

Mitochondrial Protein SLIRP Affects Biosynthesis of Cytochrome c Oxidase Subunits in HEK293T Cells
 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
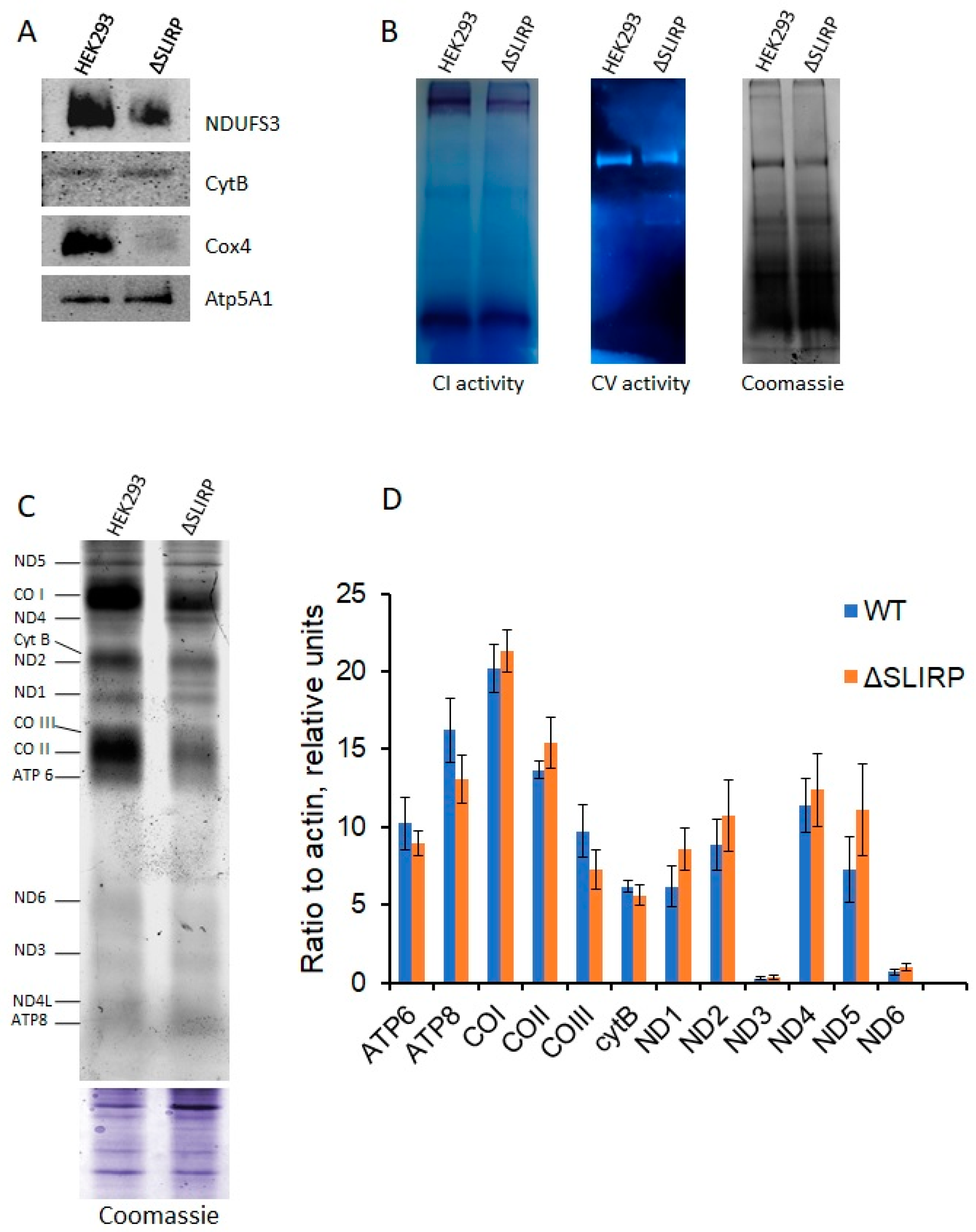
2. Results
2.1. Knockout of SLIRP Gene Impairs Mitochondrial Fitness and Complex I and IV Activities
2.2. The Absence of SLIRP Leads to Deficiency in OXPHOS Complexes I and IV Caused by Imbalance in Mitochondrial Protein Synthesis
2.3. SLIRP Co-Sediments Together with Small Subunits of Mitochondrial Ribosome
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Media, and Growth Conditions
4.2. Generation of Knockout Cell Line
4.3. Seahorse XF HS MINI Analysis
4.4. Analysis of Mitochondrial Translation Products
4.5. Western Blot
4.6. Blue-Native Electrophoresis
4.7. Quantitatively RT-PCR
4.8. Fractionation of Mitochondrial Ribosomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed]
- Keith, L.A.; Jeffrey, D.P. Evolution of mitochondrial gene content: Gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 2003, 29, 380–395. [Google Scholar]
- Leiva, L.E.; Katz, A. Regulation of Leaderless mRNA Translation in Bacteria. Microorganisms 2022, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Remes, C.; Khawaja, A.; Pearce, S.F.; Dinan, A.M.; Gopalakrishna, S.; Cipullo, M.; Kyriakidis, V.; Zhang, J.; Dopico, X.C.; Yukhnovets, O.; et al. Translation initiation of leaderless and polycistronic transcripts in mammalian mitochondria. Nucleic Acids Res. 2023, 51, 891–907. [Google Scholar] [CrossRef]
- Lee, M.; Matsunaga, N.; Akabane, S.; Yasuda, I.; Ueda, T.; Takeuchi-Tomita, N. Reconstitution of mammalian mitochondrial translation system capable of correct initiation and long polypeptide synthesis from leaderless mRNA. Nucleic Acids Res. 2021, 49, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Christian, B.E.; Spremulli, L.L. Preferential Selection of the 5′-Terminal Start Codon on Leaderless mRNAs by Mammalian Mitochondrial Ribosomes. J. Biol. Chem. 2010, 285, 28379–28386. [Google Scholar] [CrossRef] [PubMed]
- Ayyub, S.A.; Varshney, U. Translation initiation in mammalian mitochondria—A prokaryotic perspective. RNA Biol. 2020, 17, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.N.; Wilkinson, K.A.; Hung, K.T.; Weeks, K.M.; Spremulli, L.L. Lack of secondary structure characterizes the 5′ ends of mammalian mitochondrial mRNAs. RNA 2008, 14, 862–871. [Google Scholar] [CrossRef]
- Kummer, E.; Leibundgut, M.; Rackham, O.; Lee, R.G.; Boehringer, D.; Filipovska, A.; Ban, N. Unique features of mammalian mitochondrial translation initiation revealed by cryo-EM. Nature 2018, 560, 263–267. [Google Scholar] [CrossRef]
- Manna, S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie 2015, 113, 93–99. [Google Scholar] [CrossRef]
- Borna, N.N.; Kishita, Y.; Kohda, M.; Lim, S.C.; Shimura, M.; Wu, Y.; Mogushi, K.; Yatsuka, Y.; Harashima, H.; Hisatomi, Y.; et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics 2019, 20, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Pujol, G.; Ortigoza-Escobar, J.D.; Paredes-Fuentes, A.J.; Jou, C.; Ugarteburu, O.; Gort, L.; Yubero, D.; García-Cazorla, A.; O’Callaghan, M.; Campistol, J.; et al. Leigh syndrome is the main clinical characteristic of PTCD3 deficiency. Brain Pathol. 2023, 33, e13134. [Google Scholar] [CrossRef] [PubMed]
- Siira, S.J.; Spåhr, H.; Shearwood, A.-M.J.; Ruzzenente, B.; Larsson, N.-G.; Rackham, O.; Filipovska, A. LRPPRC-mediated folding of the mitochondrial transcriptome. Nat. Commun. 2017, 8, 1532. [Google Scholar] [CrossRef] [PubMed]
- Ruzzenente, B.; Metodiev, M.D.; Wredenberg, A.; Bratic, A.; Park, C.B.; Cámara, Y.; Milenkovic, D.; Zickermann, V.; Wibom, R.; Hultenby, K.; et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs: LRPPRC regulates mitochondrial translation. EMBO J. 2012, 31, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Hatchell, E.C.; Colley, S.M.; Beveridge, D.J.; Epis, M.R.; Stuart, L.M.; Giles, K.M.; Redfern, A.D.; Miles, L.E.C.; Barker, A.; MacDonald, L.M.; et al. SLIRP, a Small SRA Binding Protein, Is a Nuclear Receptor Corepressor. Mol. Cell 2006, 22, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Chujo, T.; Ohira, T.; Sakaguchi, Y.; Goshima, N.; Nomura, N.; Nagao, A.; Suzuki, T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res. 2012, 40, 8033–8047. [Google Scholar] [CrossRef]
- Guo, L.; Engelen, B.P.H.; Hemel, I.M.G.M.; De Coo, I.F.M.; Vreeburg, M.; Sallevelt, S.C.E.H.; Hellebrekers, D.M.E.I.; Jacobs, E.H.; Sadeghi-Niaraki, F.; Van Tienen, F.H.J.; et al. Pathogenic SLIRP variants as a novel cause of autosomal recessive mitochondrial encephalomyopathy with complex I and IV deficiency. Eur. J. Hum. Genet. 2021, 29, 1789–1795. [Google Scholar] [CrossRef]
- Aibara, S.; Singh, V.; Modelska, A.; Amunts, A. Structural basis of mitochondrial translation. eLife 2020, 9, e58362. [Google Scholar] [CrossRef]
- Udagawa, T.; Shimizu, Y.; Ueda, T. Evidence for the Translation Initiation of Leaderless mRNAs by the Intact 70 S Ribosome without its Dissociation into Subunits in Eubacteria. J. Biol. Chem. 2004, 279, 8539–8546. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.-P.; Schägger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Wang, X.; Auwerx, J. Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baleva, M.V.; Piunova, U.; Chicherin, I.; Vasilev, R.; Levitskii, S.; Kamenski, P. Mitochondrial Protein SLIRP Affects Biosynthesis of Cytochrome c Oxidase Subunits in HEK293T Cells. Int. J. Mol. Sci. 2024, 25, 93. https://doi.org/10.3390/ijms25010093
Baleva MV, Piunova U, Chicherin I, Vasilev R, Levitskii S, Kamenski P. Mitochondrial Protein SLIRP Affects Biosynthesis of Cytochrome c Oxidase Subunits in HEK293T Cells. International Journal of Molecular Sciences. 2024; 25(1):93. https://doi.org/10.3390/ijms25010093
Chicago/Turabian StyleBaleva, Mariia V., Uliana Piunova, Ivan Chicherin, Ruslan Vasilev, Sergey Levitskii, and Piotr Kamenski. 2024. "Mitochondrial Protein SLIRP Affects Biosynthesis of Cytochrome c Oxidase Subunits in HEK293T Cells" International Journal of Molecular Sciences 25, no. 1: 93. https://doi.org/10.3390/ijms25010093
APA StyleBaleva, M. V., Piunova, U., Chicherin, I., Vasilev, R., Levitskii, S., & Kamenski, P. (2024). Mitochondrial Protein SLIRP Affects Biosynthesis of Cytochrome c Oxidase Subunits in HEK293T Cells. International Journal of Molecular Sciences, 25(1), 93. https://doi.org/10.3390/ijms25010093