Abstract
A hallmark of Alzheimer’s disease (AD) are the proteinaceous aggregates formed by the amyloid-beta peptide (Aβ) that is deposited inside the brain as amyloid plaques. The accumulation of aggregated Aβ may initiate or enhance pathologic processes in AD. According to the amyloid hypothesis, any agent that has the capability to inhibit Aβ aggregation and/or destroy amyloid plaques represents a potential disease-modifying drug. In 2023, a humanized IgG1 monoclonal antibody (lecanemab) against the Aβ-soluble protofibrils was approved by the US FDA for AD therapy, thus providing compelling support to the amyloid hypothesis. To acquire a deeper insight on the in vivo Aβ aggregation, various animal models, including aged herbivores and carnivores, non-human primates, transgenic rodents, fish and worms were widely exploited. This review is based on the recent data obtained using transgenic animal AD models and presents experimental verification of the critical role in Aβ aggregation seeding of the interactions between zinc ions, Aβ with the isomerized Asp7 (isoD7-Aβ) and the α4β2 nicotinic acetylcholine receptor.
1. Introduction
A most common type of dementia in older adults, Alzheimer’s disease (AD), is a severe neurodegenerative pathology manifested by progressive cognitive decline, such as memory loss and suppressed logical thinking [1]. Currently, AD is defined by the following neuropathological profile: (1) deposition of amyloid-beta peptide (Aβ) aggregates in the form of diffuse and amyloid (neuritic, senile) plaques [2] and (2) the presence of intraneuronal neurofibrillary tangles and neuropil filaments (in dystrophic neurites) comprised of aggregated hyperphosphorylated tau protein [3,4]. These pathomorphological features are observed in certain areas of the brain [5,6], but are particularly common in the hippocampus, an area of the brain critical for learning and memory, where amyloid plaques, neurofibrillary tangles and neuropil threads appear at the earliest stages of AD [7,8]. In addition to classical neuropathological features, AD is characterized by systemic abnormalities and disorders of brain metabolism that appear at the molecular and biochemical levels and include cholinergic failure [9], neuroinflammation, activation of apoptosis, mitochondrial dysfunction, metabolic disorders and chronic oxidative stress [10].
Hereditary AD variants, which are characterized by the early onset and more rapid progression, account for less than 1% of all cases of this pathology and are associated with mutations [11,12] that lead to an excess of physiologically normal Aβ levels. Sporadic AD variants, which are characterized by later onset and a relatively slow progression, affect over 95% of patients and, similar to the inherited variants, are closely connected with the abnormal aggregation of endogenous Aβ [2]. Compared with the patients with late-onset Alzheimer’s disease, patients with familial AD variants have more amyloid plaques and more developed cerebral amyloid angiopathy [13].
Normal endogenous Aβ is a small polypeptide molecule of 39–43 amino acid residues [14]. Aβ is produced by proteolysis of the amyloid precursor protein (APP) [14] and is present in both the brain tissue and peripheral organs [15]. The physiological roles of Aβ may include regulation of the synaptic function, protection against infection, repair of the damaged areas in the blood–brain barrier, and compensatory role for the effects of injury [16]. In the process of the AD pathogenesis, soluble dimers and oligomers of Aβ appear in biological fluids of the body. These Aβ species, when they bind to cellular receptors, cause the dysfunction and degeneration of synapses [17]. Presumably in the later stages of AD, Aβ oligomers stay in dynamic equilibrium with aggregated Aβ molecules of the amyloid plaques [18]. The most commonly occurring sequence in amyloid plaques Aβ42 contains 42 amino acid residues [19]. In addition to Aβ and its chemically modified isoforms [20], amyloid plaques include many other components, such as proteoglycans, carbohydrate-binding proteins of the innate immune system, nucleic acids, biometal ions, lipids, and transport proteins [21]. It is believed that such components can seriously affect the processes of Aβ aggregation in AD pathogenesis [22].
Analysis of the morphology of amyloid fibrils isolated from the brain tissue of patients diagnosed with AD showed that, despite the polymorphism of the fibrils, they all consist of protofilaments with a similar structure [23]. The spatial structure of soluble Aβ monomers and oligomers cannot be obtained experimentally under the physiologically relevant conditions due to the spontaneous aggregation of Aβ at the concentrations required for modern physicochemical methods [24]. It is generally accepted that the conformational transformation and aggregation of monomeric Aβ molecules occurs via the nucleation mechanism [25,26,27]. According to this mechanism, a slow and thermodynamically unfavorable nucleation phase is followed by the fast polymerization phase [28]. In the nucleation phase, the stage that determines the integral rate of the entire Aβ aggregation is the formation of a stable nucleus of the polymerized protein. The nucleus must necessarily contain Aβ in an oligomeric state, albeit additional molecular agents, which along with Aβ are present in amyloid plaques, seem to play highly important role in the appearance of the seed of pathological Aβ aggregation [29].
The primary current hypotheses on the etiology of AD are (reviewed in [30]) (1) “amyloid cascade” [31]; (2) “protein aging” [32]; (3) “cholinergic deficit” [9]; (4) “zinc dyshomeostasis” [33]; and (5) “inflammatory” [34]. Yet, the prevailing experimental evidence on the pathological physiology of AD supports the amyloid cascade hypothesis for AD pathogenesis [31,35]. This hypothesis postulates that the accumulation of Aβ aggregates in the brain (cerebral amyloidogenesis) triggers a signaling cascade that causes pathological transformation of the tau protein, neuroinflammation, and neurodegeneration. Consequently, the appearance and spread of the extracellular Aβ aggregates (amyloid plaques) in brain tissue is one of the main pathological signs for both sporadic and hereditary variants of AD and, possibly, constitutes the primary pathogenetic process of AD [36].
The recently introduced Amyloid Cascade Hypothesis 2.0 (ACH2.0) interpretation of AD states that extracellular Aβ in general and Aβ plaques in particular are largely benign and, possibly, even physiologically protective, and that AD is actually driven by intraneuronal Aβ (iAβ) [37,38,39,40]. The causative role of iAβ is the central postulate of the ACH2.0, which envisions AD as a two-stage disease. In the first pre-symptomatic stage, AβPP-derived Aβ accumulates to critical levels in a decades-long process that induces the activation of the second, devastating symptomatic AD stage that is anchored and driven by an agent which is independent of AβPP and sustains and perpetuates its own production [37,38]. The crucial role of the AβPP proteolytic/secretory pathway in only the first, pre-symptomatic stage of AD explains why the drugs targeting extracellular AβPP-derived Aβ or its production by the AβPP proteolysis did not and indeed could not have any effect on symptomatic AD patients. The progression of the disease is driven at this stage by iAβ produced independently of AβPP and the drugs fail despite effectively fulfilling their mechanistic purpose. By the same logic, the overall success of the same drugs in the animal models suggests that no second AD stage occurs there, consistent with the inability to obtain full spectrum of AD pathology in those experimental systems [40]. However, the formation of amyloid plaques seems to occur through the same molecular mechanism in both humans and the animal models of Alzheimer’s disease.
From the time of the formulation of the amyloid hypothesis of AD in the early 1990s [31] and for many years, all drugs aimed at the prevention of Aβ aggregation failed in clinical trials [41]. As a result, until 2015 most pharmaceutical companies significantly limited further development of the anti-amyloid drugs aimed at the treatment of AD, and the amyloid hypothesis was discredited among a significant part of researchers [42,43]. However, full approval by the US FDA of the anti-Aβ antibody lecanemab (marketed as Leqembi) [44] in July 2023 marked a turning point in Alzheimer’s disease research and demonstrated the clinical benefits of anti-amyloid therapy [45,46,47].
This review addresses the advances in understanding the molecular mechanism of the formation of amyloid plaque in the nervous system of model animals and offers an analysis of the possibilities of using the structural determinants of amyloid-beta as drug targets for anti-amyloid therapy for Alzheimer’s disease.
2. Animal Models of Alzheimer’s Disease
The vast majority of AD cases have late onset, but their clinical and histopathological features are common with the early autosomal-dominant AD variants, which are the main target of animal modeling [48,49]. Autosomal-dominant variants of AD are associated with mutations in the genes that encode proteins involved in the generation of Aβ, including the APP gene, encoding amyloid-beta precursor protein (APP), as well as the PSEN1 and PSEN2 genes, encoding presenilin-1 (PS1) and presenilin-2 (PS2). APP is a transmembrane protein with the purported but as yet unclear roles in several aspects of the neuronal homeostasis. Most APP mutations cluster near the sites typically cleaved by proteases: α-, β-, and γ-secretases. PS1 and PS2 are components of the gamma-secretase complex responsible for the proteolytic cleavage of APP. AD-associated mutations in the genes APP, PSEN1, and PSEN2 promote Aβ formation by favoring proteolytic processing of APP via the β- or γ-secretase pathway rather than via the non-amyloidogenic α-secretase pathway. In the amyloidogenic pathway, APP cleavage is initiated by β-secretase. Subsequent intramembrane cleavage by γ-secretase results in the formation of 40- and 42-amino acid amyloid peptides (Aβ40 and Aβ42, respectively) The longer forms (Aβ42 or longer) display a greater tendency to self-aggregation [50]. These proteolytic processes and subsequent modifications result in the appearance of Aβ in the form of dimers, tetramers, oligomers, protofibrils, and amyloid fibrils [51].
The recognized in vivo AD models can be divided into spontaneous, interventional, and genetically modified [52,53]. Aβ accumulation and tau hyperphosphorylation can occur spontaneously in non-human primates (NHPs). For example, baboons show only a formation of neurofibrillary tangles, while macaques demonstrate amyloid deposition without evidence of tau pathology. However, these NHPs have a long lifespan, and spontaneous AD-like symptoms and pathological changes are usually observed only in the older adults. Consequently, although the spontaneous AD models using NHPs have research value, various factors such as high maintenance costs, low reproductive potential, handling problems, and risk of zoonotic transmission limit the use of these models. In the interventional models, chemicals are used to induce symptoms and pathological changes similar to those observed in the AD pathogenesis. In particular, in the interventional AD models, various chemicals are utilized to induce neuroinflammation [34].
The earliest animal AD models were created by disrupting the cholinergic system of the basal forebrain in various mammalian species using surgical techniques [54], neurotoxins, immunotoxins, and pharmacological methods. The target species included mice and rats, rabbits and monkeys [55]. These models reproduced some symptoms of AD, such as memory impairment, and were useful for testing the effectiveness of cholinesterase inhibitors, which may furnish some symptomatic relief at the early stages of AD. In these models, of course, neither plaques nor neurofibrillary tangles (NFT) were formed, nor did they reflect the development of the complex biochemical and cellular changes in AD. As a result, such models gradually lost their relevance in the AD research.
The spontaneous animal AD models, unlike transgenic AD models, do not include mutations associated with the hereditary AD variants, but are based on developing the pathology, which is accompanied by a decline in cognitive functions in old age. Therefore, spontaneous models can be used to evaluate the therapeutic approaches and to diagnose sporadic AD, which is the predominant form of AD. Amyloid plaques are found in the brains of the aged animals [56,57], indicating that this pathology is a common accompaniment of aging not only in humans, but also among other species. Neuritic plaques and cerebrovascular amyloid deposits were found in the aged monkeys, dogs and polar bears but were rarely found in aged rodents [58]. Larger animals naturally develop amyloid plaques and/or NFT as they age, and these features may be normal in larger animals. They were found in many species of primates, as well as in a number of large herbivores and carnivores. This propensity to form plaques appears to be due, at least in part, to the sequence conservation of the amyloid-beta peptide in most mammals [59] with the exception of rodents (mice and rats) [60]. In contrast to the simple demonstration of amyloid deposits, though the tau-associated tangle pathology was demonstrated in some species [61], it is much less common there than in the human brain. Thus, spontaneous animal AD models could potentially be useful to bridge the gap between the promising rodent studies and failed human clinical trials [62].
Evidently, there is no ideal animal model of Alzheimer’s disease, and any given model only reflects certain aspects of the condition of a patient with AD. Mice species are most commonly used due to their ease of breeding and genetic manipulation, as well as the relatively low cost of maintenance [48]. However, wild-type mice do not develop tau or Aβ pathology, possibly because the rat/mouse amyloid-beta differs from human amyloid-beta and that of most other vertebrates by three amino acid substitutions in the metal-binding domain 1–16, which significantly affects Aβ structural and functional properties [63]. AD models where the genetically altered (transgenic) rodents are utilized have provided fundamental insights into the molecular mechanisms of inherited variants of Alzheimer’s disease [64]. Additional vertebrate species used for modeling employing transgenic approaches include the rat (better suited for behavioral testing than mice), the sea lamprey (which has giant driven neurons), and the zebrafish with its transparent larvae that make neurons easier to visualize. Invertebrate species that are also used include the fruit fly, and the roundworm (both of which are suitable for screening of the developed drug’s effectiveness). Neurodegeneration has been successfully modeled in pigs and sheep, with the features of AD pathology modeled in transgenic pigs [65].
Currently, there are approximately two hundred spontaneous and genetically modified AD models [66], which are based on utilizing various animals, including transgenic mammals (which overexpress human genes involved in the formation of amyloid plaques and neurofibrillary tangles [67]), as well as transgenic flies [68], worms [69], and fish [70]. Mice is used in the majority of the animal AD models, including transgenic, knockout, and injection models [71]. Wild-type mice do not form senile plaques or neurofibrillary tangles even in old age and therefore cannot be used as model animals for AD. Based on the genetic evidence that nearly all mutations associated with inherited AD variants are associated with alterations in Aβ production or aggregation, advances in genetic engineering technologies have enabled the development of mouse models using the APP and PSEN1 genes. Such models are characterized in the first place by the presence of fibrillar and diffuse amyloid plaques. The most popular models include mice of the following lines: PDAPP, Tg2576, APP23, J20, TgCRND8, PS2APP, APPswe/PSEN1dE9 (APP/PS1), Tg-ArcSwe, 5xFAD, A7, and AppNL-G-F. Mice where the neuronal dysfunctions are developed in association with the hyperphosphorylated forms of tau, as a result of transgenes with mutations in MAPT, include the following strains: JNPL3, PS19, and rTg4510. The 3xTg mice were engineered by co-injection of the two genes carrying the linked mutations [APP with the Swedish mutation (KM670/671NL) and MAPT with the P301L mutation]. As a result, these mice develop progressive neuropathology, with intracellular and extracellular Aβ deposits and the aggregates of phosphorylated tau characterized by conformational changes.
3. Molecular Factors Affecting Amyloid Plaque Formation in Transgenic Animal Models of Alzheimer’s Disease
3.1. Zinc
A number of observations indicate that Aβ interactions with zinc ions are involved in the pathogenesis of AD [72]: (1) amyloid plaques contain abnormally high amounts of zinc ions [73]; (2) zinc binds to Aβ and causes its rapid aggregation [74], possibly by modulating the conformational transformation of Aβ and the population shifts in the equilibrium of Aβ polymorphic states [75]; (3) in postmortem brain tissue samples from the patients diagnosed with AD, areas of increased zinc concentration coincide with the sites of amyloid plaque formation [76]; (4) brain regions most affected by the AD pathology contain dense innervation by zinc-containing axons, while brain regions less affected by pathology contain negligible amounts of zinc-containing terminals [77]. Zinc is the second most abundant micronutrient in the human brain (after iron) [78] and serves as an essential structural and catalytic component for more than 10% of the proteins encoded in the human genome [79]. Under physiological conditions, zinc is involved in modulating synaptic plasticity, can regulate neurogenesis, neuron migration and differentiation, and plays a role in neurotransmission [80]; it also has neuroprotective properties and can protect against oxidative stress [81,82]. Hence, the physiological functions of zinc and Aβ largely overlap, and during the pathogenesis of AD, zinc and Aβ constitute the main components of the amyloid plaques and are in direct contact with each other.
The concentration of zinc ions in the amyloid plaques reaches 1 mM [73]. It is believed that the source of zinc that accumulates in the plaques is glutamatergic synapses [83]. Free zinc ions are present in high concentrations (up to 0.3 mM) in the presynaptic vesicles of glutamatergic neurons of the limbic system and cerebral cortex [84]. Upon excitation, zinc ions at a concentration of 10–100 μM enter the synaptic cleft of neurons along with glutamate molecules, where they interact with NMDA and AMPA receptors, modulating their function [85]. To confirm the important role of zinc located in the synaptic cleft, the formation of amyloid plaques was analyzed in transgenic mice with the zinc transporter ZnT3 protein inactivated, and it was found that, in such mice, plaque formation was sharply reduced [83].
The ability of Aβ molecules to bind zinc ions in vitro was first demonstrated in 1994 using Aβ40 as an example [74]. For a long time, it was not possible to establish the mechanism of interaction of Aβ with zinc ions owing to the rapid zinc-induced aggregation of both the most common, in the organism, full-length variants of Aβ, i.e., Aβ40 and Aβ42, and of the truncated at the C- terminus Aβ isoforms, particularly Aβ28 [74,86,87,88,89,90]. However, the N-terminal fragment 1–16 (Aβ16), which is also present in humans as an independent peptide [91], is an autonomous metal-binding domain of Aβ that is stable in vitro [92]. The structure of this domain was determined in the free state and in complex with a zinc ion under physiologically relevant conditions [93]. Then, the molecular mechanism of interaction between zinc ions and Aβ [94] has been established: (1) firstly, zinc ion is chelated by side groups of the amino acid residues Glu11, His13 and His14; (2) followed by folding of the N-terminal region 1–8 of Aβ16, and further by the formation of an additional coordination bond between the zinc ion and the side group of the His6 amino acid residue; this results in the appearance of a well-ordered compact structure of the entire 1–16 domain.
Importantly, the 11-EVHH-14 region of Aβ acts not only as the primary zinc ion recognition site, but also controls the processes of zinc-induced Aβ dimerization [95,96] and oligomerization [97]. The 11-EVHH-14 region has a relatively rigid backbone conformation in soluble Aβ monomers [93,98] and zinc-bound dimers [97]. This site corresponds to the β-strand β2 from the N-terminal arch of the Aβ amyloid fibrils purified from Alzheimer’s brain tissue and is solvent-exposed and accessible for interactions with the external molecules [23]. The secondary structure of the 11-EVHH-14 region is a left-handed polyproline II-type helix [99], which has an increased propensity to participate in the protein–protein interactions [100]. On the strength of these data, the 11-EVHH-14 site represents a structural determinant of Aβ.
3.2. α4β2 Nicotinic Acetylcholine Receptor
It was established using bioinformatic approaches that the extracellular N-terminal domain of the α4 subunit of α4β2-nAChR includes the fragment 35-His-Ala-Glu-Glu-38 (35-HAEE-38), ion-complementary to the 11-EVHH-14 region of Aβ. The fragment 35-HAEE-38 (conservative for humans, mice and chicken) forms the interaction interface of α4β2-nAChR with Aβ [101]. It was shown by molecular modeling that this interaction is stabilized by ion bridges between the complementary charged side groups of the H/E and E/H amino acid pairs. Thus, Aβ can interact with α4β2-nAChR via the 11-EVHH-14: 35-HAEE-38 interface. Tetrapeptide Ac-HAEE-NH2, which is the synthetic analog of the receptor side of this interface, was proven to bind Aβ and to efficiently repair the Aβ-dependent loss of cholinergic function in α4β2-nAChR-transfected oocytes [101]. The data suggest that Aβ:α4β2-nAChR interaction is mediated by the charge complementary interface (Aβ) 11-EVHH-14:35-HAEE-38 (α4), and that the interaction may be involved in Aβ aggregation seeding. So, one can rationally suggest that the absence of α4β2-nAChR would suppress amyloid plaque formation.
Indeed, study [102] reports a number of protective effects caused by the loss of α4*-nAChRs on the neuropathological alterations that develop over time in Tg2576 (APPswe) mice, a widely studied mouse model of AD that expresses a human APP transgene carrying the amyloidogenic Swedish mutation. The principal effect of α4KO was to markedly reduce the load of Aβ plaques in neocortical areas. The plaques were reduced in number, but their size distribution was unchanged, suggesting that fewer plaques are initially formed, but once they are seeded, their maturation is not altered by the loss of α4*-nAChRs. This evidence supports the hypothesis that α4*-nAChRs are directly involved in the seeding of the plaques without affecting plaque maturation and the specific Aβ isoform processing [102].
3.3. Amyloid-β with the Isomerized Asp7 (isoD7-Aβ)
One of the main signs of the pathogenesis of AD is cerebral amyloidogenesis (CA), the process of forming amyloid plaques in the patient’s brain [3,4]. The ability of Aβ aggregates isolated postmortem from the brains of patients diagnosed with AD to induce the development of cerebral amyloidosis was first shown in monkeys that were intracerebrally injected with the corresponding autopsy material [103,104]. Further, in a series of studies on transgenic rodent models of AD [105,106,107,108], it was established that the molecular agent that causes the formation of amyloid plaques in the brain tissue is the conformationally or chemically modified version of Aβ [105,106,107,108,109,110,111,112]. It was also shown that, upon induction of CA, amyloid plaques form in model animals within 24 h, followed in 1–2 days by the activation of microglial cells and their migration to the plaques, which is accompanied by local changes in the axons and dendrites of nearby neurons [113]. The spread of amyloid plaques throughout the brain occurs through the extracellular pathway [114]. As a result of incubation in hippocampal slice culture, synthetic Aβ is converted into a pathogenic agent that causes CA in vivo via a seeding mechanism (in the presence of small amounts of the brain homogenate from the AD patients) [115].
The seeding mechanism of CA initiation in AD involves an important change in the native structure of endogenous Aβ due to the interaction with pathogenic Aβ molecules [29] present in amyloid plaques. This mechanism is corroborated by the fact that small amounts of the pathologically altered Aβ molecules contained in the material obtained from patients diagnosed with AD cause the transition of native Aβ molecules to pathological state [116,117].
It was established that injections of homogenized brain preparations from patients diagnosed with AD lead to a sharply accelerated CA in model animals [29]. Of note, amyloid-β with isomerized Asp7 (isoD7-Aβ) accounts for over 50% of all Aβ molecules in the amyloid plaques [118] and, accordingly, such homogenates contain significant amounts of isoD7-Aβ. IsoD7-Aβ is formed spontaneously due to “protein aging” [32] of both the circulating and aggregated Aβ molecules [14]. It was shown that the accumulation of isoD7-Aβ in brain tissue is a common process in elderly subjects, but patients diagnosed with AD have significantly higher levels of isoD7-Aβ [119]. IsoD7-Aβ42 was also shown to be toxic to neuronal cells [120,121]. IsoD7-Aβ cytotoxicity is coupled to protein phosphorylation [122] and to the induction of oxidative stress, actin polymerization, and stress fiber formation [123]. Molecular mechanism of the in vitro formation of zinc-induced oligomers of natural Aβ isoforms [97] imply that isoD7-Aβ can act as a nucleation seed of aggregation of the endogenous native Aβ molecules in the presence of high concentrations of free zinc in vivo, e.g., in the clefts of cholinergic neurons, where the level of zinc ions reaches 300 µM [85,124].
Indeed, in study [125], it was shown that, unlike Aβ42, isoD7-Aβ42 when administered intravenously to males of transgenic mouse strain B6C3-Tg(APPswe, PSEN1-dE9)85Dbo/j (APP/PS1) caused a sharp acceleration in the development of CA. The experimental groups included male animals that were raised in specific pathogen-free conditions and received monthly intravenous injections of isoD7-Aβ42 (at doses of 100 μg) starting at 2 months of age. Then, the amount of fibrillar amyloid plaques in the hippocampus, cortex and thalamic nuclei was determined in the brain samples from 4- and 10-month-old animals. In contrast to the 4-month cohorts of mice, in which the number of amyloid plaques in the hippocampus and cerebral cortex was approximately 9 and 4 times higher in the animals injected with isoD7-Aβ42 than in the intact control animals, the 10-month cohorts showed for these areas of the brain a smaller difference of about 3 and 1.5 times, respectively. In the thalamic nuclei, the difference between the injected and intact 10-month-old transgenic mice was again very high, by 5.2 times, while the 4-month-old transgenic mice did not have amyloid plaques in this area of the brain. Unlike transgenic mice, intravenous injection of isoD7-Aβ42 to the wild-type mice did not cause the formation of amyloid plaques in the animals, regardless of their age. Thus, it was shown that isoD7-Aβ42 plays the role as a nucleus for the aggregation of endogenous Aβ molecules during the development of CA in a transgenic model of AD [125].
It was later found that intracerebral administration of the metal-binding domain 1-16 of isoD7-Aβ to the transgenic 5xFAD mice substantially enhanced CA [126]. At the age of 3 months, two groups of male 5xFAD transgenic mice were given a single intracerebral injection of isoD7-Aβ16 or Aβ16 solution using the stereotaxic procedure. Brains were removed 1 month after the stereotaxis procedure, and the 8-μm-thick sagittal sections of the brain were analyzed histochemically using Congo red staining. The mean number of congophilic amyloid plaques in the group of transgenic mice treated with the isoD7-Aβ16 was 19.9 ± 2.0, while the mean number of the congophilic amyloid plaques in the group of animals treated with Aβ16, was 12.9 ± 2.1. Hence, isoD7-Aβ16 was identified as a minimal seed for the zinc-dependent aggregation of endogenous Aβ molecules [126].
It was demonstrated that passive immunization of the transgenic 5xFAD mice by the specific monoclonal antibodies recognizing isoD7-Aβ resulted in a significant reduction of isoD7-Aβ and total Aβ in the brain [127]. Amelioration of cognitive impairment was demonstrated by the Morris water maze, elevated plus maze, pole, and contextual fear conditioning tests. Consequently, the antibody-mediated targeting of isoD7-Aβ peptides leads to the attenuation of the AD-like amyloid pathology.
4. Molecular Tools Switching On/Off the Aggregation of Endogenous Aβ Molecules in a Transgenic Model of Alzheimer’s Disease
4.1. Neither Zinc Nor isoD7-Aβ, but a Mixture of Them Triggers Amyloidogenesis
Transgenic nematodes C. elegans that overexpress human Aβ are a popular animal model in the studies of cerebral amyloidogenesis in AD [128,129,130,131]. As these nematodes age, fibrillar amyloid plaques appear in various tissues of the body, and various functional abnormalities are observed. However, the average life span expectancy of animals remains almost unchanged, which indicates only a limited effect of the constitutive amyloid plaques on the main functions of nematodes. One explanation for the relative harmlessness of the amyloidogenesis for nematodes may be the insufficient amounts of molecular components that are present in the human amyloid plaques, namely, zinc ions and isoD7-Aβ. Under the normal nematode life conditions, the concentration of zinc ions in their body cannot reach peak values [132,133]. Also, the occurrence of isoD7-Aβ in nematodes is unlikely, since spontaneous isomerization of Asp7 within Aβ requires substantial time [134], exceeding the life span of these animals.
The study [135] analyzed changes in the three main experimental characteristics associated with the development of amyloidogenesis in transgenic nematodes Caenorhabditis elegance CL2120 (integral volume of amyloid plaques in tissues; degree of muscle paralysis; and life expectancy) when several molecular agents (Aβ, isoD7-Aβ, and zinc ions), which are present in amyloid plaques of the patients diagnosed with AD were added to a nutrient medium. Any single agent by itself had no effect on the animals. A binary mixture of Aβ and zinc ions was also harmless to nematodes. However, the concurrent addition of zinc ions and isoD7-Aβ led to a sharp increase in the amyloid load, an increase in the degree of paralysis and a shortening of the lifespan of animals. Therefore, it was demonstrated in vivo that the isoD7-Aβ/zinc complex has a determinative role in triggering the chain process of zinc-induced aggregation of endogenous Aβ molecules, associated with the severe functional consequences for the life of experimental animals.
4.2. In Transgenic Nematodes, nAChR α4-Derived Peptide HAEE Neutralizes the Aggregation Seeding Effect of the Zinc and isoD7-Aβ Mixture
On the basis of the molecular mechanism of zinc-dependent oligomerization of Aβ isoforms [97], it can be stipulated that molecular agents that can, together with the 11-EVHH-14 sites of Aβ chelate the common zinc ion, will inhibit zinc-dependent oligomerization of Aβ. Indeed, it was shown [135] that the presence of the tetrapeptide Acetyl-HAEE-NH2 (HAEE) completely neutralizes the negative effects of the mixture of zinc ions and isoD7-Aβ on the quality of life of nematodes Caenorhabditis elegance CL2120. Moreover, it was determined from the biosensor experiments based on the SPR effect, the NMR spectroscopy, and molecular modeling that both the Aβ and isoD7-Aβ form a stable non-covalent complex with HAEE, in which one zinc ion is located at the intermolecular interface 11-EVHH-14:HAEE. Previously, using a transgenic mouse model of AD, it was shown that intravenous injections of HAEE highly limit the development of CA in experimental animals [136].
5. Conclusions
Taken together, the above data (summarized in Table 1) point at the fundamental role of the non-covalent complexes of zinc ion and isoD7-Aβ as a necessary and sufficient molecular tool that, with the participation of the α4β2 nicotinic acetylcholine receptor, triggers a chain process of pathological aggregation of the endogenous Aβ molecules. The following integral scenario of the molecular mechanism of amyloid plaque formation in transgenic models of AD is suggested (Figure 1). Phase 1. Due to unknown reasons (most likely stress and aging), isoD7-Aβ appears in the brain. Phase 2. The 11-EVHH-14 region of this chemically modified Aβ isoform interacts with the 35-HAEE-38 region of the α4 subunit of the α4β2-nAChR, resulting in the formation of an “amyloid matrix”, a stable complex on the outer surface of the neuron formed by the isoD7-Aβ and α4β2-nAChR. Phase 3. To the “amyloid matrix”, when there is a surge in the concentration of zinc ions in the synaptic cleft, a molecule of the intact Aβ “sticks” according to the zinc-dependent mechanism. The process continues: (i) from the Aβ molecule, the fragment 11-EVHH-14 participates in the interface, and from the “amyloid matrix”, the residues His6 and His13 of the isoD7-Aβ molecule also participate; (ii) an insoluble aggregate appears, on its outer side there is a molecule of endogenous Aβ, which acquires a pathological conformation (due to interaction with the isoD7-Aβ from the “amyloid matrix”); (iii) according to the same scheme, endogenous Aβ molecules newly arriving from the extracellular space “stick” to the Aβ molecules already aggregated on the initial “amyloid matrix”, and the amyloid plaque grows (to a certain canonical volume).

Table 1.
Molecular agents involved in the formation of amyloid plaques in transgenic animal models of Alzheimer’s disease.
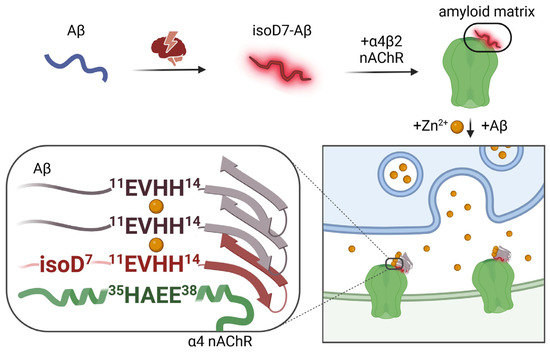
Figure 1.
Proposed scenario of the molecular mechanism of amyloid plaque formation in transgenic models of AD.
From this mechanism, it follows that the most effective and safe way to destroy such plaques is to utilize structural analogs (peptides or peptidomimetics) of the 35-5HAEE-38 region of the α4 subunit of α4β2-nAChR, e.g., the substance Ac-HAEE-NH2, the potential therapeutic effect of which has already been confirmed in the animal AD models [135,136]. When interacting with an amyloid plaque, such analogs will consistently destroy intermolecular interfaces involving the 11-EVHH-14 fragments of Aβ due to their specific binding to the 11-EVHH-14 regions of the aggregated Aβ molecules. In this case, the “amyloid matrix” itself, located at the foundation of this pathological pyramid, will eventually be destroyed, and the neuron, which was burdened with amyloid plaques for many years or even decades, will return to normal life.
Author Contributions
Conceptualization, S.A.K.; writing—original draft preparation, S.A.K. and O.I.K.; writing—review and editing, A.A.A., V.A.M. and A.A.M.; funding acquisition, A.A.M. All authors have read and agreed to the published version of the manuscript.
Funding
The work was performed employing the unique scientific facility “Scanning ion-conductance microscope with a confocal module” (registration number 2512530), and was financially supported by the Ministry of Education and Science of the Russian Federation, Agreement No. 075-15-2022-264.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Figure 1 and the graphical abstract were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Golde, T.E. Alzheimer’s disease—The journey of a healthy brain into organ failure. Mol. Neurodegener. 2022, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Jagust, W.; Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Haeberlein, S.B.; Holtzman, D.M.; Jessen, F.; Karlawish, J.; Liu, E.; Molinuevo, J.L.; et al. “Alzheimer’s disease” is neither “Alzheimer’s clinical syndrome” nor “dementia”. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Schobel, S.A.; Buxton, R.B.; Witter, M.P.; Barnes, C.A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E.; Bohl, J. Staging of Alzheimer-Related Cortical Destruction. Eur. Neurol. 1993, 33, 403–408. [Google Scholar] [CrossRef]
- Wahlund, L.-O.; Julin, P.; Lindqvist, J.; Scheltens, P. Visual assessment of medial temporal lobe atrophy in demented and healthy control subjects: Correlation with volumetry. Psychiatry Res. Neuroimaging 1999, 90, 193–199. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Polis, B.; Samson, A.O. A New Perspective on Alzheimer’s Disease as a Brain Expression of a Complex Metabolic Disorder. In Alzheimer’s Disease [Internet]; Wisniewski, T., Ed.; Codon Publications: Brisbane, Australia, 2019. [Google Scholar] [CrossRef]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Selkoe, D.J. Biochemistry of Amyloid β-Protein and Amyloid Deposits in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.-J. A systemic view of Alzheimer disease—insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Strittmatter, S.M. Binding Sites for Amyloid-β Oligomers and Synaptic Toxicity. Cold Spring Harb. Perspect. Med. 2017, 7, a024075. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef]
- Mukherjee, S.; Perez, K.A.; Lago, L.C.; Klatt, S.; McLean, C.A.; Birchall, I.E.; Barnham, K.J.; Masters, C.L.; Roberts, B.R. Quantification of N-terminal amyloid-β isoforms reveals isomers are the most abundant form of the amyloid-β peptide in sporadic Alzheimer’s disease. Brain Commun. 2021, 3, fcab028. [Google Scholar] [CrossRef]
- Walker, L.C. Aβ Plaques. Free Neuropathol. 2020, 1, 31. [Google Scholar] [CrossRef]
- Stewart, K.L.; Radford, S.E. Amyloid plaques beyond Aβ: A survey of the diverse modulators of amyloid aggregation. Biophys. Rev. 2017, 9, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [PubMed]
- Takano, K. Amyloid beta conformation in aqueous environment. Curr. Alzheimer Res. 2008, 5, 540–547. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 2011, 70, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Friesen, M.; Meyer-Luehmann, M. Aβ Seeding as a Tool to Study Cerebral Amyloidosis and Associated Pathology. Front. Mol. Neurosci. 2019, 12, 233. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef]
- Kozin, S.A.; Makarov, A.A. The Convergence of Alzheimer’s Disease Pathogenesis Concepts. Mol. Biol. 2019, 53, 896–903. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Orpiszewski, J.; Schormann, N.; Kluve-Beckerman, B.; Liepnieks, J.J.; Benson, M.D. Protein aging hypothesis of Alzheimer disease. FASEB J. 2000, 14, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; DeKosky, S.T.; Galasko, D. Alzheimer’s disease: The right drug, the right time. Science 2018, 362, 1250–1251. [Google Scholar] [CrossRef] [PubMed]
- Volloch, V.; Rits-Volloch, S. The Amyloid Cascade Hypothesis 2.0: On the Possibility of Once-in-a-Lifetime-Only Treatment for Prevention of Alzheimer’s Disease and for Its Potential Cure at Symptomatic Stages. J. Alzheimer’s Dis. Rep. 2022, 6, 369–399. [Google Scholar] [CrossRef] [PubMed]
- Volloch, V.; Rits-Volloch, S. Principles of Design of Clinical Trials for Prevention and Treatment of Alzheimer’s Disease and Aging-Associated Cognitive Decline in the ACH2.0 Perspective: Potential Outcomes, Challenges, and Solutions. J. Alzheimer’s Dis. Rep. 2023, 7, 921–955. [Google Scholar] [CrossRef]
- Volloch, V.; Rits-Volloch, S. The Amyloid Cascade Hypothesis 2.0: Generalization of the Concept. J. Alzheimer’s Dis. Rep. 2023, 7, 21–35. [Google Scholar] [CrossRef]
- Volloch, V.; Rits-Volloch, S. The Amyloid Cascade Hypothesis 2.0 for Alzheimer’s Disease and Aging-Associated Cognitive Decline: From Molecular Basis to Effective Therapy. Int. J. Mol. Sci. 2023, 24, 12246. [Google Scholar] [CrossRef]
- Hillen, H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1154. [Google Scholar] [CrossRef]
- Kurkinen, M.; Fułek, M.; Fułek, K.; Beszłej, J.A.; Kurpas, D.; Leszek, J. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: Should We Change Our Thinking? Biomolecules 2023, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Mummery, C. An anti-amyloid therapy works for Alzheimer’s disease: Why has it taken so long and what is next? Brain 2023, 146, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Day, G.S. Anti-amyloid therapies for Alzheimer disease: Finally, good news for patients. Mol. Neurodegener. 2023, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Bodea, L.G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef]
- Li, C.; Briner, A.; Götz, J. The search for improved animal models of Alzheimer’s disease and novel strategies for therapeutic intervention. Future Med. Chem. 2019, 11, 1853–1857. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Zhang, Y. Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Scearce-Levie, K.; Sanchez, P.E.; Lewcock, J.W. Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat. Rev. Drug Discov. 2020, 19, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Toledano, A.; Álvarez, M.I. Lesion-Induced Vertebrate Models of Alzheimer Dementia. In Animal Models of Dementia; De Deyn, P.P., Van Dam, D., Eds.; Humana Press: Totowa, NJ, USA, 2011; pp. 295–345. [Google Scholar] [CrossRef]
- Ridley, R.M.; Murray, T.K.; Johnson, J.A.; Baker, H.F. Learning impairment following lesion of the basal nucleus of Meynert in the marmoset: Modification by cholinergic drugs. Brain Res. 1986, 376, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Fiock, K.L.; Smith, J.D.; Crary, J.F.; Hefti, M.M. β-amyloid and tau pathology in the aging feline brain. J. Comp. Neurol. 2020, 528, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.J.; McKean, N.E.; Henty, K.; Portelius, E.; Blennow, K.; Rudiger, S.R.; Bawden, C.S.; Handley, R.R.; Verma, P.J.; Faull, R.L.M.; et al. Alzheimer’s disease markers in the aged sheep (Ovis aries). Neurobiol. Aging 2017, 58, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.S.; Supalkar, K.V.; Sadar, S.S.; Vyawahare, N.S. Animal models of Alzheimer’s disease: An originof innovativetreatments and insight to the disease’s etiology. Brain Res. 2023, 1814, 148449. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, E.M.; Chaney, M.O.; Norris, F.H.; Pascual, R.; Little, S.P. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Mol. Brain Res. 1991, 10, 299–305. [Google Scholar] [CrossRef]
- Benedikz, E.; Kloskowska, E.; Winblad, B. The rat as an animal model of Alzheimer’s disease. J. Cell Mol. Med. 2009, 13, 1034–1042. [Google Scholar] [CrossRef]
- Gunn-Moore, D.; Kaidanovich-Beilin, O.; Gallego Iradi, M.C.; Gunn-Moore, F.; Lovestone, S. Alzheimer’s disease in humans and other animals: A consequence of postreproductive life span and longevity rather than aging. Alzheimer’s Dement. 2018, 14, 195–204. [Google Scholar] [CrossRef]
- Zeiss, C.J. Utility of spontaneous animal models of Alzheimer’s disease in preclinical efficacy studies. Cell Tissue Res. 2020, 380, 273–286. [Google Scholar] [CrossRef]
- Istrate, A.N.; Tsvetkov, P.O.; Mantsyzov, A.B.; Kulikova, A.A.; Kozin, S.A.; Makarov, A.A.; Polshakov, V.I. NMR solution structure of rat Aβ(1-16): Toward understanding the mechanism of rats’ resistance to Alzheimer’s disease. Biophys. J. 2012, 102, 136–143. [Google Scholar] [CrossRef]
- Myers, A.; McGonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81. [Google Scholar] [CrossRef]
- Lee, S.E.; Hyun, H.; Park, M.R.; Choi, Y.; Son, Y.J.; Park, Y.G.; Jeong, S.G.; Shin, M.Y.; Ha, H.J.; Hong, H.S.; et al. Production of transgenic pig as an Alzheimer’s disease model using a multi-cistronic vector system. PLoS ONE 2017, 12, e0177933. [Google Scholar] [CrossRef]
- Vitek, M.P.; Araujo, J.A.; Fossel, M.; Greenberg, B.D.; Howell, G.R.; Rizzo, S.J.S.; Seyfried, N.T.; Tenner, A.J.; Territo, P.R.; Windisch, M.; et al. Translational animal models for Alzheimer’s disease: An Alzheimer’s Association Business Consortium Think Tank. Alzheimer’s Dement. 2020, 6, e12114. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Papanikolopoulou, K.; Skoulakis, E.M. Temporally distinct phosphorylations differentiate Tau-dependent learning deficits and premature mortality in Drosophila. Hum. Mol. Genet. 2015, 24, 2065–2077. [Google Scholar] [CrossRef][Green Version]
- Benbow, S.J.; Strovas, T.J.; Darvas, M.; Saxton, A.; Kraemer, B.C. Synergistic toxicity between tau and amyloid drives neuronal dysfunction and neurodegeneration in transgenic C. elegans. Hum. Mol. Genet. 2020, 29, 495–505. [Google Scholar] [CrossRef]
- Javed, I.; Peng, G.; Xing, Y.; Yu, T.; Zhao, M.; Kakinen, A.; Faridi, A.; Parish, C.L.; Ding, F.; Davis, T.P.; et al. Inhibition of amyloid beta toxicity in zebrafish with a chaperone-gold nanoparticle dual strategy. Nat. Commun. 2019, 10, 3780. [Google Scholar] [CrossRef]
- Yokoyama, M.; Kobayashi, H.; Tatsumi, L.; Tomita, T. Mouse Models of Alzheimer’s Disease. Front. Mol. Neurosci. 2022, 15, 912995. [Google Scholar] [CrossRef]
- Craddock, T.J.; Tuszynski, J.A.; Chopra, D.; Casey, N.; Goldstein, L.E.; Hameroff, S.R.; Tanzi, R.E. The zinc dyshomeostasis hypothesis of Alzheimer’s disease. PLoS ONE 2012, 7, e33552. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H., Jr.; Paradis, M.D.; Tanzi, R.E. Modulation of A beta adhesiveness and secretase site cleavage by zinc. J. Biol. Chem. 1994, 269, 12152–12158. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.; Ma, B.; Nussinov, R. Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. USA 2010, 107, 9490–9495. [Google Scholar] [CrossRef]
- Miller, L.M.; Wang, Q.; Telivala, T.P.; Smith, R.J.; Lanzirotti, A.; Miklossy, J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 2006, 155, 30–37. [Google Scholar] [CrossRef]
- Suh, S.W.; Jensen, K.B.; Jensen, M.S.; Silva, D.S.; Kesslak, P.J.; Danscher, G.; Frederickson, C.J. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 2000, 852, 274–278. [Google Scholar] [CrossRef]
- DeBenedictis, C.A.; Raab, A.; Ducie, E.; Howley, S.; Feldmann, J.; Grabrucker, A.M. Concentrations of Essential Trace Metals in the Brain of Animal Species-A Comparative Study. Brain Sci. 2020, 10, 460. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Krall, R.F.; Tzounopoulos, T.; Aizenman, E. The Function and Regulation of Zinc in the Brain. Neuroscience 2021, 457, 235–258. [Google Scholar] [CrossRef]
- Prasad, A.S. Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp. Gerontol. 2008, 43, 370–377. [Google Scholar] [CrossRef]
- Tapiero, H.; Tew, K.D. Trace elements in human physiology and pathology: Zinc and metallothioneins. Biomed. Pharmacother. 2003, 57, 399–411. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef]
- Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Huang, X.; Cuajungco, M.P.; Atwood, C.S.; Moir, R.D.; Tanzi, R.E.; Bush, A.I. Alzheimer’s disease, beta-amyloid protein and zinc. J. Nutr. 2000, 130 (Suppl. S5), 1488s–1492s. [Google Scholar] [CrossRef]
- Liu, S.T.; Howlett, G.; Barrow, C.J. Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the A beta peptide of Alzheimer’s disease. Biochemistry 1999, 38, 9373–9378. [Google Scholar] [CrossRef]
- Miura, T.; Suzuki, K.; Kohata, N.; Takeuchi, H. Metal binding modes of Alzheimer’s amyloid beta-peptide in insoluble aggregates and soluble complexes. Biochemistry 2000, 39, 7024–7031. [Google Scholar] [CrossRef]
- Yang, D.S.; McLaurin, J.; Qin, K.; Westaway, D.; Fraser, P.E. Examining the zinc binding site of the amyloid-beta peptide. Eur. J. Biochem. 2000, 267, 6692–6698. [Google Scholar] [CrossRef]
- Portelius, E.; Price, E.; Brinkmalm, G.; Stiteler, M.; Olsson, M.; Persson, R.; Westman-Brinkmalm, A.; Zetterberg, H.; Simon, A.J.; Blennow, K. A novel pathway for amyloid precursor protein processing. Neurobiol. Aging 2011, 32, 1090–1098. [Google Scholar] [CrossRef]
- Kozin, S.A.; Zirah, S.; Rebuffat, S.; Hoa, G.H.; Debey, P. Zinc binding to Alzheimer’s Abeta(1-16) peptide results in stable soluble complex. Biochem. Biophys. Res. Commun. 2001, 285, 959–964. [Google Scholar] [CrossRef]
- Zirah, S.; Kozin, S.A.; Mazur, A.K.; Blond, A.; Cheminant, M.; Segalas-Milazzo, I.; Debey, P.; Rebuffat, S. Structural changes of region 1-16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 2006, 281, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.O.; Kulikova, A.A.; Golovin, A.V.; Tkachev, Y.V.; Archakov, A.I.; Kozin, S.A.; Makarov, A.A. Minimal Zn(2+) binding site of amyloid-β. Biophys. J. 2010, 99, L84–L86. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Mezentsev, Y.V.; Kulikova, A.A.; Indeykina, M.I.; Golovin, A.V.; Ivanov, A.S.; Tsvetkov, P.O.; Makarov, A.A. Zinc-induced dimerization of the amyloid-β metal-binding domain 1–16 is mediated by residues 11–14. Mol. BioSyst. 2011, 7, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, A.A.; Tsvetkov, P.O.; Indeykina, M.I.; Popov, I.A.; Zhokhov, S.S.; Golovin, A.V.; Polshakov, V.I.; Kozin, S.A.; Nudler, E.; Makarov, A.A. Phosphorylation of Ser8 promotes zinc-induced dimerization of the amyloid-β metal-binding domain. Mol. BioSyst. 2014, 10, 2590–2596. [Google Scholar] [CrossRef] [PubMed]
- Istrate, A.N.; Kozin, S.A.; Zhokhov, S.S.; Mantsyzov, A.B.; Kechko, O.I.; Pastore, A.; Makarov, A.A.; Polshakov, V.I. Interplay of histidine residues of the Alzheimer’s disease Aβ peptide governs its Zn-induced oligomerization. Sci. Rep. 2016, 6, 21734. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.M.; Nuttall, S.D.; Robert, R.; Caine, J.M.; Dolezal, O.; Hattarki, M.; Pearce, L.A.; Davydova, N.; Masters, C.L.; Varghese, J.N.; et al. Structural studies of the tethered N-terminus of the Alzheimer’s disease amyloid-β peptide. Proteins Struct. Funct. Bioinform. 2013, 81, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, A.A.; Anashkina, A.A.; Makarov, A.A. Left-handed polyproline-II helix revisited: Proteins causing proteopathies. J. Biomol. Struct. Dyn. 2017, 35, 2701–2713. [Google Scholar] [CrossRef]
- Adzhubei, A.A.; Sternberg, M.J.; Makarov, A.A. Polyproline-II helix in proteins: Structure and function. J. Mol. Biol. 2013, 425, 2100–2132. [Google Scholar] [CrossRef]
- Barykin, E.P.; Garifulina, A.I.; Tolstova, A.P.; Anashkina, A.A.; Adzhubei, A.A.; Mezentsev, Y.V.; Shelukhina, I.V.; Kozin, S.A.; Tsetlin, V.I.; Makarov, A.A. Tetrapeptide Ac-HAEE-NH(2) Protects α4β2 nAChR from Inhibition by Aβ. Int. J. Mol. Sci. 2020, 21, 6272. [Google Scholar] [CrossRef]
- Vilella, A.; Romoli, B.; Bodria, M.; Pons, S.; Maskos, U.; Zoli, M. Evidence for a protective effect of the loss of α4-containing nicotinic acetylcholine receptors on Aβ-related neuropathology in Tg2576 mice. Front. Neurosci. 2023, 17, 1097857. [Google Scholar] [CrossRef]
- Baker, H.F.; Ridley, R.M.; Duchen, L.W.; Crow, T.J.; Bruton, C.J. Induction of beta (A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol. Neurobiol. 1994, 8, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.M.; Baker, H.F.; Windle, C.P.; Cummings, R.M. Very long term studies of the seeding of beta-amyloidosis in primates. J. Neural. Transm. 2006, 113, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Langer, F.; Eisele, Y.S.; Fritschi, S.K.; Staufenbiel, M.; Walker, L.C.; Jucker, M. Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. J. Neurosci. 2011, 31, 14488–14495. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Duran-Aniotz, C.; Castilla, J.; Estrada, L.D.; Soto, C. De novo induction of amyloid-β deposition in vivo. Mol. Psychiatry 2012, 17, 1347–1353. [Google Scholar] [CrossRef]
- Rosen, R.F.; Fritz, J.J.; Dooyema, J.; Cintron, A.F.; Hamaguchi, T.; Lah, J.J.; LeVine III, H.; Jucker, M.; Walker, L.C. Exogenous seeding of cerebral β-amyloid deposition in βAPP-transgenic rats. J. Neurochem. 2012, 120, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Giles, K.; Grillo, S.K.; Lemus, A.; DeArmond, S.J.; Prusiner, S.B. Bioluminescence imaging of Abeta deposition in bigenic mouse models of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2011, 108, 2528–2533. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Bolmont, T.; Heikenwalder, M.; Langer, F.; Jacobson, L.H.; Yan, Z.X.; Roth, K.; Aguzzi, A.; Staufenbiel, M.; Walker, L.C.; et al. Induction of cerebral beta-amyloidosis: Intracerebral versus systemic Abeta inoculation. Proc. Natl. Acad. Sci. USA 2009, 106, 12926–12931. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Obermüller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef]
- Liu, K.Y.; Villain, N.; Ayton, S.; Ackley, S.F.; Planche, V.; Howard, R.; Thambisetty, M. Key questions for the evaluation of anti-amyloid immunotherapies for Alzheimer’s disease. Brain Commun. 2023, 5, fcad175. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Mezias, C.; Raj, A. Analysis of Amyloid-β Pathology Spread in Mouse Models Suggests Spread Is Driven by Spatial Proximity, Not Connectivity. Front. Neurol. 2017, 8, 653. [Google Scholar] [CrossRef] [PubMed]
- Novotny, R.; Langer, F.; Mahler, J.; Skodras, A.; Vlachos, A.; Wegenast-Braun, B.M.; Kaeser, S.A.; Neher, J.J.; Eisele, Y.S.; Pietrowski, M.J.; et al. Conversion of Synthetic Aβ to In Vivo Active Seeds and Amyloid Plaque Formation in a Hippocampal Slice Culture Model. J. Neurosci. 2016, 36, 5084–5093. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J.; et al. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Petrushanko, I.Y.; Yegorov, Y.E.; Simonenko, O.V.; Vishnyakova, K.S.; Kulikova, A.A.; Tsvetkov, P.O.; Makarov, A.A.; Kozin, S.A. Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells. Cell Death Dis. 2013, 4, e939. [Google Scholar] [CrossRef]
- Yurinskaya, M.M.; Mitkevich, V.A.; Kozin, S.A.; Evgen’ev, M.B.; Makarov, A.A.; Vinokurov, M.G. HSP70 protects human neuroblastoma cells from apoptosis and oxidative stress induced by amyloid peptide isoAsp7-Aβ(1-42). Cell Death Dis. 2015, 6, e1977. [Google Scholar] [CrossRef]
- Zatsepina, O.G.; Kechko, O.I.; Mitkevich, V.A.; Kozin, S.A.; Yurinskaya, M.M.; Vinokurov, M.G.; Serebryakova, M.V.; Rezvykh, A.P.; Evgen’ev, M.B.; Makarov, A.A. Amyloid-β with isomerized Asp7 cytotoxicity is coupled to protein phosphorylation. Sci. Rep. 2018, 8, 3518. [Google Scholar] [CrossRef]
- Kolmogorov, V.S.; Erofeev, A.S.; Barykin, E.P.; Timoshenko, R.V.; Lopatukhina, E.V.; Kozin, S.A.; Gorbacheva, L.R.; Salikhov, S.V.; Klyachko, N.L.; Mitkevich, V.A.; et al. Scanning Ion-Conductance Microscopy for Studying β-Amyloid Aggregate Formation on Living Cell Surfaces. Anal. Chem. 2023, 95, 15943–15949. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.M.; Smart, T.G. A physiological role for endogenous zinc in rat hippocampal synaptic neurotransmission. Nature 1991, 349, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally applied synthetic peptide isoAsp7-Abeta(1-42) triggers cerebral beta-amyloidosis. Neurotox. Res. 2013, 24, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, A.A.; Cheglakov, I.B.; Kukharsky, M.S.; Ovchinnikov, R.K.; Kozin, S.A.; Makarov, A.A. Intracerebral Injection of Metal-Binding Domain of Aβ Comprising the Isomerized Asp7 Increases the Amyloid Burden in Transgenic Mice. Neurotox. Res. 2016, 29, 551–557. [Google Scholar] [CrossRef]
- Gnoth, K.; Piechotta, A.; Kleinschmidt, M.; Konrath, S.; Schenk, M.; Taudte, N.; Ramsbeck, D.; Rieckmann, V.; Geissler, S.; Eichentopf, R.; et al. Targeting isoaspartate-modified Aβ rescues behavioral deficits in transgenic mice with Alzheimer’s disease-like pathology. Alzheimer’s Res. Ther. 2020, 12, 149. [Google Scholar] [CrossRef]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef]
- Chen, X.; Barclay, J.W.; Burgoyne, R.D.; Morgan, A. Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem. Cent. J. 2015, 9, 65. [Google Scholar] [CrossRef]
- Dostal, V.; Link, C.D. Assaying β-amyloid toxicity using a transgenic C. elegans model. J. Vis. Exp. 2010, 44, e2252. [Google Scholar] [CrossRef]
- Ewald, C.Y.; Li, C. Understanding the molecular basis of Alzheimer’s disease using a Caenorhabditis elegans model system. Brain Struct. Funct. 2010, 214, 263–283. [Google Scholar] [CrossRef]
- Earley, B.J.; Mendoza, A.D.; Tan, C.H.; Kornfeld, K. Zinc homeostasis and signaling in the roundworm C. elegans. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118882. [Google Scholar] [CrossRef]
- Kumar, J.; Barhydt, T.; Awasthi, A.; Lithgow, G.J.; Killilea, D.W.; Kapahi, P. Zinc Levels Modulate Lifespan through Multiple Longevity Pathways in Caenorhabditis elegans. PLoS ONE 2016, 11, e0153513. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Watanabe, A.; Ogawara, M.; Mori, H.; Shirasawa, T. Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Arch. Biochem. Biophys. 2000, 381, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Barykin, E.P.; Eremina, S.; Pani, B.; Katkova-Zhukotskaya, O.; Polshakov, V.I.; Adzhubei, A.A.; Kozin, S.A.; Mironov, A.S.; Makarov, A.A.; et al. Zn-dependent β-amyloid Aggregation and its Reversal by the Tetrapeptide HAEE. Aging Dis. 2023, 14, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.O.; Cheglakov, I.B.; Ovsepyan, A.A.; Mediannikov, O.Y.; Morozov, A.O.; Telegin, G.B.; Kozin, S.A. Peripherally Applied Synthetic Tetrapeptides HAEE and RADD Slow Down the Development of Cerebral β-Amyloidosis in AβPP/PS1 Transgenic Mice. J. Alzheimer’s Dis. 2015, 46, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Eldik, L.V.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Ratovitski, T.; van Lare, J.; Lee, M.K.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Price, D.L.; Sisodia, S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- McColl, G.; Roberts, B.R.; Pukala, T.L.; Kenche, V.B.; Roberts, C.M.; Link, C.D.; Ryan, T.M.; Masters, C.L.; Barnham, K.J.; Bush, A.I.; et al. Utility of an improved model of amyloid-beta (Aβ1-42) toxicity in Caenorhabditis elegansfor drug screening for Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 57. [Google Scholar] [CrossRef]
- Bugrova, A.E.; Strelnikova, P.A.; Indeykina, M.I.; Kononikhin, A.S.; Zakharova, N.V.; Brzhozovskiy, A.G.; Barykin, E.P.; Pekov, S.I.; Gavrish, M.S.; Babaev, A.A.; et al. The Dynamics of β-Amyloid Proteoforms Accumulation in the Brain of a 5xFAD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 23, 27. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).