
Challenges of Robust RNAi-Mediated Gene Silencing in Aedes Mosquitoes

 , , , , , , , , and
, , , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Variable RNAi Effects in Ae. aegypti upon Oral Delivery of iRNA-Expressing Microorganisms
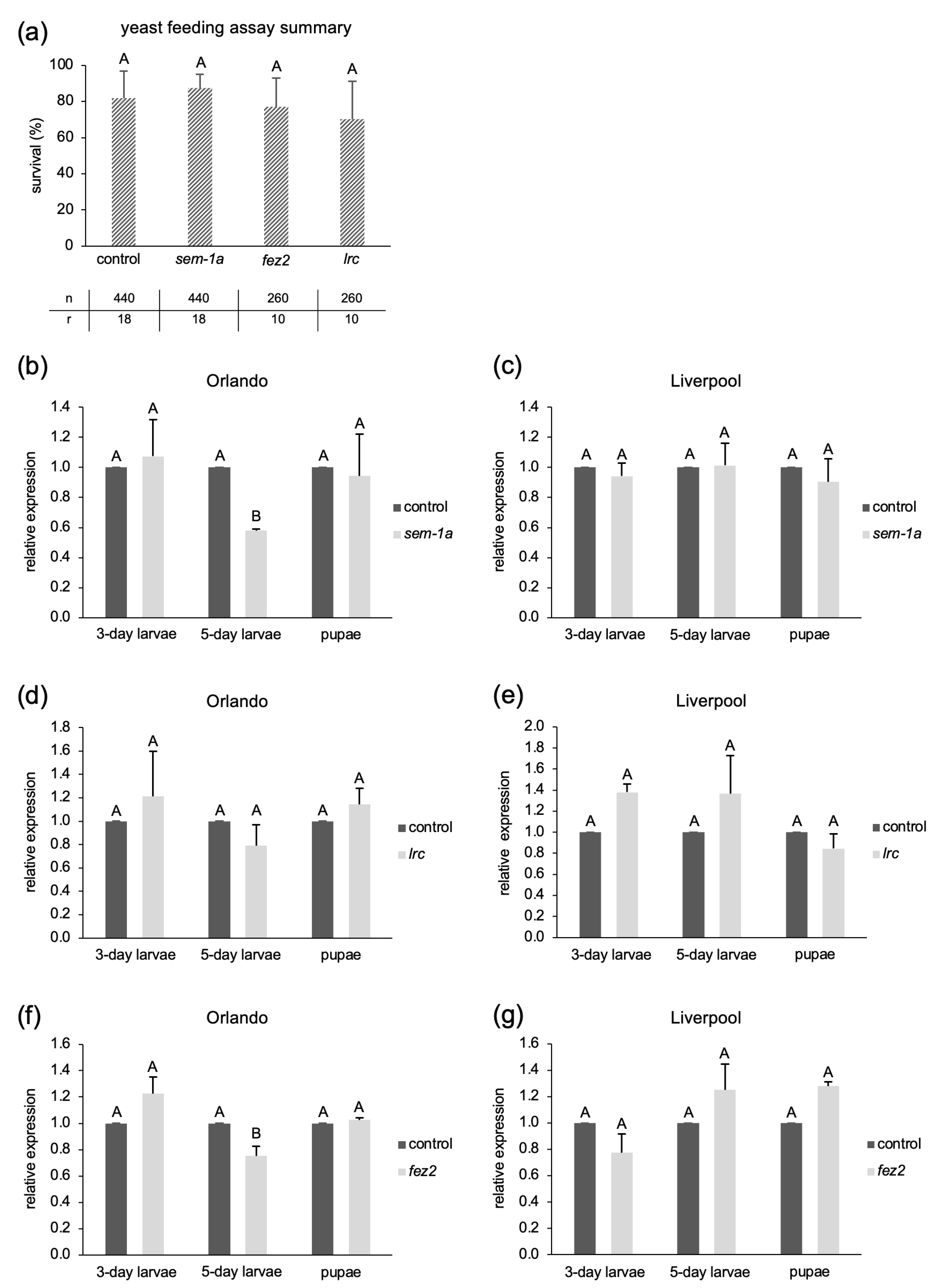
2.1.1. Feeding Larvae with shRNA-Producing Yeast Strains
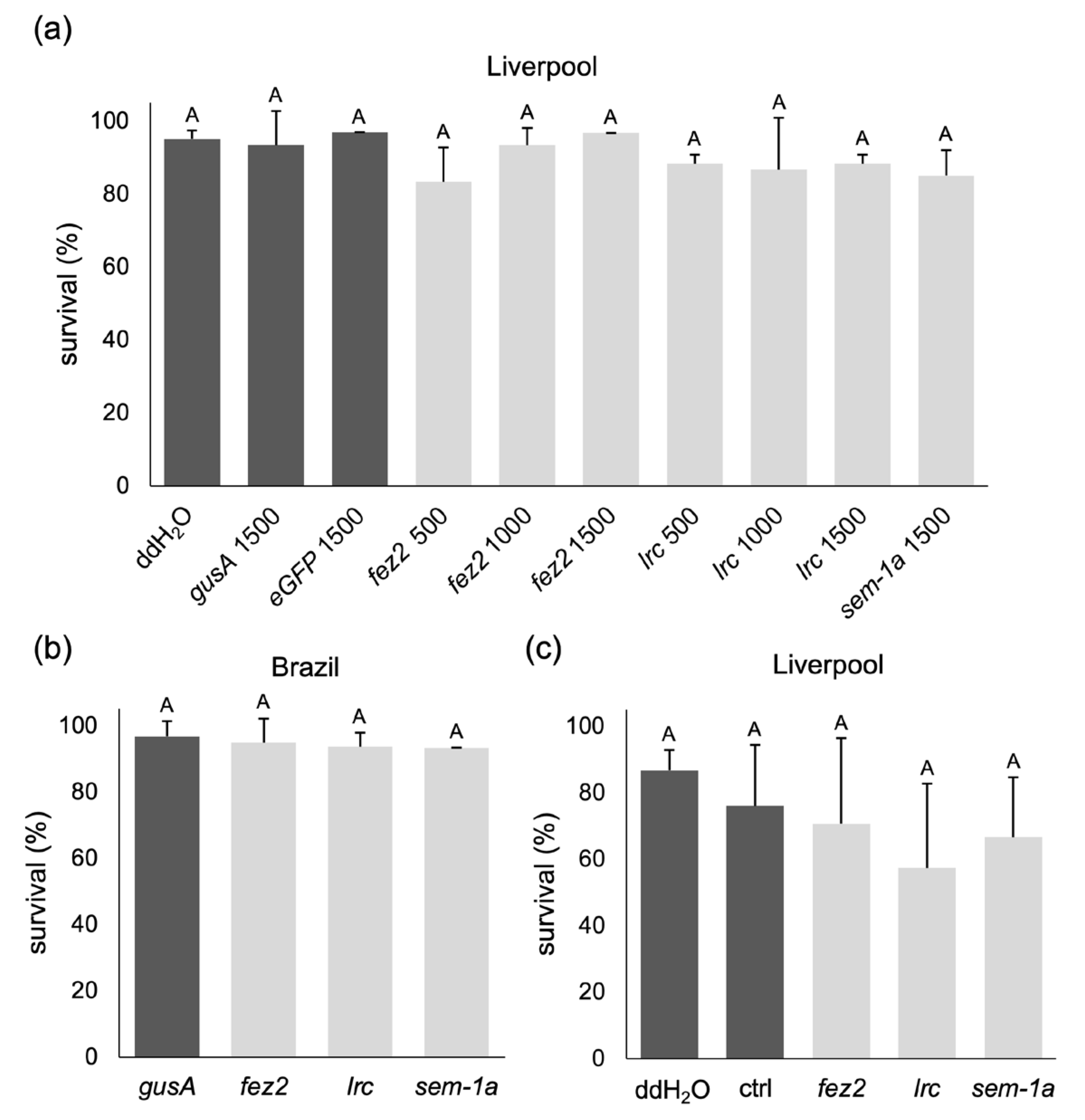
2.1.2. Feeding Larvae with Different Concentrations of dsRNA-Producing Bacteria

2.1.3. Further Enhancement of Bacteria Ingestion Does Not Improve the RNAi Effect
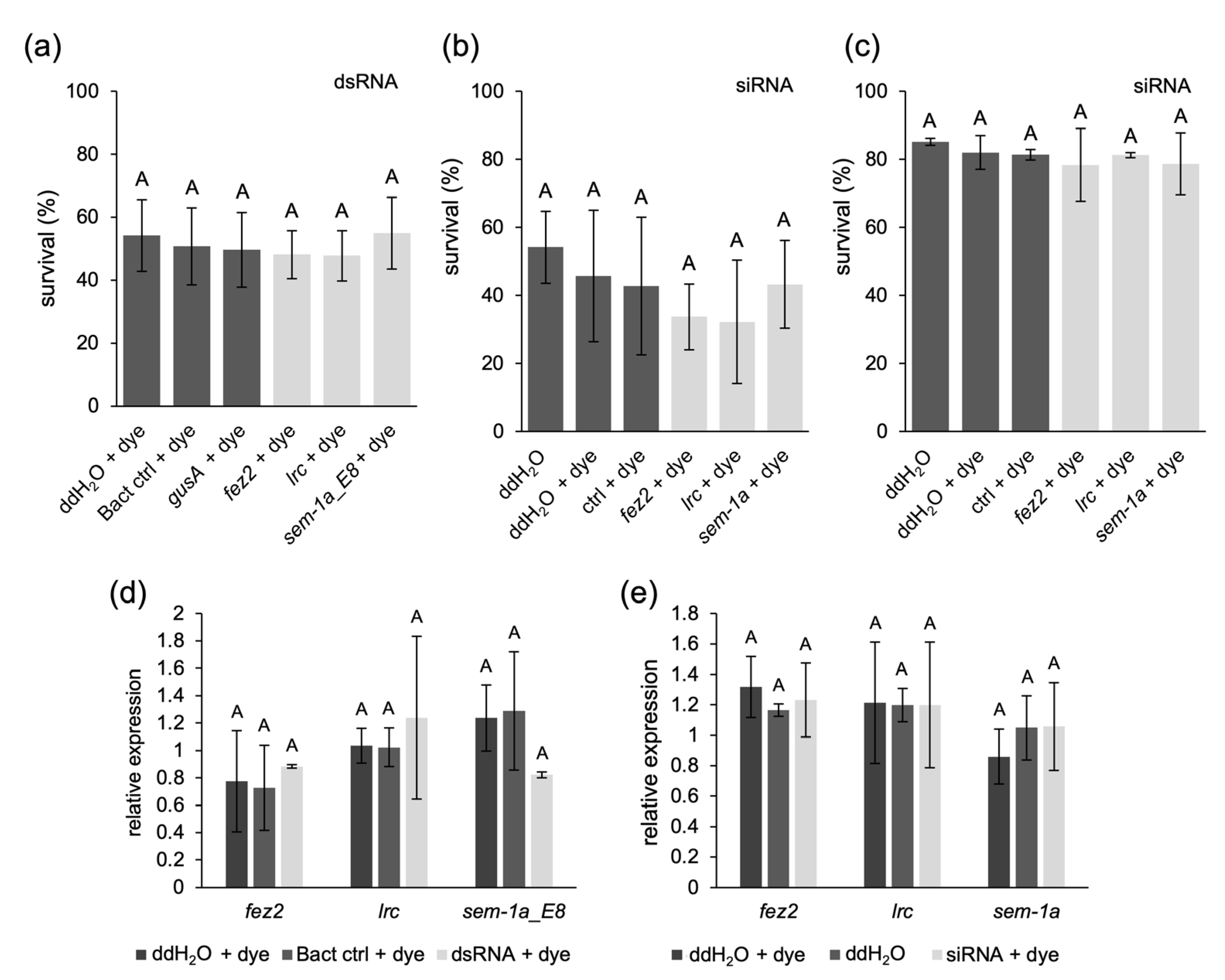
2.2. Co-Delivery of shRNAs against Gut RNases Does Not Enhance Target Gene-Specific RNAi Effects
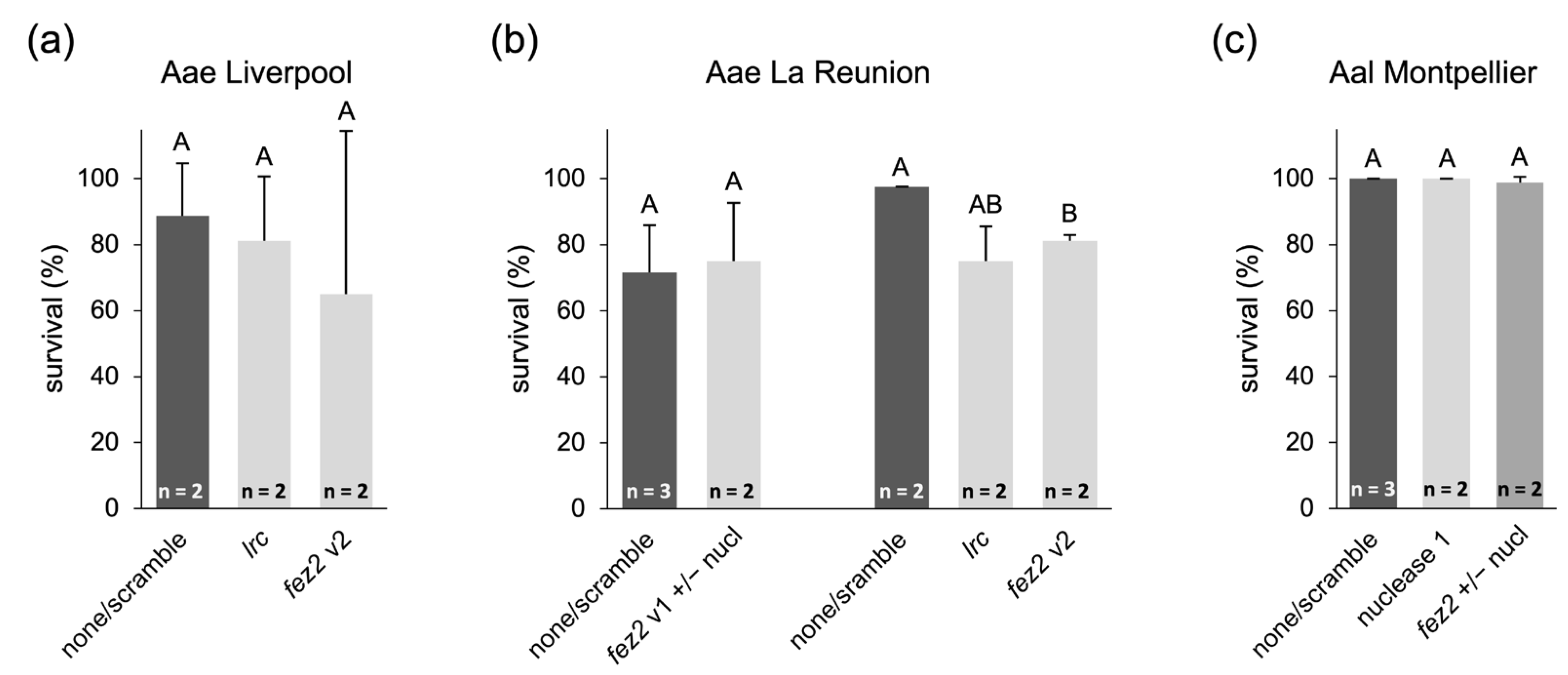
2.3. Soaking of Ae. aegypti Larvae in Concentrated dsRNA or siRNA Solutions Does Not Improve RNAi Efficiency
2.4. siRNA or dsRNA Injections of Ae. aegypti Larvae Do Not Lead to Gene Knockdown
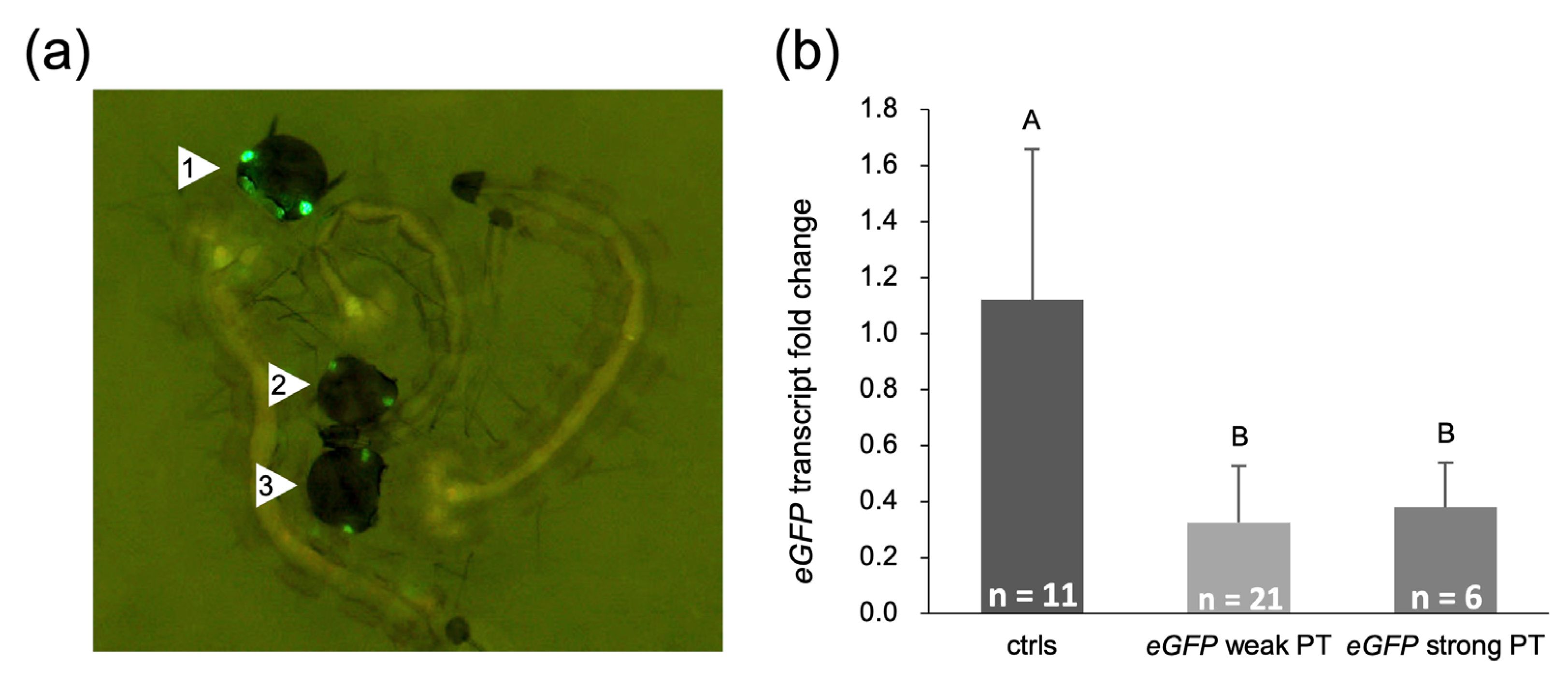
2.5. Embryonic Injection with eGFP dsRNA Reduces eGFP mRNA and Protein Levels in a Transgenic Line
3. Discussion
4. Materials and Methods
4.1. Protocols from Department of Insect Biotechnology in Plant Protection, Justus Liebig University Giessen, Germany
4.1.1. Mosquito Rearing
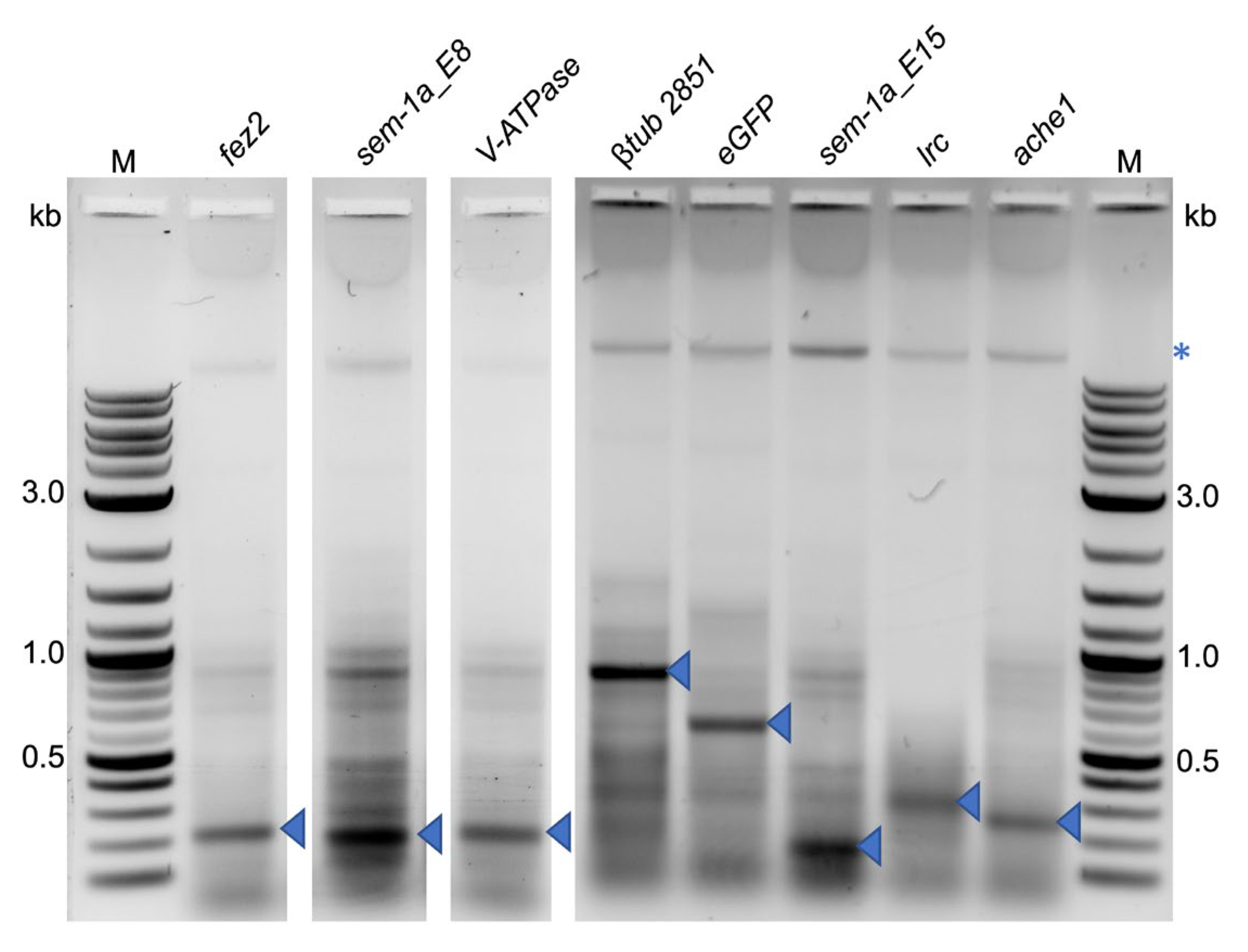
4.1.2. Cloning of dsRNA Expression Vectors for Bacterial Expression
4.1.3. In Vitro Transcription (IVT) of βtub and gusA dsRNA
4.1.4. Cloning of shRNA Expression Vectors for Expression in the Yeast Strain BY4742
4.1.5. Culturing of dsRNA-Expressing Bacteria
4.1.6. Feeding of Ae. aegypti L1 Larvae with dsRNA-Expressing Bacteria
4.1.7. Feeding of Ae. aegypti L1 Larvae with shRNA-Expressing Yeast
4.1.8. Soaking of Neonate Ae. aegypti Larvae with dsRNA or siRNA
4.1.9. RNA Extraction and RT-qPCR of Larvae/Pupae Sampled from Yeast shRNA Feeding Assays
4.1.10. Larval Injections with siRNA or dsRNA Solutions
4.1.11. Embryonic Microinjections with eGFP dsRNA
4.2. Protocols from ASTRE, CIRAD, Montpellier, France
4.2.1. Mosquito Rearing
4.2.2. shRNA Cloning for Bacterial Expression
4.2.3. Culturing of shRNA-Expressing Bacteria and Preparation of Food Pellets
4.2.4. Feeding of Ae. aegypti and Ae. albopictus Larvae with shRNA-Expressing Bacteria
4.3. Protocols from Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, USA
4.3.1. Mosquito Rearing
4.3.2. Ae. aegypti Doublesex and GFP dsRNA Synthesis by IVT
4.3.3. Ae. aegypti Larval Soaking in dsx dsRNA
4.3.4. Total RNA Extraction and RT-qPCR of dsx dsRNA-Treated Ae. aegypti Larvae
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burand, J.P.; Hunter, W.B. RNAi: Future in insect management. J. Invertebr. Pathol. 2013, 112, S68–S74. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Joga, M.R.; Zotti, M.J.; Smagghe, G.; Christiaens, O. RNAi efficiency, systemic properties, and novel delivery methods for pest insect control: What we know so far. Front. Physiol. 2016, 7, 553. [Google Scholar] [CrossRef]
- Ortolá, B.; Daròs, J.-A. RNA interference in insects: From a natural mechanism of gene expression regulation to a biotechnological crop protection promise. Biology 2024, 13, 137. [Google Scholar] [CrossRef]
- Karkare, S.; Daniel, S.; Bhatnagar, D. RNA interference silencing the transcriptional message. Appl. Biochem. Biotech. 2004, 119, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ketting, R.F.; Fischer, S.E.J.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H.A. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Palli, S.R. RNAi turns 25: Contributions and challenges in insect science. Front. Insect Sci. 2023, 3, 1209478. [Google Scholar] [CrossRef]
- Tuschl, T. RNA interference and small interfering RNAs. ChemBioChem 2001, 2, 239–245. [Google Scholar] [CrossRef]
- Yadav, M.; Dahiya, N.; Sehrawat, N. Mosquito gene targeted RNAi studies for vector control. Funct. Integr. Genom. 2023, 23, 180. [Google Scholar] [CrossRef]
- Rono, M.K.; Whitten, M.M.; Oulad-Abdelghani, M.; Levashina, E.A.; Marois, E. The major yolk protein vitellogenin interferes with the anti-plasmodium response in the malaria mosquito Anopheles gambiae. PLoS Biol. 2010, 8, e1000434. [Google Scholar] [CrossRef]
- Olmo, R.P.; Ferreira, A.G.A.; Izidoro-Toledo, T.C.; Aguiar, E.R.G.R.; Faria, I.J.S.; Souza, K.P.R.; Osório, K.P.; Kuhn, L.; Hammann, P.; Andrade, E.G.; et al. Control of dengue virus in the midgut of Aedes aegypti by ectopic expression of the dsRNA-binding protein Loqs2. Nat. Microbiol. 2018, 3, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Olmo, R.P.; Todjro, Y.M.H.; Aguiar, E.R.G.R.; de Almeida, J.P.P.; Ferreira, F.V.; Armache, J.N.; de Faria, I.J.S.; Ferreira, A.G.A.; Amadou, S.C.G.; Silva, A.T.S.; et al. Mosquito vector competence for dengue is modulated by insect-specific viruses. Nat. Microbiol. 2023, 8, 135–149. [Google Scholar] [CrossRef]
- He, L.; Huang, Y.; Tang, X. RNAi-based pest control: Production, application and the fate of dsRNA. Front. Bioeng. Biotechnol. 2022, 10, 1080576. [Google Scholar] [CrossRef] [PubMed]
- Mehlhorn, S.; Hunnekuhl, V.S.; Geibel, S.; Nauen, R.; Bucher, G. Establishing RNAi for basic research and pest control and identification of the most efficient target genes for pest control: A brief guide. Front. Zool. 2021, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Christiaens, O.; Sweet, J.; Dzhambazova, T.; Urru, I.; Smagghe, G.; Kostov, K.; Arpaia, S. Implementation of RNAi-based arthropod pest control: Environmental risks, potential for resistance and regulatory considerations. J. Pest Sci. 2022, 95, 1–15. [Google Scholar] [CrossRef]
- Christiaens, O.; Whyard, S.; Velez, A.M.; Smagghe, G. Double-stranded RNA technology to control insect pests: Current status and challenges. Front. Plant Sci. 2020, 11, 451. [Google Scholar] [CrossRef]
- Whyard, S.; Erdelyan, C.N.; Partridge, A.L.; Singh, A.D.; Beebe, N.W.; Capina, R. Silencing the buzz: A new approach to population suppression of mosquitoes by feeding larvae double-stranded RNAs. Parasit. Vectors 2015, 8, 96. [Google Scholar] [CrossRef]
- Schetelig, M.F.; Milano, A.; Saccone, G.; Handler, A.M. Male only progeny in Anastrepha suspensa by RNAi-induced sex reversion of chromosomal females. Insect Biochem. Mol. Biol. 2012, 42, 51–57. [Google Scholar] [CrossRef]
- Pane, A.; Salvemini, M.; Delli Bovi, P.; Polito, C.; Saccone, G. The transformer gene in Ceratitis capitata provides a genetic basis for selecting and remembering the sexual fate. Development 2002, 129, 3715–3725. [Google Scholar] [CrossRef]
- Mysore, K.; Sun, L.; Tomchaney, M.; Sullivan, G.; Adams, H.; Piscoya, A.S.; Severson, D.W.; Syed, Z.; Duman-Scheel, M. siRNA-mediated silencing of doublesex during female development of the dengue vector mosquito Aedes aegypti. PLoS Negl. Trop. Dis. 2015, 9, e0004213. [Google Scholar] [CrossRef]
- Cruz, C.; Tayler, A.; Whyard, S. RNA interference-mediated knockdown of male fertility genes in the Queensland fruit fly Bactrocera tryoni (Diptera: Tephritidae). Insects 2018, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Hoang, K.P.; Teo, T.M.; Ho, T.X.; Le, V.S. Mechanisms of sex determination and transmission ratio distortion in Aedes aegypti. Parasit. Vectors 2016, 9, 49. [Google Scholar] [CrossRef]
- Coy, M.R.; Sanscrainte, N.D.; Chalaire, K.C.; Inberg, A.; Maayan, I.; Glick, E.; Paldi, N.; Becnel, J.J. Gene silencing in adult Aedes aegypti mosquitoes through oral delivery of double-stranded RNA. J. Appl. Entomol. 2012, 136, 741–748. [Google Scholar] [CrossRef]
- Bouyer, J.; Marois, E. Genetic Control of Vectors. In Prevention and Control of Pests and Vector-Borne Diseases in the Livestock Industry. Emerging Pests and Vector-Borne Diseases in Europe; Smallegange, R., Takken, W., Bouyer, J., Garros, C., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2018; Volume 5, pp. 435–451. [Google Scholar]
- Lutrat, C.; Giesbrecht, D.; Marois, E.; Whyard, S.; Baldet, T.; Bouyer, J. Sex sorting for pest control: It’s raining men! Trends Parasitol. 2019, 35, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.T.; Mamai, W.; Wang, X.; Zhu, J.; Li, Y.; Liu, J.; Tang, Q.; Huang, Y.; Zhang, J.; Zhou, J.; et al. Developing an automatic mosquito sex sorter for mass production of sterile males in support of area-wide release for vector control. Sci. Robot. 2024; in press. [Google Scholar]
- Mamai, W.; Bueno-Masso, O.; Wallner, T.; Nikièma, S.A.; Meletiou, S.; Deng, L.; Balestrino, F.; Yamada, H.; Bouyer, J. Efficiency assessment of a novel automatic mosquito pupae sex separation system in support of area-wide male-based release strategies. Sci. Rep. 2024, 14, 9170. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.E.; Clarke, D.W.; Criswell, V.; Desnoyer, M.; Cornel, D.; Deegan, B.; Gong, K.; Hopkins, K.C.; Howell, P.; Hyde, J.S. Efficient production of male Wolbachia-infected Aedes aegypti mosquitoes enables large-scale suppression of wild populations. Nature Biotechnol. 2020, 38, 482–492. [Google Scholar] [CrossRef]
- Hapairai, L.K.; Mysore, K.; Chen, Y.; Harper, E.I.; Scheel, M.P.; Lesnik, A.M.; Sun, L.; Severson, D.W.; Wei, N.; Duman-Scheel, M. Lure-and-kill yeast interfering RNA larvicides targeting neural genes in the human disease vector mosquito Aedes aegypti. Sci. Rep. 2017, 7, 13223. [Google Scholar] [CrossRef]
- Mysore, K.; Andrews, E.; Li, P.; Duman-Scheel, M. Chitosan/siRNA nanoparticle targeting demonstrates a requirement for single-minded during larval and pupal olfactory system development of the vector mosquito Aedes aegypti. BMC Dev. Biol. 2014, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Mysore, K.; Flannery, E.; Leming, M.T.; Tomchaney, M.; Shi, L.; Sun, L.; O’Tousa, J.E.; Severson, D.W.; Duman-Scheel, M. Role of semaphorin-1a in the developing visual system of the disease vector mosquito Aedes aegypti. Dev. Dyn. 2014, 243, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Mysore, K.; Hapairai, L.K.; Sun, L.; Harper, E.I.; Chen, Y.; Eggleson, K.K.; Realey, J.S.; Scheel, N.D.; Severson, D.W.; Wei, N.; et al. Yeast interfering RNA larvicides targeting neural genes induce high rates of Anopheles larval mortality. Malar. J. 2017, 16, 461. [Google Scholar] [CrossRef] [PubMed]
- Mysore, K.; Li, P.; Wang, C.-W.; Hapairai, L.K.; Scheel, N.D.; Realey, J.S.; Sun, L.; Severson, D.W.; Wei, N.; Duman-Scheel, M. Characterization of a broad-based mosquito yeast interfering RNA larvicide with a conserved target site in mosquito semaphorin-1a genes. Parasit. Vectors 2019, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Wong, S.; Ryan, C.P.; Whyard, S. Oral delivery of double-stranded RNA in larvae of the yellow fever mosquito, Aedes aegypti: Implications for pest mosquito control. J. Insect Sci. 2013, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- Hapairai, L.K.; Mysore, K.; Sun, L.; Li, P.; Wang, C.-W.; Scheel, N.D.; Lesnik, A.; Scheel, M.P.; Igiede, J.; Wei, N.; et al. Characterization of an adulticidal and larvicidal interfering RNA pesticide that targets a conserved sequence in mosquito G protein-coupled dopamine 1 receptor genes. Insect Biochem. Mol. Biol. 2020, 120, 103359. [Google Scholar] [CrossRef] [PubMed]
- Mysore, K.; Hapairai, L.K.; Wei, N.; Realey, J.S.; Scheel, N.D.; Severson, D.W.; Duman-Scheel, M. Preparation and use of a yeast shRNA delivery system for gene silencing in mosquito larvae. In Insect Genomics. Methods and Protocols; Brown, S.J., Pfrender, M.E., Eds.; Humana New York: New York, NY, USA, 2019; Volume 1858, pp. 213–231. [Google Scholar]
- Mysore, K.; Li, P.; Wang, C.-W.; Hapairai, L.K.; Scheel, N.D.; Realey, J.S.; Sun, L.; Roethele, J.B.; Severson, D.W.; Wei, N.; et al. Characterization of a yeast interfering RNA larvicide with a target site conserved in the synaptotagmin gene of multiple disease vector mosquitoes. PLoS Negl. Trop. Dis. 2019, 13, e0007422. [Google Scholar] [CrossRef]
- Blitzer, E.J.; Vyazunova, I.; Lan, Q. Functional analysis of AeSCP-2 using gene expression knockdown in the yellow fever mosquito, Aedes aegypti. Insect Mol. Biol. 2005, 14, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Bona, A.C.D.; Chitolina, R.F.; Fermino, M.L.; de Castro Poncio, L.; Weiss, A.; Lima, J.B.P.; Paldi, N.; Bernardes, E.S.; Henen, J.; Maori, E. Larval application of sodium channel homologous dsRNA restores pyrethroid insecticide susceptibility in a resistant adult mosquito population. Parasit. Vectors 2016, 9, 397. [Google Scholar] [CrossRef]
- Figueira-Mansur, J.; Ferreira-Pereira, A.; Mansur, J.F.; Franco, T.A.; Alvarenga, E.S.L.; Sorgine, M.H.F.; Neves, B.C.; Melo, A.C.A.; Leal, W.S.; Masuda, H.; et al. Silencing of P-glycoprotein increases mortality in temephos-treated Aedes aegypti larvae. Insect Mol. Biol. 2013, 22, 648–658. [Google Scholar] [CrossRef]
- Lopez, S.B.G.; Guimarães-Ribeiro, V.; Rodriguez, J.V.G.; Dorand, F.A.P.S.; Salles, T.S.; Sá-Guimarães, T.E.; Alvarenga, E.S.L.; Melo, A.C.A.; Almeida, R.V.; Moreira, M.F. RNAi-based bioinsecticide for Aedes mosquito control. Sci. Rep. 2019, 9, 4038. [Google Scholar] [CrossRef] [PubMed]
- Meleshkevitch, E.A.; Voronov, D.A.; Miller, M.M.; Penneda, M.; Fox, J.M.; Metzler, R.; Boudko, D.Y. A novel eukaryotic Na+ methionine selective symporter is essential for mosquito development. Insect Biochem. Mol. Biol. 2013, 43, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.R.; Kumar, P.S.; Gandhi, M.R.; Al-Dhabi, N.A.; Paulraj, M.G.; Ignacimuthu, S. Delivery of chitosan/dsRNA nanoparticles for silencing of wing development vestigial (vg) gene in Aedes aegypti mosquitoes. Int. J. Biol. Macromol. 2016, 86, 89–95. [Google Scholar] [CrossRef]
- Munawar, K.; Alahmed, A.M.; Khalil, S.M.S. Delivery methods for RNAi in mosquito larvae. J. Insect Sci. 2020, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Debnath, N.; Cui, Y.; Unrine, J.; Palli, S.R. Chitosan, carbon quantum dot, and silica nanoparticle mediated dsRNA delivery for gene silencing in Aedes aegypti: A comparative analysis. ACS Appl. Mater. Interfaces 2015, 7, 19530–19535. [Google Scholar] [CrossRef] [PubMed]
- Gillet, F.X.; Garcia, R.A.; Macedo, L.L.P.; Albuquerque, E.V.S.; Silva, M.C.M.; Grossi-de-Sa, M.F. Investigating engineered ribonucleoprotein particles to improve oral RNAi delivery in crop insect pests. Front. Physiol. 2017, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Huang, J.H.; Liu, Y.; Belles, X.; Lee, H.J. Oral delivery of dsRNA lipoplexes to German cockroach protects dsRNA from degradation and induces RNAi response. Pest Manag. Sci. 2017, 73, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, N.L.; Smagghe, G.; Sharma, R.; Oliveira, E.E.; Christiaens, O. Liposome encapsulation and EDTA formulation of dsRNA targeting essential genes increase oral RNAi-caused mortality in the Neotropical stink bug Euschistus heros. Pest Manag. Sci. 2019, 75, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Dhandapani, R.K.; Gurusamy, D.; Howell, J.L.; Palli, S.R. Development of CS-TPP-dsRNA nanoparticles to enhance RNAi efficiency in the yellow fever mosquito, Aedes aegypti. Sci. Rep. 2019, 9, 8775. [Google Scholar] [CrossRef]
- Zhang, X.; Mysore, K.; Flannery, E.; Michel, K.; Severson, D.W.; Zhu, K.Y.; Duman-Scheel, M. Chitosan/interfering RNA nanoparticle mediated gene silencing in disease vector mosquito larvae. J. Vis. Exp. 2015, e52523. [Google Scholar] [CrossRef]
- Giesbrecht, D. RNA Interference-Based Sterile Insect Technique in Mosquitoes: Overcoming Barriers to Implementation. Ph.D. Thesis, University of Manitoba, Winnipeg, MB, Canada, 2021. [Google Scholar]
- Isoe, J.; Petchampai, N.; Isoe, Y.E.; Co, K.; Mazzalupo, S.; Scaraffia, P.Y. Xanthine dehydrogenase-1 silencing in Aedes aegypti mosquitoes promotes a blood feeding–induced adulticidal activity. FASEB J. 2017, 31, 2276–2286. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Shin, D.; Mathias, D.K.; Londono-Renteria, B.; Noh, M.Y.; Colpitts, T.M.; Dinglasan, R.R.; Han, Y.S.; Hong, Y.S. Homologs of human dengue-resistance genes, FKBP1B and ATCAY, confer antiviral resistance in Aedes aegypti mosquitoes. Insects 2019, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M.; Silver, K.; Zhang, J.; Park, Y.; Zhu, K.Y. Molecular mechanisms influencing efficiency of RNA interference in insects. Pest Manag. Sci. 2019, 75, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-J.; Donahue, K.; Koh, Y.; Martin, R.R.; Choi, M.-Y. Microbial-based double-stranded RNA production to develop cost-effective RNA interference application for insect pest management. Int. J. Insect Sci. 2019, 11, 1179543319840323. [Google Scholar] [CrossRef] [PubMed]
- Ongvarrasopone, C.; Roshorm, Y.; Panyim, S. A simple and cost effective method to generate dsRNA for RNAi studies in invertebrates. Sci. Asia 2007, 33, 35–39. [Google Scholar] [CrossRef]
- Timmons, L.; Court, D.L.; Fire, A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 2001, 263, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo Prates, L.H.; Merlau, M.; Rühl-Teichner, J.; Schetelig, M.F.; Häcker, I. An optimized/scale up-ready protocol for extraction of bacterially produced dsRNA at good yield and low costs. Int. J. Mol. Sci. 2023, 24, 9266. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.A.; Roberts, J.K. Progress towards RNAi-mediated insect pest management. In Advances in Insect Physiology; Dhadialla, T.S., Gill, S.S., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 47, pp. 249–295. [Google Scholar]
- Haugen, M.; Flannery, E.; Tomchaney, M.; Mori, A.; Behura, S.K.; Severson, D.W.; Duman-Scheel, M. Semaphorin-1a is required for Aedes aegypti embryonic nerve cord development. PLoS ONE 2011, 6, e21694. [Google Scholar] [CrossRef]
- Dalaisón-Fuentes, L.I.; Pascual, A.; Crespo, M.; Andrada, N.L.; Welchen, E.; Catalano, M.I. Knockdown of double-stranded RNases (dsRNases) enhances oral RNA interference (RNAi) in the corn leafhopper, Dalbulus maidis. Pestic. Biochem. Physiol. 2023, 196, 105618. [Google Scholar] [CrossRef]
- Giesbrecht, D.; Heschuk, D.; Wiens, I.; Boguski, D.; LaChance, P.; Whyard, S. RNA interference is enhanced by knockdown of double-stranded RNases in the yellow fever mosquito Aedes aegypti. Insects 2020, 11, 327. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Taning, C.N.T.; Smagghe, G.; Christiaens, O. Silencing of double-stranded ribonuclease improves oral RNAi efficacy in southern green stinkbug Nezara viridula. Insects 2021, 12, 115. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Romoli, O.; Henrion-Lacritick, A.; Blanc, H.; Frangeul, L.; Saleh, M.C. Limitations in harnessing oral RNA interference as an antiviral strategy in Aedes aegypti. iScience 2024, 27, 109261. [Google Scholar] [CrossRef]
- Coon, K.L.; Valzania, L.; McKinney, D.A.; Vogel, K.J.; Brown, M.R.; Strand, M.R. Bacteria-mediated hypoxia functions as a signal for mosquito development. Proc. Natl. Acad. Sci. USA 2017, 114, E5362–E5369. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, M.; Laureti, M.; Levée, T.; Terry, S.; Kohl, A.; Pondeville, E. Improved transient silencing of gene expression in the mosquito female Aedes aegypti. Insect Mol. Biol. 2021, 30, 355–365. [Google Scholar] [CrossRef]
- Airs, P.M.; Kudrna, K.E.; Lubinski, B.; Phanse, Y.; Bartholomay, L.C. A comparative analysis of RNAi trigger uptake and distribution in mosquito vectors of disease. Insects 2023, 14, 556. [Google Scholar] [CrossRef] [PubMed]
- Mysore, K.; Njoroge, T.M.; Stewart, A.T.M.; Winter, N.; Hamid-Adiamoh, M.; Sun, L.; Feng, R.S.; James, L.D.; Mohammed, A.; Severson, D.W.; et al. Characterization of a novel RNAi yeast insecticide that silences mosquito 5-HT1 receptor genes. Sci. Rep. 2023, 13, 22511. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.T.M.; Mysore, K.; Njoroge, T.M.; Winter, N.; Feng, R.S.; Singh, S.; James, L.D.; Singkhaimuk, P.; Sun, L.; Mohammed, A.; et al. Demonstration of RNAi yeast insecticide activity in semi-field larvicide and attractive targeted sugar bait trials conducted on Aedes and Culex mosquitoes. Insects 2023, 14, 950. [Google Scholar] [CrossRef]
- Arjunan, N.; Thiruvengadam, V.; Sushil, S. Nanoparticle-mediated dsRNA delivery for precision insect pest control: A comprehensive review. Mol. Biol. Rep. 2024, 51, 355. [Google Scholar] [CrossRef]
- Pugsley, C.E.; Isaac, R.E.; Warren, N.J.; Cayre, O.J. Recent advances in engineered nanoparticles for RNAi-mediated crop protection against insect pests. Front. Agron. 2021, 3, 652981. [Google Scholar] [CrossRef]
- Yan, S.; Ren, B.-Y.; Shen, J. Nanoparticle-mediated double-stranded RNA delivery system: A promising approach for sustainable pest management. Insect Sci. 2021, 28, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Dönitz, J.; Schmitt-Engel, C.; Grossmann, D.; Gerischer, L.; Tech, M.; Schoppmeier, M.; Klingler, M.; Bucher, G. iBeetle-Base: A database for RNAi phenotypes in the red flour beetle Tribolium castaneum. Nucleic Acids Res. 2014, 43, D720–D725. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.L.; Walker, W.B. Saliva of Lygus lineolaris digests double stranded ribonucleic acids. J. Insect Physiol. 2012, 58, 391–396. [Google Scholar] [CrossRef]
- Christiaens, O.; Swevers, L.; Smagghe, G. DsRNA degradation in the pea aphid (Acyrthosiphon pisum) associated with lack of response in RNAi feeding and injection assay. Peptides 2014, 53, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Garbutt, J.S. RNA Interference in Insects: Persistence and Uptake of Double-Stranded RNA and Activation of RNAi Genes. Ph.D. Thesis, University of Bath, Bath, UK, 2011. [Google Scholar]
- Garbutt, J.S.; Bellés, X.; Richards, E.H.; Reynolds, S.E. Persistence of double-stranded RNA in insect hemolymph as a potential determiner of RNA interference success: Evidence from Manduca sexta and Blattella germanica. J. Insect Physiol. 2013, 59, 171–178. [Google Scholar] [CrossRef]
- Wynant, N.; Santos, D.; Verdonck, R.; Spit, J.; Van Wielendaele, P.; Vanden Broeck, J. Identification, functional characterization and phylogenetic analysis of double stranded RNA degrading enzymes present in the gut of the desert locust, Schistocerca gregaria. Insect Biochem. Mol. Biol. 2014, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wynant, N.; Verlinden, H.; Breugelmans, B.; Simonet, G.; Vanden Broeck, J. Tissue-dependence and sensitivity of the systemic RNA interference response in the desert locust, Schistocerca gregaria. Insect Biochem. Mol. Biol. 2012, 42, 911–917. [Google Scholar] [CrossRef]
- Stephenson, C.J.; Coatsworth, H.; Kang, S.; Lednicky, J.A.; Dinglasan, R.R. Transmission potential of floridian Aedes aegypti mosquitoes for dengue virus serotype 4: Implications for estimating local dengue risk. mSphere 2021, 6, e0027121. [Google Scholar] [CrossRef]
- Gloria-Soria, A.; Soghigian, J.; Kellner, D.; Powell, J.R. Genetic diversity of laboratory strains and implications for research: The case of Aedes aegypti. PLoS Negl. Trop. Dis. 2019, 13, e0007930. [Google Scholar] [CrossRef]
- Macdonald, W.W. The selection of a strain of Aedes aegypti susceptible to infection with semi-periodic Brugia malayi. Ann. Trop. Med. Parasitol. 1962, 56, 368–372. [Google Scholar] [CrossRef]
- Matthews, B.J.; Dudchenko, O.; Kingan, S.B.; Koren, S.; Antoshechkin, I.; Crawford, J.E.; Glassford, W.J.; Herre, M.; Redmond, S.N.; Rose, N.H.; et al. Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 2018, 563, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Nene, V.; Wortman, J.R.; Lawson, D.; Haas, B.; Kodira, C.; Tu, Z.; Loftus, B.; Xi, Z.; Megy, K.; Grabherr, M.; et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science 2007, 316, 1718–1723. [Google Scholar] [CrossRef] [PubMed]
- Häcker, I.; Rehling, T.; Schlosser, H.; Mayorga-Ch, D.; Heilig, M.; Yan, Y.; Armbruster, P.A.; Schetelig, M.F. Improved piggyBac transformation with capped transposase mRNA in pest insects. Int. J. Mol. Sci. 2023, 24, 15155. [Google Scholar] [CrossRef] [PubMed]
- Arziman, Z.; Horn, T.; Boutros, M. E-RNAi: A web application to design optimized RNAi constructs. Nucleic Acids Res. 2005, 33 (Suppl. S2), W582–W588. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, G.J.; Fanning, G.C. Design and cloning strategies for constructing shRNA expression vectors. BMC Biotechnol. 2006, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Dzaki, N.; Ramli, K.N.; Azlan, A.; Ishak, I.H.; Azzam, G. Evaluation of reference genes at different developmental stages for quantitative real-time PCR in Aedes aegypti. Sci. Rep. 2017, 7, 43618. [Google Scholar] [CrossRef] [PubMed]
- Zhiyong, X.; Cynthia, C.H.K.; Dobson, S.L. Wolbachia establishment and invasion in an Aedes aegypti laboratory population. Science 2005, 310, 326–328. [Google Scholar]
- Box, G.E.P.; Cox, D.R. An analysis of transformations. J. R. Stat. Soc. Series B Stat. Methodol. 1964, 26, 211–252. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueiredo Prates, L.H.; Fiebig, J.; Schlosser, H.; Liapi, E.; Rehling, T.; Lutrat, C.; Bouyer, J.; Sun, Q.; Wen, H.; Xi, Z.; et al. Challenges of Robust RNAi-Mediated Gene Silencing in Aedes Mosquitoes. Int. J. Mol. Sci. 2024, 25, 5218. https://doi.org/10.3390/ijms25105218
Figueiredo Prates LH, Fiebig J, Schlosser H, Liapi E, Rehling T, Lutrat C, Bouyer J, Sun Q, Wen H, Xi Z, et al. Challenges of Robust RNAi-Mediated Gene Silencing in Aedes Mosquitoes. International Journal of Molecular Sciences. 2024; 25(10):5218. https://doi.org/10.3390/ijms25105218
Chicago/Turabian StyleFigueiredo Prates, Lucas Henrique, Jakob Fiebig, Henrik Schlosser, Eleni Liapi, Tanja Rehling, Célia Lutrat, Jeremy Bouyer, Qiang Sun, Han Wen, Zhiyong Xi, and et al. 2024. "Challenges of Robust RNAi-Mediated Gene Silencing in Aedes Mosquitoes" International Journal of Molecular Sciences 25, no. 10: 5218. https://doi.org/10.3390/ijms25105218
APA StyleFigueiredo Prates, L. H., Fiebig, J., Schlosser, H., Liapi, E., Rehling, T., Lutrat, C., Bouyer, J., Sun, Q., Wen, H., Xi, Z., Schetelig, M. F., & Häcker, I. (2024). Challenges of Robust RNAi-Mediated Gene Silencing in Aedes Mosquitoes. International Journal of Molecular Sciences, 25(10), 5218. https://doi.org/10.3390/ijms25105218