

Melatonin Inhibits Hypoxia-Induced Alzheimer’s Disease Pathogenesis by Regulating the Amyloidogenic Pathway in Human Neuroblastoma Cells


Abstract
1. Introduction
2. Results
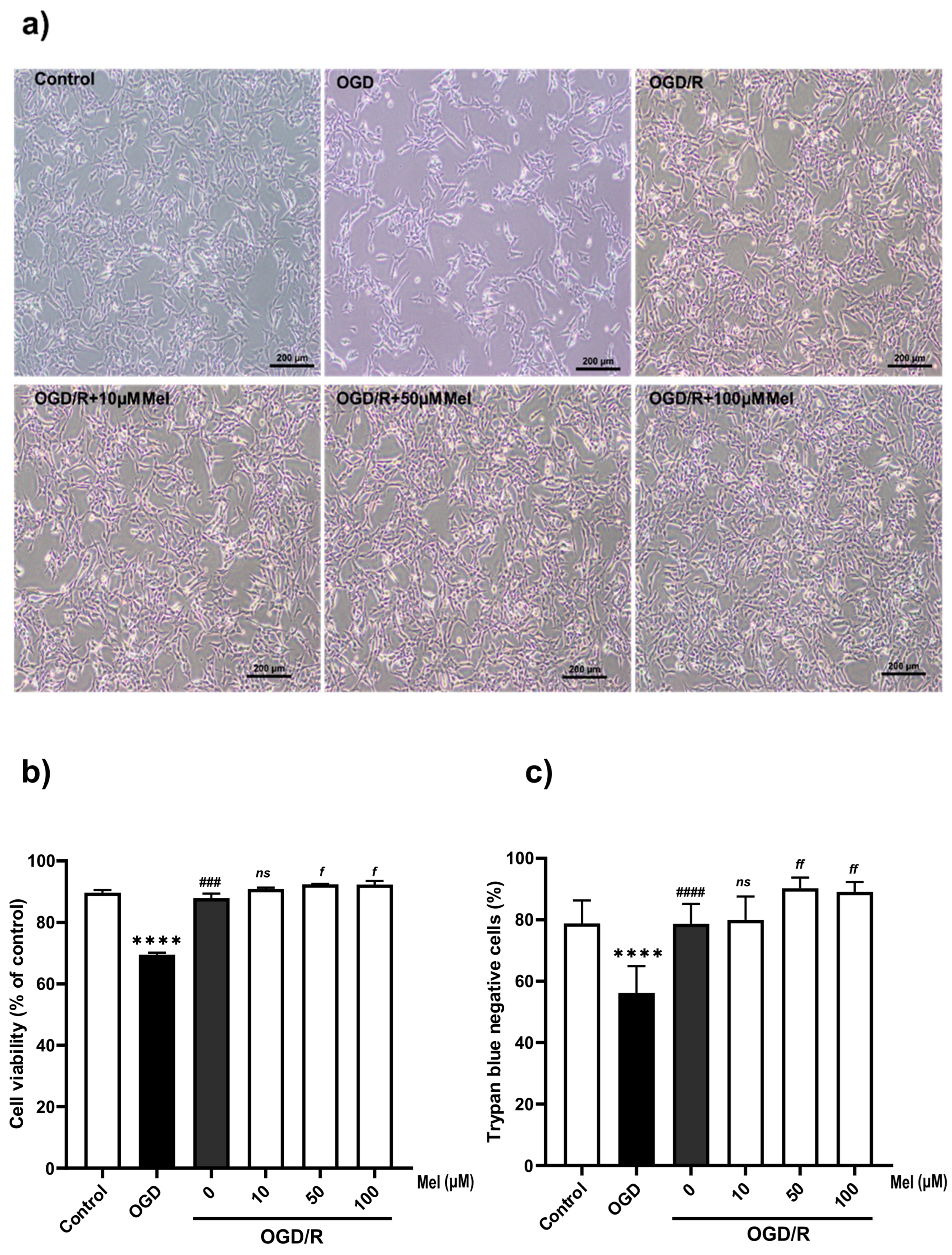
2.1. Effect of Melatonin on the Viability of SH-SY5Y Cells under OGD/R Conditions
2.2. Melatonin Affects the Nuclear Translocation and mRNA Transcription of HIF-1α in SH-SY5Y Cells under OGD/R Conditions
2.3. Effect of Melatonin on the Transcription Level of Amyloidogenic Processing Enzymes in the SH-SY5Y Cells under OGD/R Conditions
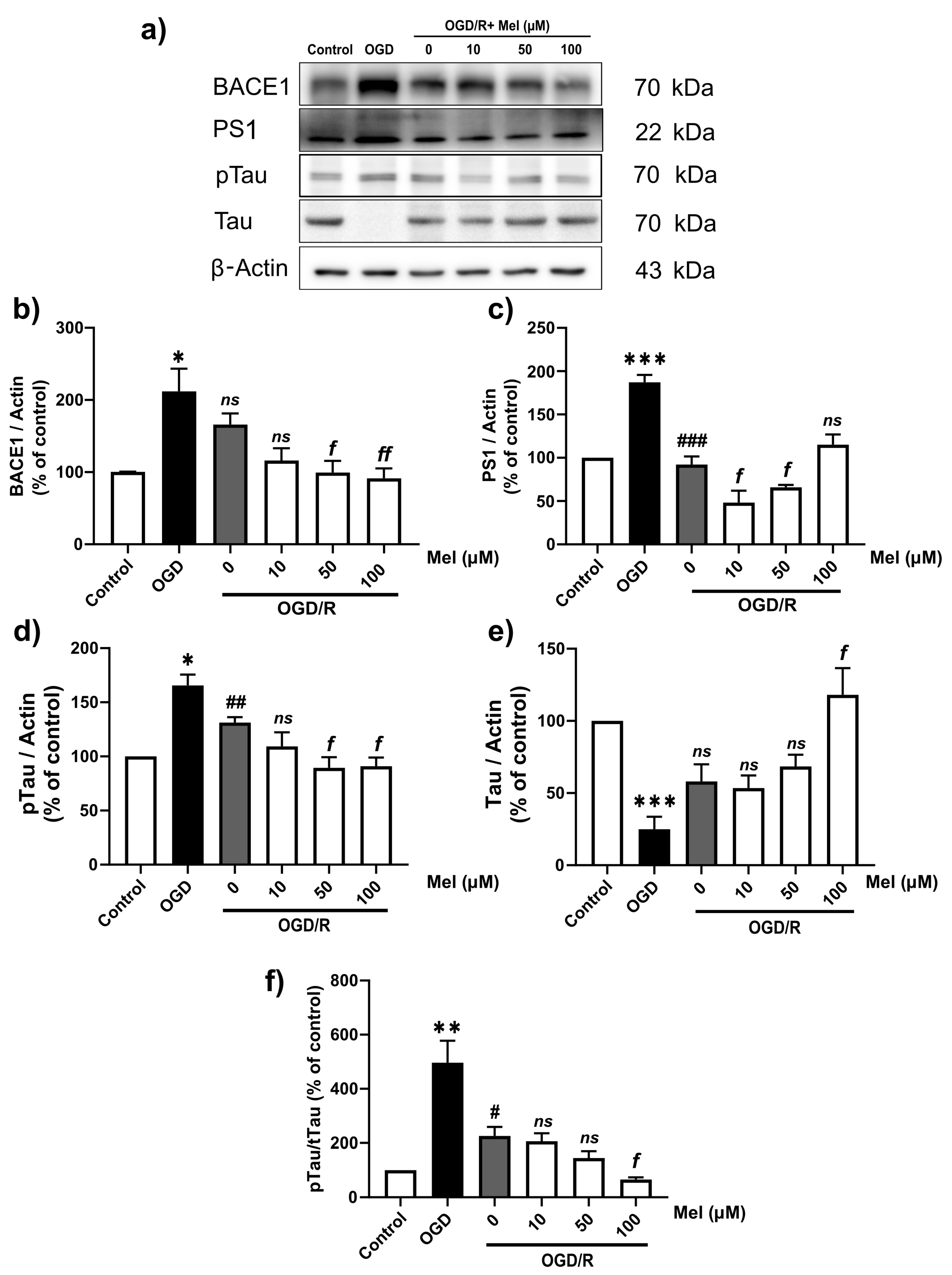
2.4. Effect of Melatonin on the Amyloid Pathway and Tau Protein in SH-SY5Y Cells under OGD/R Conditions
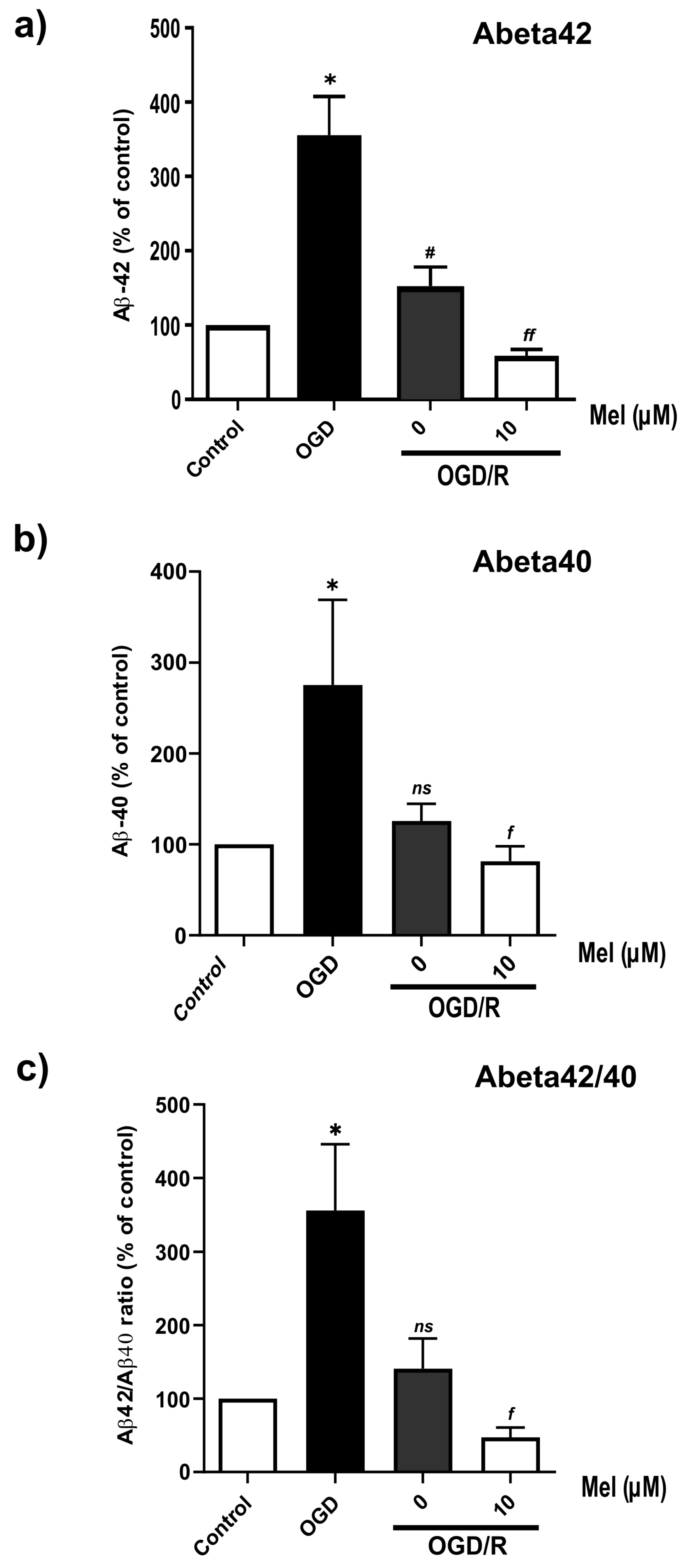
2.5. Effect of Melatonin on the Production of Amyloid Beta in the SH-SY5Y Cells under OGD/R Conditions
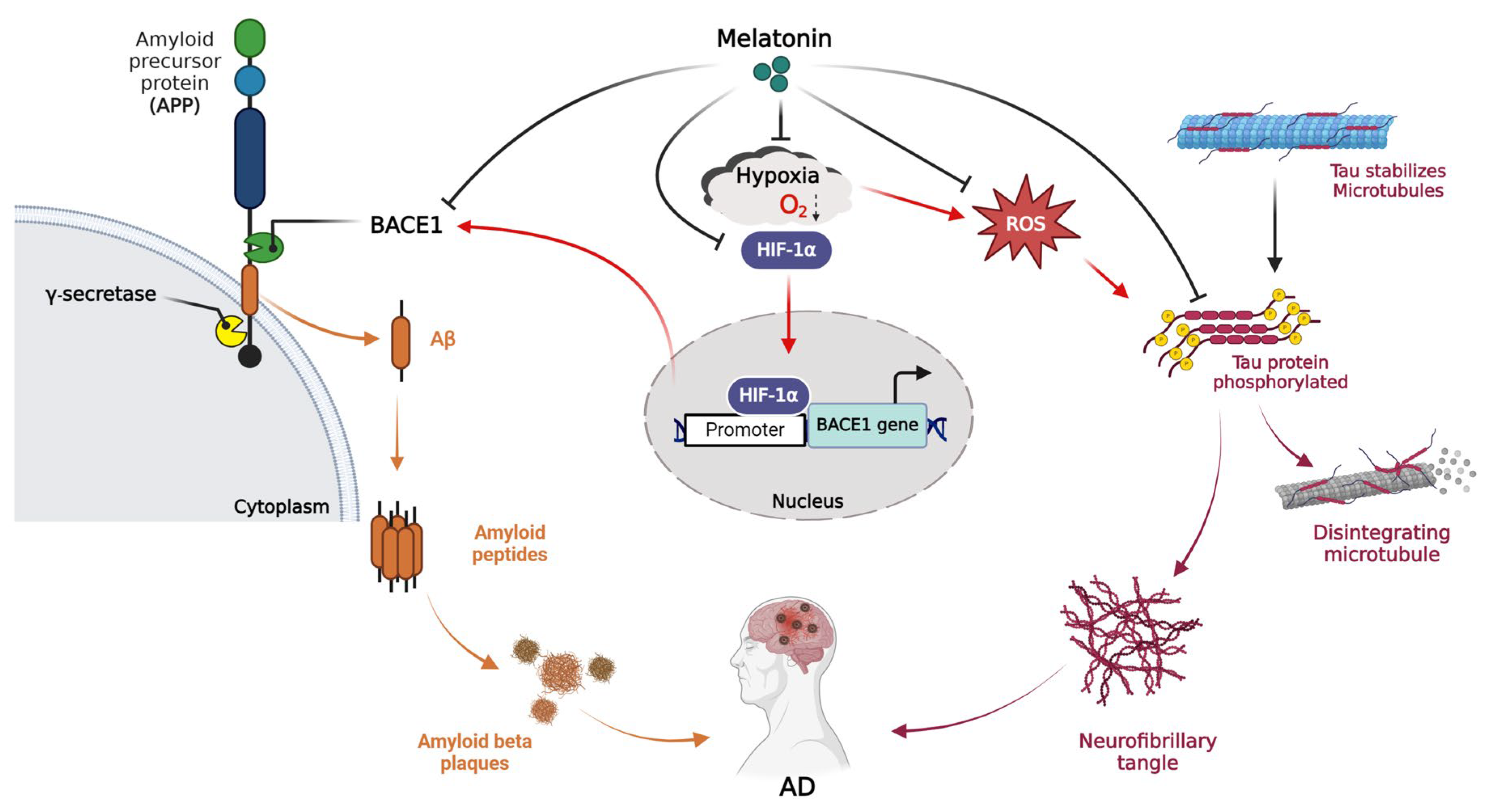
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments under Oxygen–Glucose Deprivation (OGD) and Reoxygenation (OGD/R) Conditions
4.2. Trypan Blue Assay
4.3. Cell Viability Assay
4.4. Quantitative RT-PCR
4.5. Western Blot Analysis
4.6. Enzyme-Linked Immunosorbent Assay
4.7. Immunocytochemistry
4.8. Luciferase Assay
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hathidara, M.Y.; Saini, V.; Malik, A.M. Stroke in the Young: A Global Update. Curr. Neurol. Neurosci. Rep. 2019, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.K. Ischemic Stroke. Am. J. Med. 2021, 134, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr. Pharm. Des. 2020, 26, 4246–4260. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Geetha, K.M. Neurotherapeutic applications of nanomedicine for treating Alzheimer’s disease. J. Control. Release 2020, 325, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. [23]—Characterization of Molecular Pathology of Alzheimer’s Disease. In Methods in Neurosciences; Conn, P.M., Ed.; Academic Press: Cambridge, MA, USA, 1989; Volume 1, pp. 425–443. [Google Scholar]
- Zhang, S.; Wang, P.; Ren, L.; Hu, C.; Bi, J. Protective effect of melatonin on soluble Aβ1–42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus. Alzheimer’s Res. Ther. 2016, 8, 40. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 1359–1370. [Google Scholar] [CrossRef]
- Chi, N.F.; Chien, L.N.; Ku, H.L.; Hu, C.J.; Chiou, H.Y. Alzheimer disease and risk of stroke: A population-based cohort study. Neurology 2013, 80, 705–711. [Google Scholar] [CrossRef]
- Tolppanen, A.-M.; Lavikainen, P.; Solomon, A.; Kivipelto, M.; Soininen, H.; Hartikainen, S. Incidence of stroke in people with Alzheimer disease. Neurology 2013, 80, 353–358. [Google Scholar] [CrossRef]
- Gamaldo, A.; Moghekar, A.; Kilada, S.; Resnick, S.M.; Zonderman, A.B.; O’Brien, R. Effect of a clinical stroke on the risk of dementia in a prospective cohort. Neurology 2006, 67, 1363–1369. [Google Scholar] [CrossRef]
- Menon, R.S.; Kidwell, C.S. Neuroimaging demonstration of evolving small vessel ischemic injury in cerebral amyloid angiopathy. Stroke 2009, 40, e675–e677. [Google Scholar] [CrossRef] [PubMed]
- Reijmer, Y.D.; van Veluw, S.J.; Greenberg, S.M. Ischemic brain injury in cerebral amyloid angiopathy. J. Cereb. Blood Flow Metab. 2016, 36, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Gregory, J.; Kuchibhotla, K.V.; Fine, S.; Wei, Y.; Ayata, C.; Frosch, M.P.; Greenberg, S.M.; Bacskai, B.J. Cerebrovascular lesions induce transient β-amyloid deposition. Brain 2011, 134, 3697–3707. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Yamashita, T.; Tian, F.; Li, X.; Liu, X.; Shi, X.; Nakano, Y.; Tsunoda, K.; Nomura, E.; Sasaki, R.; et al. Chronic cerebral hypoperfusion alters amyloid-β transport related proteins in the cortical blood vessels of Alzheimer’s disease model mouse. Brain Res. 2019, 1723, 146379. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of Amyloid Precursor Protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Shiota, S.; Takekawa, H.; Matsumoto, S.-e.; Takeda, K.; Nurwidya, F.; Yoshioka, Y.; Takahashi, F.; Hattori, N.; Tabira, T.; Mochizuki, H.; et al. Chronic Intermittent Hypoxia/Reoxygenation Facilitate Amyloid-β Generation in Mice. J. Alzheimer’s Dis. 2013, 37, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; McClatchy, D.B.; Maher, P.; Liang, Z.; Diedrich, J.K.; Soriano-Castell, D.; Goldberg, J.; Shokhirev, M.; Yates, J.R., 3rd; Schubert, D.; et al. Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell Death Dis. 2020, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hampton, M.J.W.; Barajas, M.B.; Riess, M.L. Development of a Cell Co-Culture Model to Mimic Cardiac Ischemia/Reperfusion In Vitro. J. Vis. Exp. 2021, 176, e62913. [Google Scholar]
- Kuan, C.-Y.; Chen, H.-R.; Gao, N.; Kuo, Y.-M.; Chen, C.-W.; Yang, D.; Kinkaid, M.M.; Hu, E.; Sun, Y.-Y. Brain-targeted hypoxia-inducible factor stabilization reduces neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 2021, 148, 105200. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef]
- Marciante, A.B.; Howard, J.; Kelly, M.N.; Moreno, J.S.; Allen, L.L.; Gonzalez-Rothi, E.J.; Mitchell, G.S. Dose-dependent phosphorylation of endogenous Tau by intermittent hypoxia in rat brain. J. Appl. Physiol. 2022, 133, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Y.W.; Zhang, X.; Liu, R.; Zhang, X.; Hong, S.; Xia, K.; Xia, J.; Zhang, Z.; Xu, H. Transcriptional regulation of APH-1A and increased gamma-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J. 2006, 20, 1275–1277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.-F.; Xu, H.; Zhang, Y.-W. Hypoxia-inducible Factor 1α (HIF-1α)-mediated Hypoxia Increases BACE1 Expression and β-Amyloid Generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef] [PubMed]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef] [PubMed]
- García, J.J.; López-Pingarrón, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; García-Gil, F.A.; Tan, D.-X.; Reiter, R.J.; Ramírez, J.M.; Bernal-Pérez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef]
- Andrade, M.K.; Souza, L.C.; Azevedo, E.M.; Bail, E.L.; Zanata, S.M.; Andreatini, R.; Vital, M. Melatonin reduces β-amyloid accumulation and improves short-term memory in streptozotocin-induced sporadic Alzheimer’s disease model. IBRO Neurosci. Rep. 2023, 14, 264–272. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Sos, M.; Omar, R.A.; Bick, R.J.; Hickson-Bick, D.L.; Reiter, R.J.; Efthimiopoulos, S.; Robakis, N.K. Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J. Neurosci. 1997, 17, 1683–1690. [Google Scholar] [CrossRef]
- Daniels, W.M.; van Rensburg, S.J.; van Zyl, J.M.; Taljaard, J.J. Melatonin prevents beta-amyloid-induced lipid peroxidation. J. Pineal Res. 1998, 24, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, U.; Tanbek, K.; Gul, S.; Gul, M.; Koc, A.; Sandal, S. Melatonin Attenuates Cerebral Ischemia/Reperfusion Injury through Inducing Autophagy. Neuroendocrinology 2023, 113, 1035–1050. [Google Scholar] [CrossRef]
- Panmanee, J.; Nopparat, C.; Chavanich, N.; Shukla, M.; Mukda, S.; Song, W.; Vincent, B.; Govitrapong, P. Melatonin regulates the transcription of βAPP-cleaving secretases mediated through melatonin receptors in human neuroblastoma SH-SY5Y cells. J. Pineal Res. 2015, 59, 308–320. [Google Scholar] [CrossRef]
- Thangwong, P.; Jearjaroen, P.; Govitrapong, P.; Tocharus, C.; Tocharus, J. Melatonin improves cognitive function by suppressing endoplasmic reticulum stress and promoting synaptic plasticity during chronic cerebral hypoperfusion in rats. Biochem. Pharmacol. 2022, 198, 114980. [Google Scholar] [CrossRef]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol. Cell. Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef]
- Gardner, L.B.; Li, Q.; Park, M.S.; Flanagan, W.M.; Semenza, G.L.; Dang, C.V. Hypoxia Inhibits G1/S Transition through Regulation of p27 Expression. J. Biol. Chem. 2001, 276, 7919–7926. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, S.; Feron, O.; Raes, M.; Michiels, C. Intermittent hypoxia changes HIF-1alpha phosphorylation pattern in endothelial cells: Unravelling of a new PKA-dependent regulation of HIF-1alpha. Biochim. Biophys. Acta 2007, 1773, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Köhler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin α/β pathway. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2008, 1783, 394–404. [Google Scholar] [CrossRef]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.W.; Crawford, J.D.; Desmond, D.W.; Godefroy, O.; Jokinen, H.; Mahinrad, S.; Bae, H.J.; Lim, J.S.; Köhler, S.; Douven, E.; et al. Profile of and risk factors for poststroke cognitive impairment in diverse ethnoregional groups. Neurology 2019, 93, e2257–e2271. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, Y. Dementia and Death After Stroke in Older Adults During a 10-year Follow-up: Results from a Competing Risk Model. J. Nutr. Health Aging 2018, 22, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Shared Genomic and Proteomic Contribution of Amyloid and Tau Protein Characteristic of Alzheimer’s Disease to Brain Ischemia. Int. J. Mol. Sci. 2020, 21, 3186. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Participation of Amyloid and Tau Protein in Neuronal Death and Neurodegeneration after Brain Ischemia. Int. J. Mol. Sci. 2020, 21, 4599. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek-Kozioł, M.; Kocki, J.; Bogucki, J.; Januszewski, S.; Bogucka-Kocka, A.; Czuczwar, S.J. Expression of the Tau Protein and Amyloid Protein Precursor Processing Genes in the CA3 Area of the Hippocampus in the Ischemic Model of Alzheimer’s Disease in the Rat. Mol. Neurobiol. 2020, 57, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Jolkkonen, J.; Cuzzocrea, S.; Pedata, F.; Cechetto, D.; Popa-Wagner, A. Cognitive Impairment with Vascular Impairment and Degeneration. Curr. Neurovasc. Res. 2011, 8, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, X.; Yang, D.; Luo, G.; Chen, S.; Le, W. Hypoxia increases Aβ generation by altering β- and γ-cleavage of APP. Neurobiol. Aging 2009, 30, 1091–1098. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Zhang, X.; Xie, W.; Li, L.; Yang, D.; Heng, X.; Du, Y.; Doody, R.S.; Le, W. Prenatal hypoxia may aggravate the cognitive impairment and Alzheimer’s disease neuropathology in APPSwe/PS1A246E transgenic mice. Neurobiol. Aging 2013, 34, 663–678. [Google Scholar] [CrossRef]
- Ryou, M.-G.; Mallet, R.T. An In Vitro Oxygen–Glucose Deprivation Model for Studying Ischemia–Reperfusion Injury of Neuronal Cells. In Traumatic and Ischemic Injury: Methods and Protocols; Tharakan, B., Ed.; Springer: New York, NY, USA, 2018; pp. 229–235. [Google Scholar]
- Mazure, N.M.; Chen, E.Y.; Yeh, P.; Laderoute, K.R.; Giaccia, A.J. Oncogenic transformation and hypoxia synergistically act to modulate vascular endothelial growth factor expression. Cancer Res. 1996, 56, 3436–3440. [Google Scholar] [PubMed]
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P.; et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Ghobrial, I.M.; Azab, A.K. Hypoxia promotes dissemination and colonization in new bone marrow niches in Waldenström macroglobulinemia. Mol. Cancer Res. 2015, 13, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Tuder, R.M. Hypoxia-induced pulmonary vascular remodeling: A model for what human disease? J. Clin. Investig. 2000, 106, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Plagnes-Juan, E.; Geurden, I.; Panserat, S.; Marandel, L. Exposure to an acute hypoxic stimulus during early life affects the expression of glucose metabolism-related genes at first-feeding in trout. Sci. Rep. 2017, 7, 363. [Google Scholar] [CrossRef]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Taylor, C.T. Targeting the HIF pathway in inflammation and immunity. Curr. Opin. Pharmacol. 2013, 13, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Halterman, M.W.; Miller, C.C.; Federoff, H.J. Hypoxia-inducible factor-1alpha mediates hypoxia-induced delayed neuronal death that involves p53. J. Neurosci. 1999, 19, 6818–6824. [Google Scholar] [CrossRef]
- Basak, J.M.; Falk, M.; Mitchell, D.N.; Coakley, K.A.; Quillinan, N.; Orfila, J.E.; Herson, P.S. Targeting BACE1-mediated production of amyloid beta improves hippocampal synaptic function in an experimental model of ischemic stroke. J. Cereb. Blood Flow Metab. 2023, 43, 66–77. [Google Scholar] [CrossRef]
- Christensen, M.A.; Zhou, W.; Qing, H.; Lehman, A.; Philipsen, S.; Song, W. Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase, by Sp1. Mol. Cell. Biol. 2004, 24, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Roßner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression—Implications for Alzheimer’s disease. Prog. Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S. BACE1 inhibitor reduces β-amyloid production in humans. Nat. Rev. Drug Discov. 2017, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.; Salihoglu, H.; Rodrigues, E.; Herzog, E.; Blume, T.; Filser, S.; Dorostkar, M.; Shimshek, D.R.; Brose, N.; Neumann, U.; et al. BACE1 inhibition more effectively suppresses initiation than progression of β-amyloid pathology. Acta Neuropathol. 2018, 135, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient Cerebral Ischemia Induces Aberrant Neuronal Cell Cycle Re-entry and Alzheimer’s Disease-like Tauopathy in Female Rats. J. Biol. Chem. 2004, 279, 22684–22692. [Google Scholar] [CrossRef] [PubMed]
- Uzun, A.B.; Iliescu, M.G.; Stanciu, L.E.; Ionescu, E.V.; Ungur, R.A.; Ciortea, V.M.; Irsay, L.; Motoașcă, I.; Popescu, M.N.; Popa, F.L.; et al. Effectiveness of Intermittent Hypoxia-Hyperoxia Therapy in Different Pathologies with Possible Metabolic Implications. Metabolites 2023, 13, 181. [Google Scholar] [CrossRef]
- Zhi, W.; Li, K.; Wang, H.; Lei, M.; Guo, Y. Melatonin elicits protective effects on OGD/R-insulted H9c2 cells by activating PGC-1α/Nrf2 signaling. Int. J. Mol. Med. 2020, 45, 1294–1304. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | GenBank | Forward (5’3’) | Reverse (5’3’) |
|---|---|---|---|
| APP | NM_001136131.3 | GCTGGCCTGCTGGCTGAACC | GCGACGGTGTGCCAGTGAA |
| TAU | XM_054316146.1 | GACAGAGTCCAGTCGAAGATTG | AGGAGACATTGCTGAGATGC |
| BACE1 | NM_001411039.1 | AGGTTACCTTGGCGTGTGTC | GAGGCAATCTTTGCACCAAT |
| PS1 | XM_054376420.1 | AATAGAGAACGGCAGGAGCA | GCCATGAGGGCACTAATCAT |
| HIF1α | NM_001243084.2 | CTTGCTCATCAGTTGCCACTTC | GCCATTTCTGTGTGTAAGCATTTC |
| GAPDH | NM_001357943.2 | ACAACTTTGGTATCGTGGAAGG | GCCATCACGCCACAGTTTC |
| Antibodies | Catalogue Number | Dilution | Source |
|---|---|---|---|
| Rabbit anti-Aβ1-42 | 14947 | 1:2000 | Cell Signaling Technology, Inc., Danvers, MA, USA |
| Mouse anti-APP-C99 | MABN380 | 1:2000 | Merck Millipore, Temecula, CA, USA |
| Rabbit anti-PS1 | 3622S | 1:1000 | Cell Signaling Technology, Inc., Danvers, MA, USA |
| Rabbit anti-HIF1α | Ab179483 | 1:1000 | Abcam, Cambridge, UK |
| Rabbit anti-BACE1 | Ab108394 | 1:2000 | Abcam, Cambridge, UK |
| Mouse anti-pTau (Thr181) | MABN388 | 1:500 | Merck Millipore, Temecula, CA, USA |
| Mouse anti-Tau | 05348 | 1:1000 | Merck Millipore, Temecula, CA, USA |
| Mouse anti-β-Actin | MAB1501 | 1:20,000 | Merck Millipore, Temecula, CA, USA |
| Goat anti-Mouse IgG | AP142P | 1:1000–1:20,000 | Merck Millipore, Temecula, CA, USA |
| Goat anti-Rabbit IgG | AP132P | 1:1000–1:20,000 | Merck Millipore, Temecula, CA, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singrang, N.; Nopparat, C.; Panmanee, J.; Govitrapong, P. Melatonin Inhibits Hypoxia-Induced Alzheimer’s Disease Pathogenesis by Regulating the Amyloidogenic Pathway in Human Neuroblastoma Cells. Int. J. Mol. Sci. 2024, 25, 5225. https://doi.org/10.3390/ijms25105225
Singrang N, Nopparat C, Panmanee J, Govitrapong P. Melatonin Inhibits Hypoxia-Induced Alzheimer’s Disease Pathogenesis by Regulating the Amyloidogenic Pathway in Human Neuroblastoma Cells. International Journal of Molecular Sciences. 2024; 25(10):5225. https://doi.org/10.3390/ijms25105225
Chicago/Turabian StyleSingrang, Nongnuch, Chutikorn Nopparat, Jiraporn Panmanee, and Piyarat Govitrapong. 2024. "Melatonin Inhibits Hypoxia-Induced Alzheimer’s Disease Pathogenesis by Regulating the Amyloidogenic Pathway in Human Neuroblastoma Cells" International Journal of Molecular Sciences 25, no. 10: 5225. https://doi.org/10.3390/ijms25105225
APA StyleSingrang, N., Nopparat, C., Panmanee, J., & Govitrapong, P. (2024). Melatonin Inhibits Hypoxia-Induced Alzheimer’s Disease Pathogenesis by Regulating the Amyloidogenic Pathway in Human Neuroblastoma Cells. International Journal of Molecular Sciences, 25(10), 5225. https://doi.org/10.3390/ijms25105225