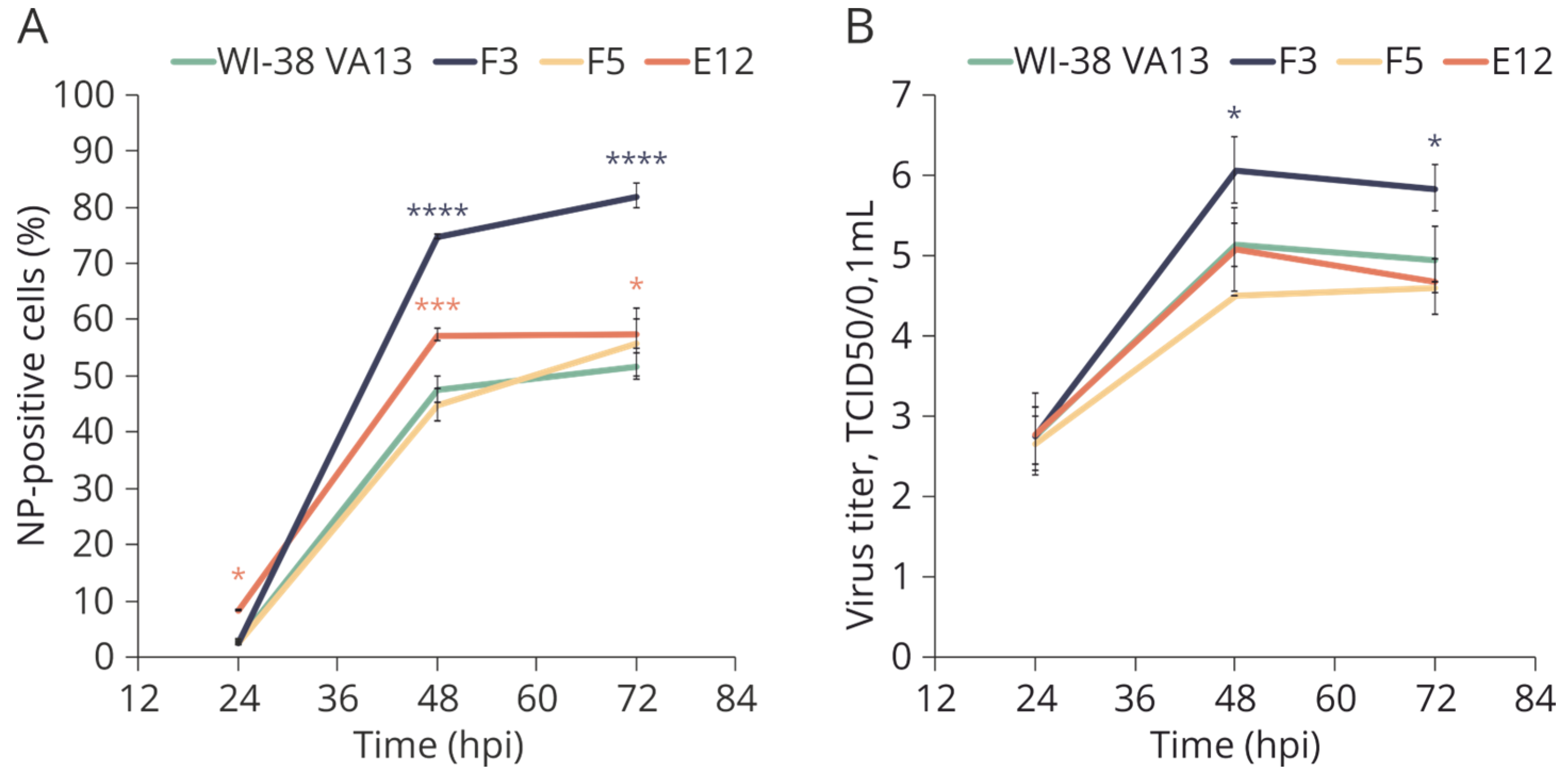
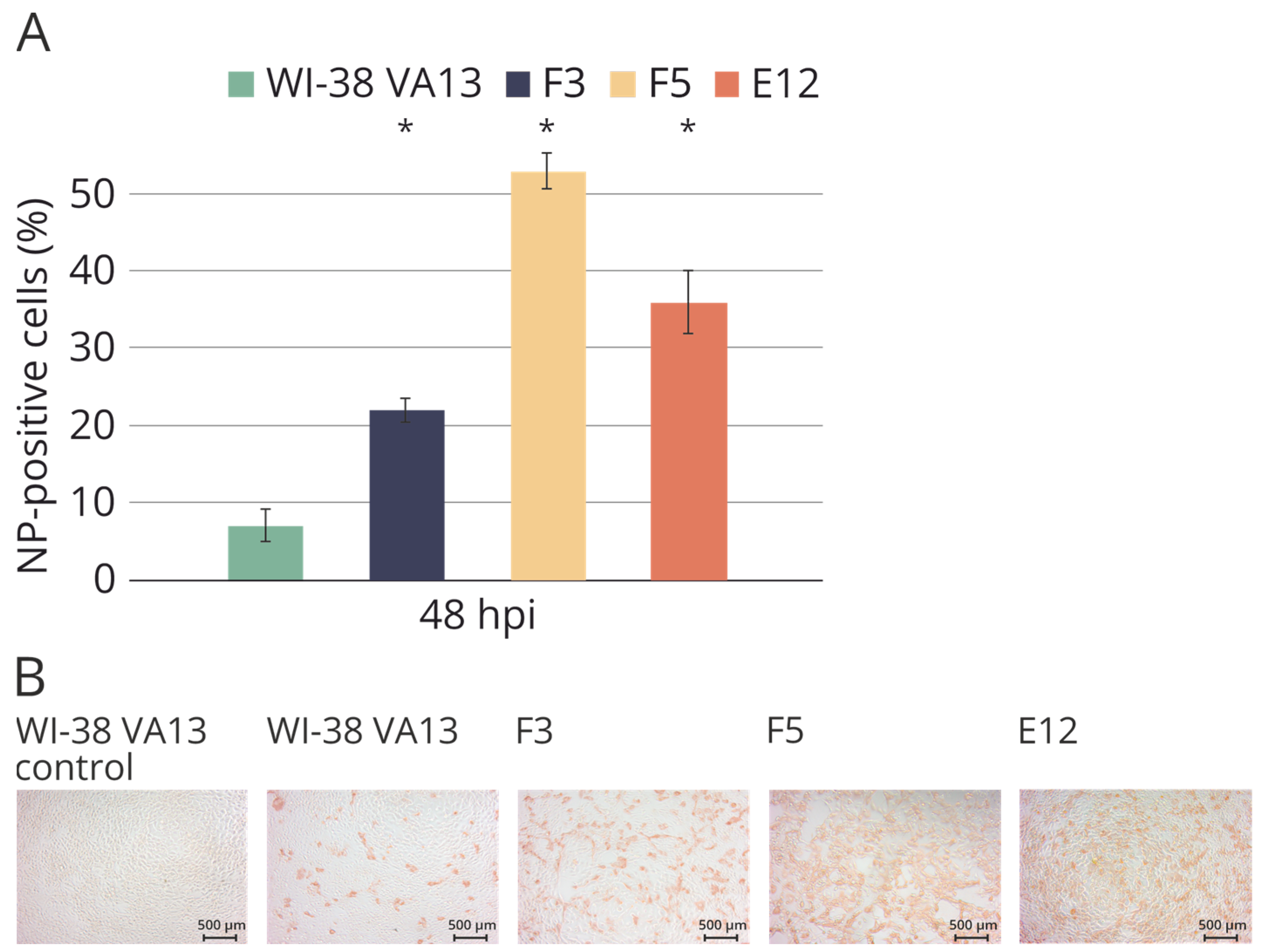
2.4. Sensitivity of WI-38 VA13 IFITM3−/− Cell Lines to Influenza Infection
3. Discussion
4. Materials and Methods
4.1. Construction of Plasmids Expressing Components of CRISPR/Cas9 Systems Directed to IFITM3 Gene Knockout
4.2. Cell Transfection and Cloning
4.3. Analysis of the Monoclonal Cell Lines for IFITM3 Gene Mutations
4.4. Detection of Deletions and Insertions in Monoclonal Cell Lines
4.5. Real-Time Cell Growth Analysis
4.6. Quantification of mRNA and Viral RNA Expression by RT-PCR
4.7. Detection of the IFITM3 Protein in the Monoclonal Cell Lines by Western Blotting
4.8. Infection of Cells with the Influenza A Virus
4.9. Analysis of Transcriptome-Sequencing Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Prim. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Somes, M.P.; Turner, R.M.; Dwyer, L.J.; Newall, A.T. Estimating the annual attack rate of seasonal influenza among unvaccinated individuals: A systematic review and meta-analysis. Vaccine 2018, 36, 3199–3207. [Google Scholar] [CrossRef] [PubMed]
- Vaccines against Influenza: WHO Position Paper—May 2022. Available online: https://www.who.int/publications/i/item/who-wer9719 (accessed on 4 October 2023).
- Ott, J.J.; Breteler, J.K.; Tam, J.S.; Hutubessy, R.C.W.; Jit, M.; De Boer, M.R. Influenza vaccines in low and middle income countries: A systematic review of economic evaluations. Hum. Vaccines Immunother. 2013, 9, 1500–1511. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, T.; Rivetti, A.; Di Pietrantonj, C.; Demicheli, V. Vaccines for preventing influenza in healthy children. Cochrane Database Syst. Rev. 2018, 2, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F. Influenza Virus Infections of the Chick Embryo by the Amniotic Route. Aust. J. Exp. Biol. Med. Sci. 1940, 18, 353–360. [Google Scholar] [CrossRef]
- Burnet, F. Growth of influenza virus in the allantoic cavity of the chick embryo. Aust. J. Exp. Biol. Med. Sci. 1941, 19, 291–295. [Google Scholar] [CrossRef]
- Skowronski, D.M.; Janjua, N.Z.; De Serres, G.; Sabaiduc, S.; Eshaghi, A.; Dickinson, J.A.; Fonseca, K.; Winter, A.L.; Gubbay, J.B.; Krajden, M.; et al. Low 2012–13 Influenza Vaccine Effectiveness Associated with Mutation in the Egg-Adapted H3N2 Vaccine Strain Not Antigenic Drift in Circulating Viruses. PLoS ONE 2014, 9, e92153. [Google Scholar] [CrossRef]
- Zost, S.J.; Parkhouse, K.; Gumina, M.E.; Kim, K.; Perez, S.D.; Wilson, P.C.; Treanor, J.J.; Sant, A.J.; Cobey, S.; Hensley, S.E. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl. Acad. Sci. USA 2017, 114, 12578–12583. [Google Scholar] [CrossRef]
- Coop, C.A.; Balanon, S.K.; White, K.M.; Whisman, B.A.; Rathkopf, M.M. Anaphylaxis from the influenza virus vaccine. Int. Arch. Allergy Immunol. 2008, 146, 85–88. [Google Scholar] [CrossRef]
- Wolff, M.W.; Reichl, U. Downstream Processing: From Egg to Cell Culture-Derived Influenza Virus Particles. Chem. Eng. Technol. 2008, 31, 846–857. [Google Scholar] [CrossRef]
- Lamb, Y.N. Cell-Based Quadrivalent Inactivated Influenza Virus Vaccine (Flucelvax® Tetra/Flucelvax Quadrivalent®): A Review in the Prevention of Influenza. Drugs 2019, 79, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Nolan, T.; Fortanier, A.C.; Leav, B.; Põder, A.; Bravo, L.C.; Szymański, H.T.; Heeringa, M.; Vermeulen, W.; Matassa, V.; Smolenov, I.; et al. Efficacy of a Cell-Culture-Derived Quadrivalent Influenza Vaccine in Children. N. Engl. J. Med. 2021, 385, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.S.; Cook, P.; Attwell, A.M.; Williams, S.P. Replicative advantage in tissue culture of egg-adapted influenza virus over tissue-culture derived virus: Implications for vaccine manufacture. Vaccine 1995, 13, 1583–1588. [Google Scholar] [CrossRef]
- Lu, B.; Zhou, H.; Chan, W.; Kemble, G.; Jin, H. Single amino acid substitutions in the hemagglutinin of influenza A/Singapore/21/04 (H3N2) increase virus growth in embryonated chicken eggs. Vaccine 2006, 24, 6691–6693. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.R. Adventitious Agents and Vaccines. Emerg. Infect. Dis. 2001, 7, 562. [Google Scholar] [CrossRef]
- Gregersen, J.P. A quantitative risk assessment of exposure to adventitious agents in a cell culture-derived subunit influenza vaccine. Vaccine 2008, 26, 3332–3340. [Google Scholar] [CrossRef] [PubMed]
- Genzel, Y.; Dietzsch, C.; Rapp, E.; Schwarzer, J.; Reichl, U. MDCK and Vero cells for influenza virus vaccine production: A one-to-one comparison up to lab-scale bioreactor cultivation. Appl. Microbiol. Biotechnol. 2010, 88, 461–475. [Google Scholar] [CrossRef]
- Gregersen, J.P.; Schmitt, H.J.; Trusheim, H.; Bröker, M. Safety of MDCK cell culture-based influenza vaccines. Future Microbiol. 2011, 6, 143–152. [Google Scholar] [CrossRef]
- Ehrlich, H.J.; Berezuk, G.; Fritsch, S.; Aichinger, G.; Singer, J.; Portsmouth, D.; Hart, M.K.; El-Amin, W.; Kistner, O.; Barrett, P.N. Clinical development of a Vero cell culture-derived seasonal influenza vaccine. Vaccine 2012, 30, 4377–4386. [Google Scholar] [CrossRef]
- Schwarzer, J.; Rapp, E.; Hennig, R.; Genzel, Y.; Jordan, I.; Sandig, V.; Reichl, U. Glycan analysis in cell culture-based influenza vaccine production: Influence of host cell line and virus strain on the glycosylation pattern of viral hemagglutinin. Vaccine 2009, 27, 4325–4336. [Google Scholar] [CrossRef]
- Hütter, J.; Rödig, J.V.; Höper, D.; Seeberger, P.H.; Reichl, U.; Rapp, E.; Lepenies, B. Toward animal cell culture-based influenza vaccine design: Viral hemagglutinin N-glycosylation markedly impacts immunogenicity. J. Immunol. 2013, 190, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Global Influenza Strategy 2019–2030. Available online: https://www.who.int/publications/i/item/9789241515320 (accessed on 10 October 2023).
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315. [Google Scholar] [CrossRef] [PubMed]
- Flerlage, T.; Boyd, D.F.; Meliopoulos, V.; Thomas, P.G.; Schultz-Cherry, S. Influenza virus and SARS-CoV-2: Pathogenesis and host responses in the respiratory tract. Nat. Rev. Microbiol. 2021, 19, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Prokhorova, D.; Zhukova, N.; Lemza, A.; Sergeeva, M.; Amirkhanov, R.; Stepanov, G. Application of the CRISPR/Cas9 System to Study Regulation Pathways of the Cellular Immune Response to Influenza Virus. Viruses 2022, 14, 437. [Google Scholar] [CrossRef] [PubMed]
- Hickford, D.; Frankenberg, S.; Shaw, G.; Renfree, M.B. Evolution of vertebrate interferon inducible transmembrane proteins. BMC Genom. 2012, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, J.; Winkler, C.A.; An, P.; Guo, J.T. IFITM genes, variants, and their roles in the control and pathogenesis of viral infections. Front. Microbiol. 2019, 10, 3228. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Lei, N.; Lu, J.; Gao, R.B.; Li, Z.; Liu, L.Q.; Sun, Y.; Guo, J.F.; Wang, D.Y.; Shu, Y.L. Interferon-induced Transmembrane Protein 3 Prevents Acute Influenza Pathogenesis in Mice. Biomed. Environ. Sci. 2020, 33, 295–305. [Google Scholar] [CrossRef]
- Jiménez-Munguía, I.; Beaven, A.H.; Blank, P.S.; Sodt, A.J.; Zimmerberg, J. Interferon-induced transmembrane protein 3 (IFITM3) and its antiviral activity. Curr. Opin. Struct. Biol. 2022, 77, 102467. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef]
- Olshansky, S.J.; Hayflick, L. The Role of the WI-38 Cell Strain in Saving Lives and Reducing Morbidity. AIMS Public Health 2017, 4, 127–138. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Steentoft, C.; Hauge, C.; Hansen, L.; Thomsen, A.L.; Niola, F.; Vester-Christensen, M.B.; Frödin, M.; Clausen, H.; Wandall, H.H.; et al. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015, 43, e59. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Shi, M.L. Coordinated regulation of IFITM1, 2 and 3 genes by an IFN-responsive enhancer through long-range chromatin interactions. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 August 2020).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Lè Ne Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Sequence analysis STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. Sequence analysis featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Regulation | Number |
|---|---|---|
| WI-38 VA13 (24 hpi vs. 0 hpi) | Upregulated | 177 |
| Downregulated | 3 | |
| F3 (24 hpi vs. 0 hpi) | Upregulated | 364 |
| Downregulated | 54 | |
| F5 (24 hpi vs. 0 hpi) | Upregulated | 397 |
| Downregulated | 43 | |
| E12 (24 hpi vs. 0 hpi) | Upregulated | 965 |
| Downregulated | 334 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eshchenko, N.; Sergeeva, M.; Zhuravlev, E.; Kudria, K.; Goncharova, E.; Komissarov, A.; Stepanov, G. A Knockout of the IFITM3 Gene Increases the Sensitivity of WI-38 VA13 Cells to the Influenza A Virus. Int. J. Mol. Sci. 2024, 25, 625. https://doi.org/10.3390/ijms25010625
Eshchenko N, Sergeeva M, Zhuravlev E, Kudria K, Goncharova E, Komissarov A, Stepanov G. A Knockout of the IFITM3 Gene Increases the Sensitivity of WI-38 VA13 Cells to the Influenza A Virus. International Journal of Molecular Sciences. 2024; 25(1):625. https://doi.org/10.3390/ijms25010625
Chicago/Turabian StyleEshchenko, Natalya, Mariia Sergeeva, Evgenii Zhuravlev, Kira Kudria, Elena Goncharova, Andrey Komissarov, and Grigory Stepanov. 2024. "A Knockout of the IFITM3 Gene Increases the Sensitivity of WI-38 VA13 Cells to the Influenza A Virus" International Journal of Molecular Sciences 25, no. 1: 625. https://doi.org/10.3390/ijms25010625
APA StyleEshchenko, N., Sergeeva, M., Zhuravlev, E., Kudria, K., Goncharova, E., Komissarov, A., & Stepanov, G. (2024). A Knockout of the IFITM3 Gene Increases the Sensitivity of WI-38 VA13 Cells to the Influenza A Virus. International Journal of Molecular Sciences, 25(1), 625. https://doi.org/10.3390/ijms25010625