
Leveraging the Fragment Molecular Orbital and MM-GBSA Methods in Virtual Screening for the Discovery of Novel Non-Covalent Inhibitors Targeting the TEAD Lipid Binding Pocket

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Results
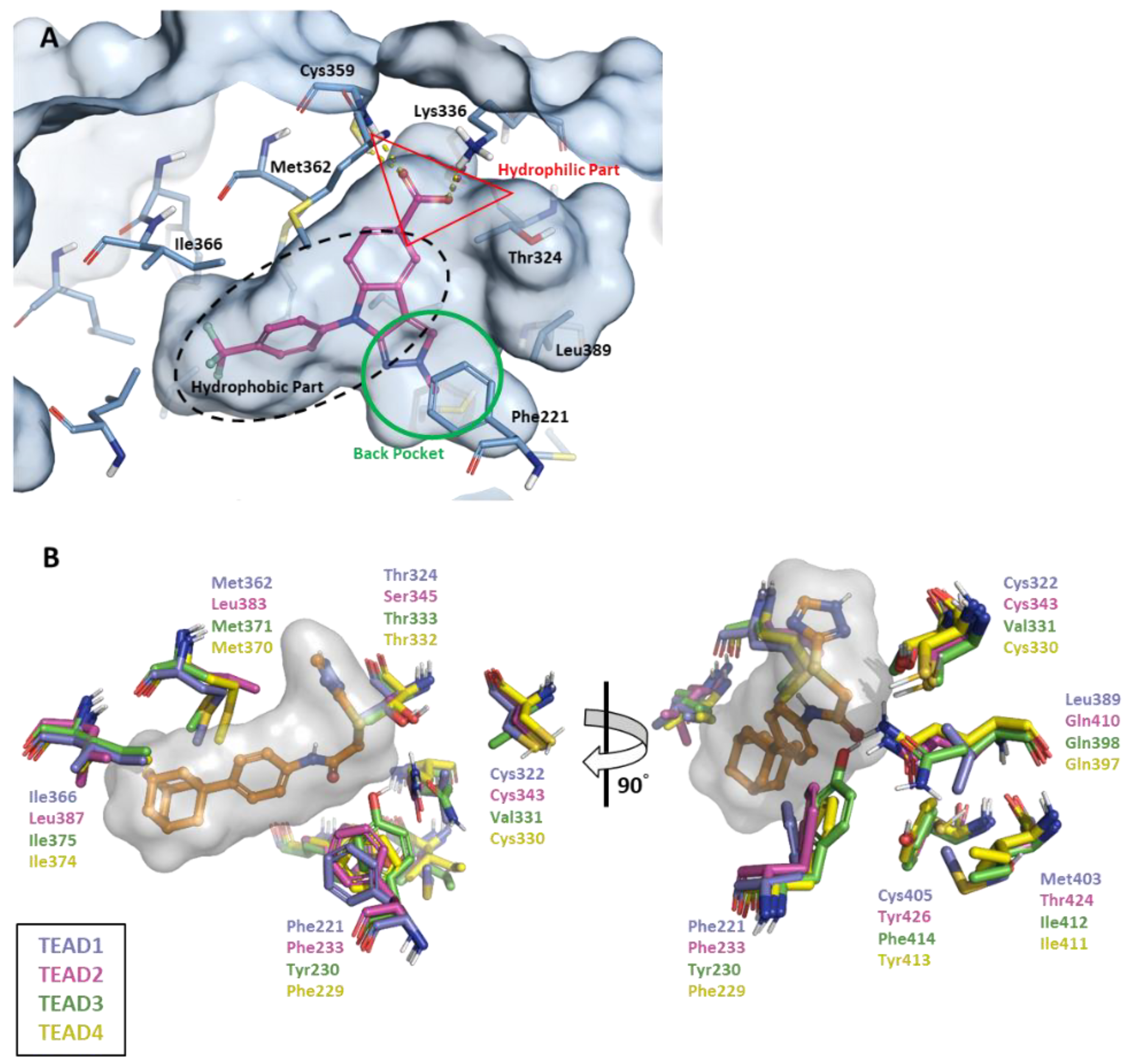
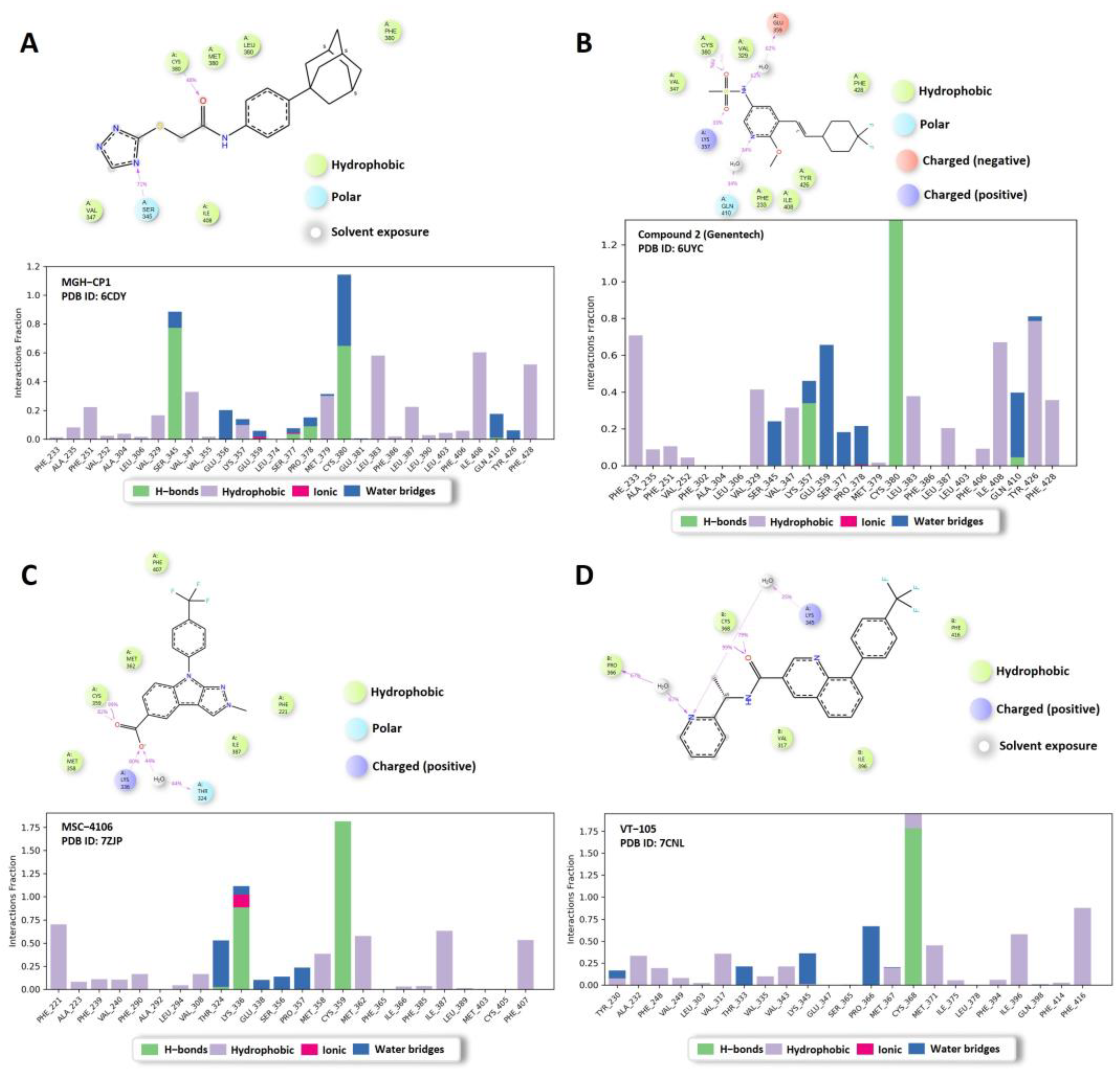
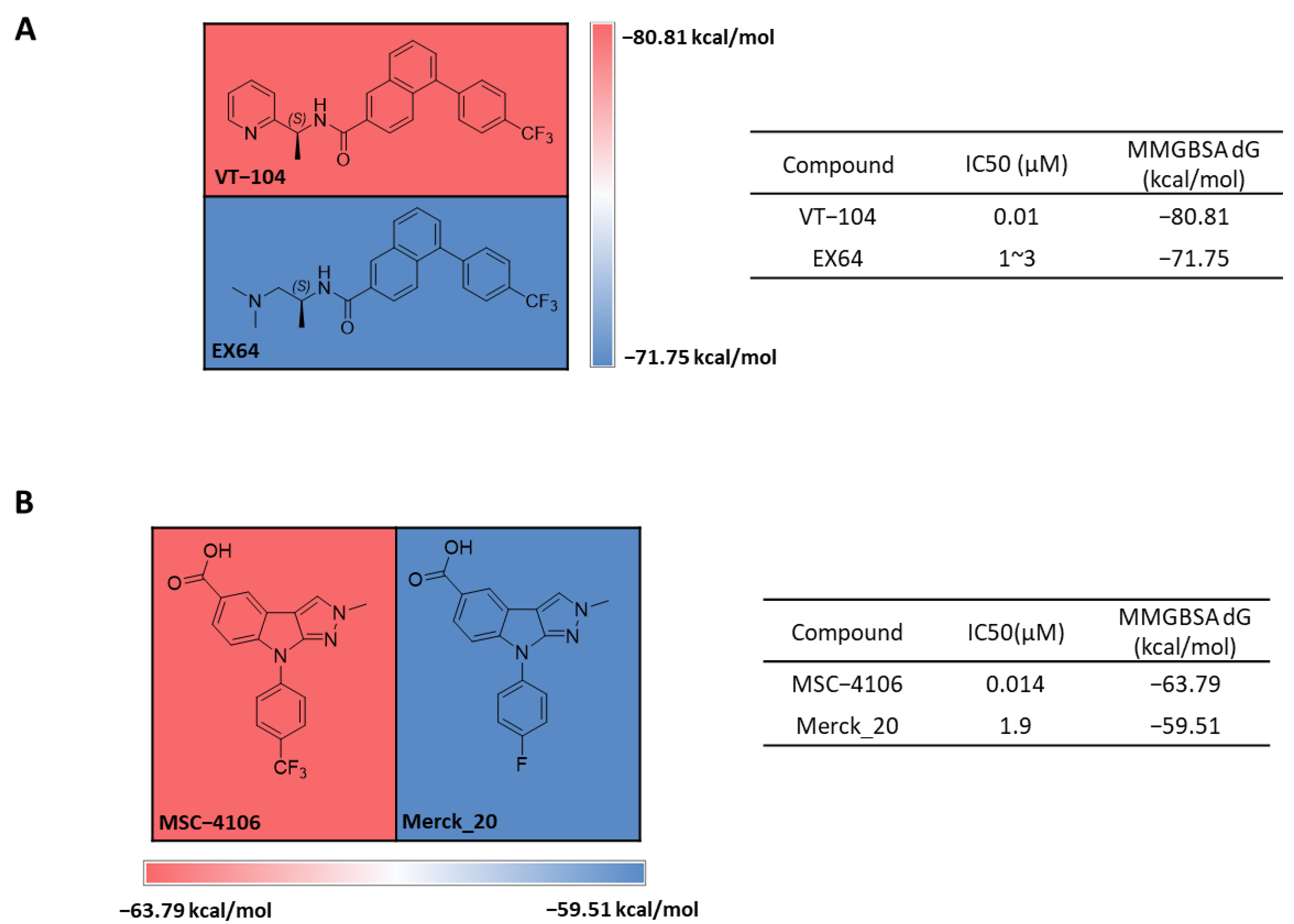
2.1. FMO Analysis of TEAD Complexes with Reference Compounds: Insights into Binding Interactions
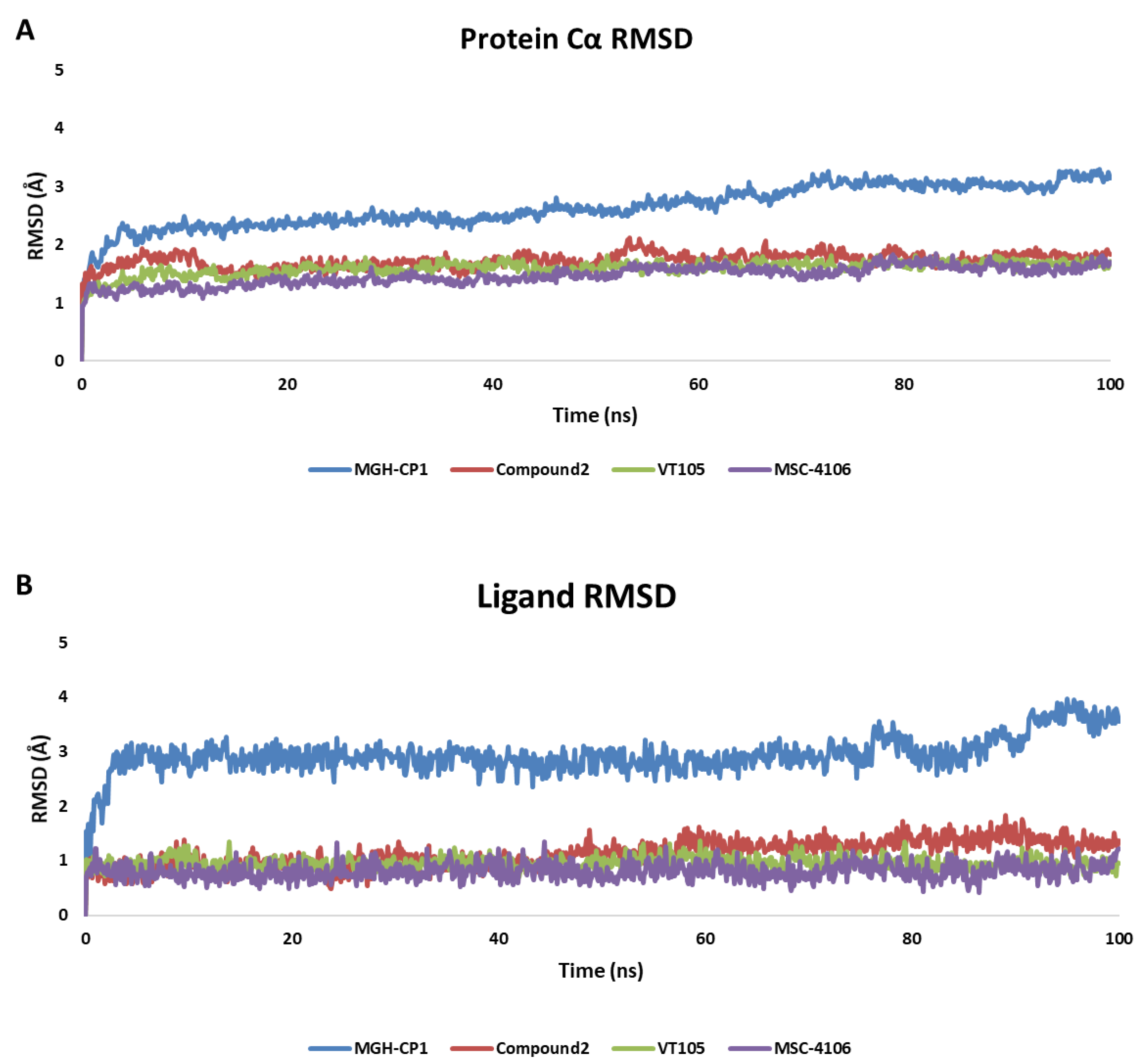
2.2. MD Analysis of TEAD Complexes with Reference Compounds: Unveiling Structural Insights
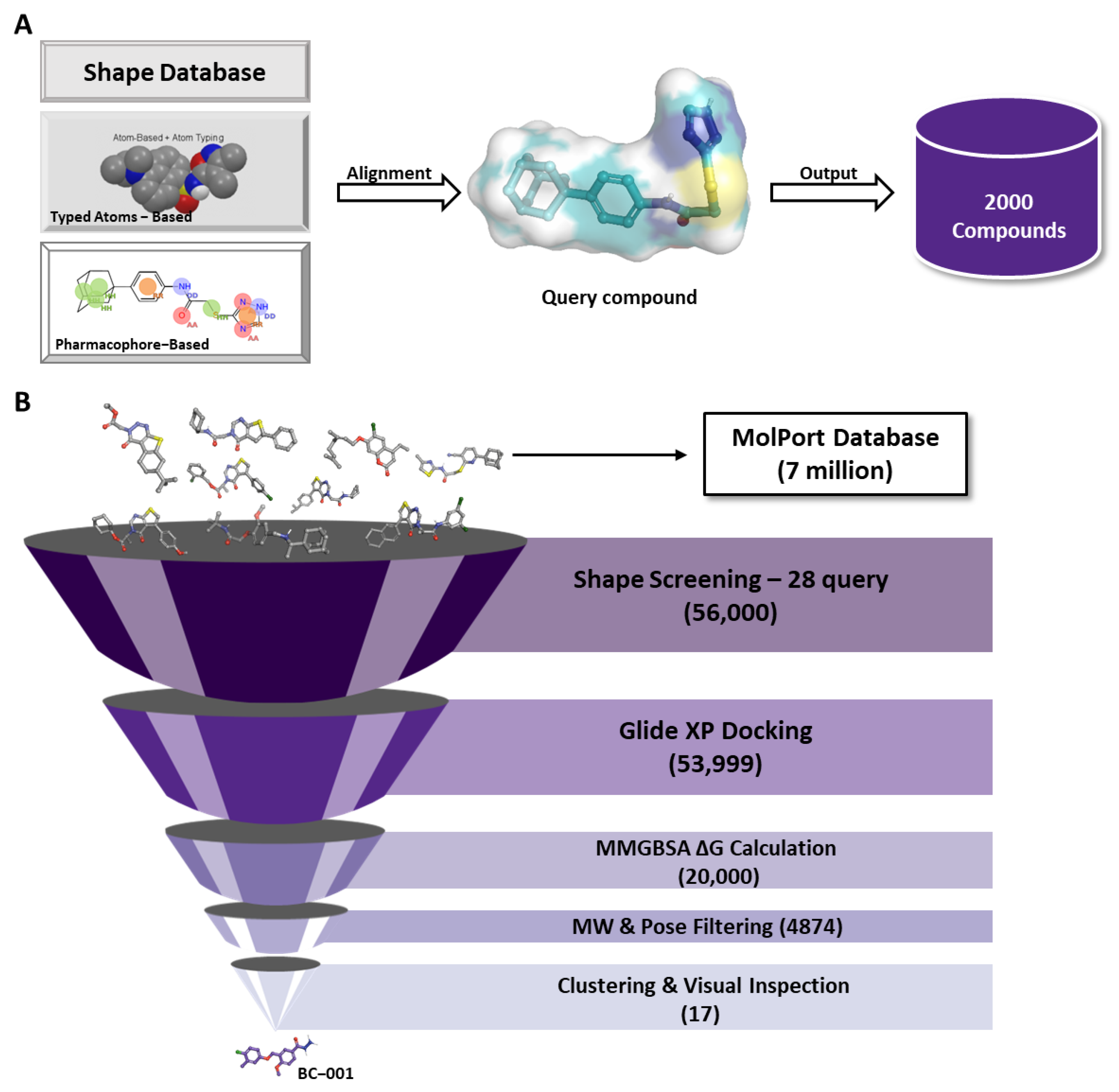
2.3. Structure-Based Virtual Screening Using MM-GBSA for Tool Compound Identification
2.4. Computational Analysis of the Compound BC-001
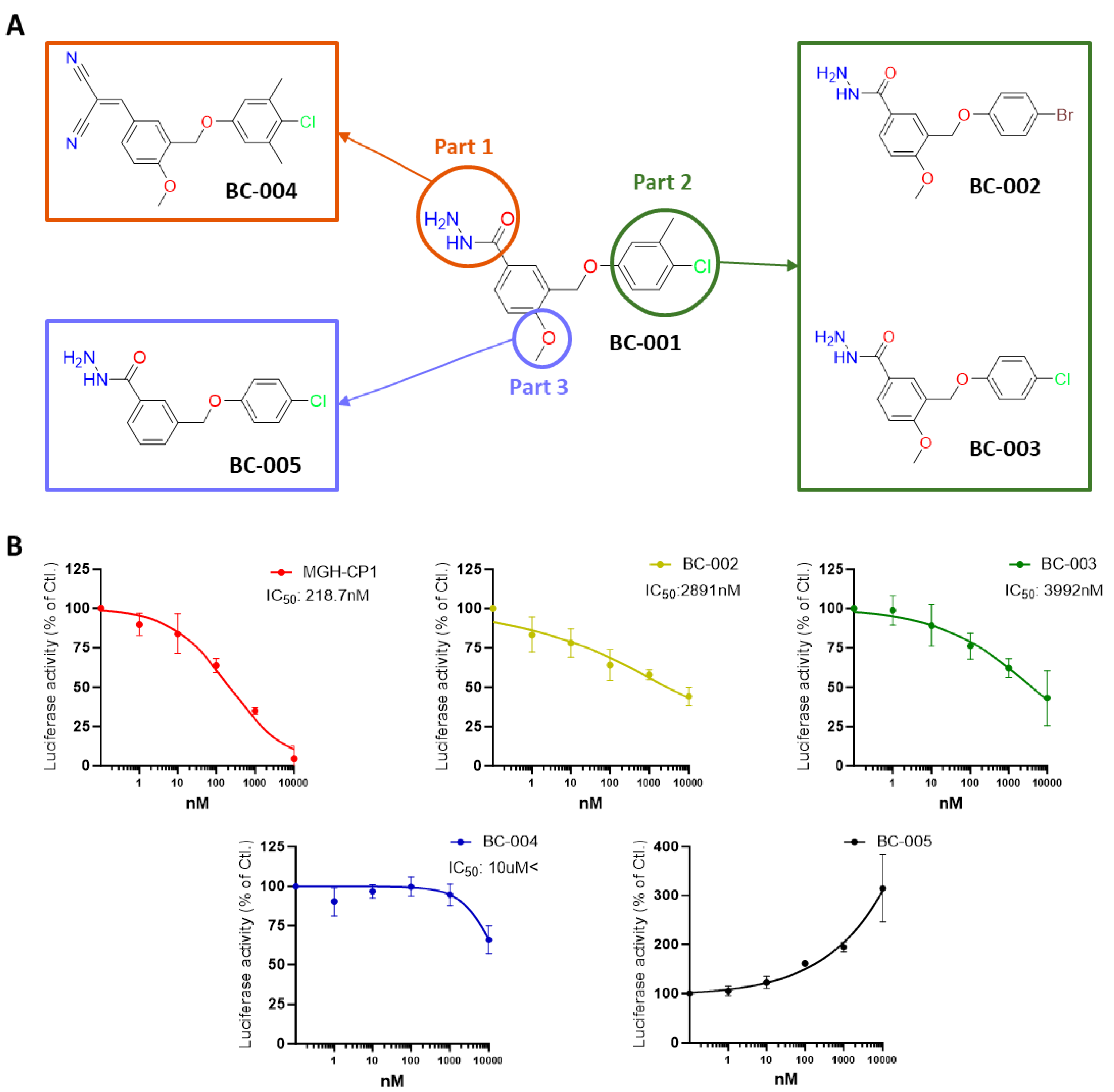
2.5. Structure–Activity Relationship Study of the Hit Compound BC-001, for Optimization
2.6. Retrospective Prediction of Binding Free Energy Using Post-MD MM-GBSA Average Values
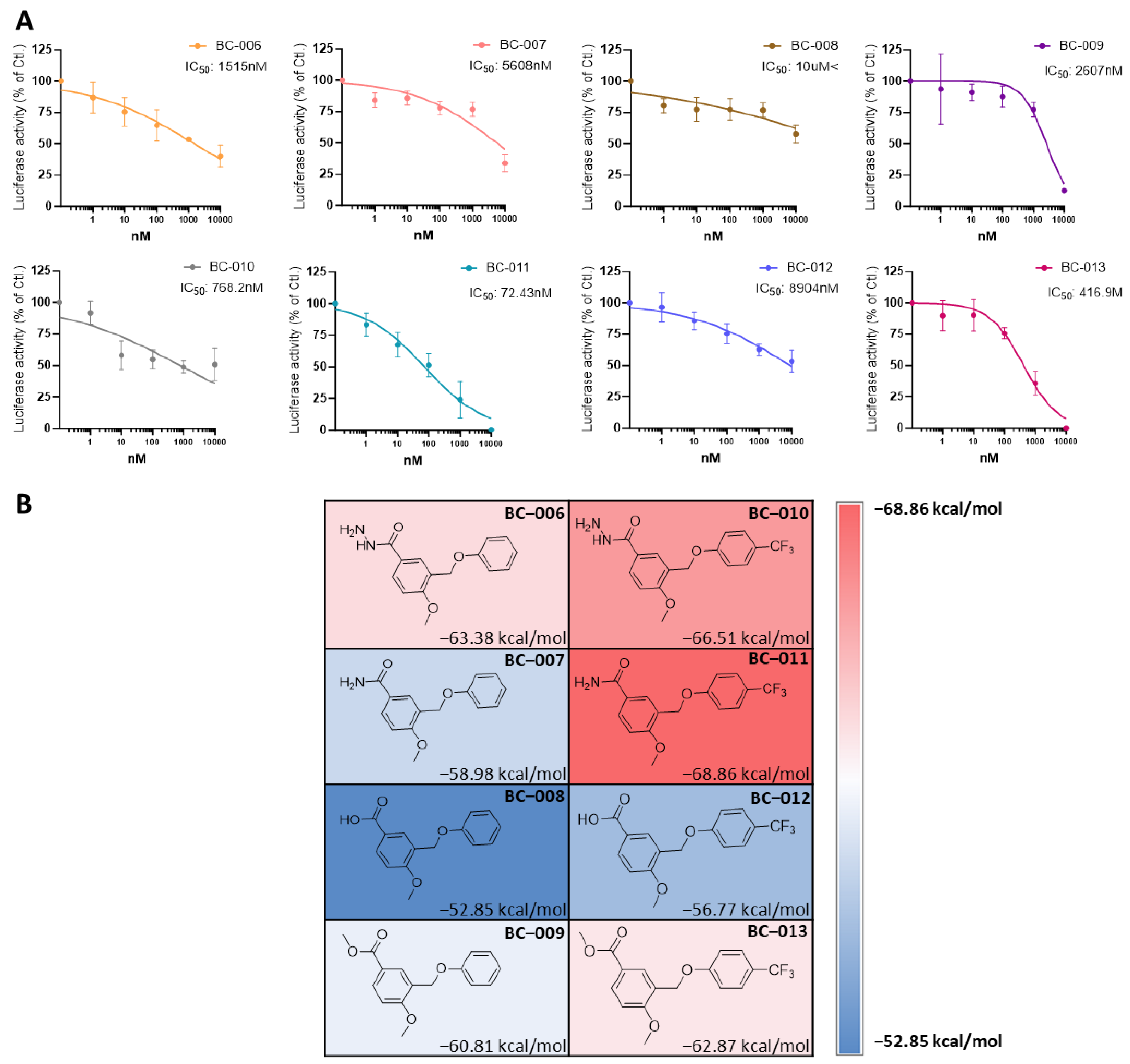
2.7. Optimizing and Synthesizing the BC-001 Scaffold: Enhancing Binding Affinity and Biological Activity
2.8. Cell Viability and TEAD Target Gene Expression Assay of BC-010, BC-011, and BC-013
3. Discussion
4. Materials and Methods
4.1. Protein Structure Preparation
4.2. FMO Calculations
4.3. Shape-Screening
4.4. Molecular Docking
4.5. MD Simulations
4.6. MM-GBSA Calculation
4.7. General Methods for Compound Synthesis and Analysis
4.8. Synthesis of Compounds
4.9. Cell Culture and Stable Cell Lines
4.10. Luciferase Assay
4.11. mRNA Expression
4.12. Cell Viability Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila Tumor Suppressor Gene Warts Encodes a Homolog of Human Myotonic Dystrophy Kinase and Is Required for the Control of Cell Shape and Proliferation. Genes. Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo Pathway and Human Cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Halder, G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Moya, I.M.; Halder, G. Hippo–YAP/TAZ Signalling in Organ Regeneration and Regenerative Medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Sahu, M.R.; Mondal, A.C. The Emerging Role of Hippo Signaling in Neurodegeneration. J. Neurosci. Res. 2020, 98, 796–814. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhao, X.; Fu, H.; Gao, Y. The Effects of YAP and Its Related Mechanisms in Central Nervous System Diseases. Front. Neurosci. 2020, 14, 595. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Rausch, V.; Hansen, C.G. The Hippo Pathway, YAP/TAZ, and the Plasma Membrane. Trends Cell Biol. 2020, 30, 32–48. [Google Scholar] [CrossRef]
- Liu, A.M.; Poon, R.T.P.; Luk, J.M. MicroRNA-375 Targets Hippo-Signaling Effector YAP in Liver Cancer and Inhibits Tumor Properties. Biochem. Biophys. Res. Commun. 2010, 394, 623–627. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, S.M.; Balbin, O.A.; Chen, G.; Nadal, E.; Kalyana-Sundaram, S.; Pan, J.; Veeneman, B.; Cao, X.; Malik, R.; Vats, P.; et al. Transcriptome Meta-Analysis of Lung Cancer Reveals Recurrent Aberrations in NRG1 and Hippo Pathway Genes. Nat. Commun. 2014, 5, 5893. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Bum-Erdene, K.; Zhou, D.; Gonzalez-Gutierrez, G.; Ghozayel, M.K.; Si, Y.; Xu, D.; Shannon, H.E.; Bailey, B.J.; Corson, T.W.; Pollok, K.E.; et al. Small-Molecule Covalent Modification of Conserved Cysteine Leads to Allosteric Inhibition of the TEAD⋅Yap Protein-Protein Interaction. Cell Chem. Biol. 2019, 26, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Dong, J. The Hippo Signaling Pathway in Drug Resistance in Cancer. Cancers 2021, 13, 318. [Google Scholar] [CrossRef] [PubMed]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Huh, H.D.; Kim, D.H.; Jeong, H.-S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Gibault, F.; Sturbaut, M.; Bailly, F.; Melnyk, P.; Cotelle, P. Targeting Transcriptional Enhanced Associate Domains (TEADs). J. Med. Chem. 2018, 61, 5057–5072. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.-L.; Xu, Y. Structural Insights into the YAP and TEAD Complex. Genes. Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and Functional Similarity between the Vgll1-TEAD and the YAP-TEAD Complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef]
- Noland, C.L.; Gierke, S.; Schnier, P.D.; Murray, J.; Sandoval, W.N.; Sagolla, M.; Dey, A.; Hannoush, R.N.; Fairbrother, W.J.; Cunningham, C.N. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure 2016, 24, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of TEAD Proteins Regulates Transcriptional Output of the Hippo Pathway. Nat. Chem. Biol. 2016, 12, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.-Y.; Kang, C.; Chia, C.S.B.; Luo, X.; Hong, W.; et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chan, S.W.; Zhang, X.; Walsh, M.; Lim, C.J.; Hong, W.; Song, H. Structural Basis of YAP Recognition by TEAD4 in the Hippo Pathway. Genes. Dev. 2010, 24, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting Hippo Pathway by Specific Interruption of YAP-TEAD Interaction Using Cyclic YAP-like Peptides. FASEB J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Salem, B.; Mesrouze, Y.; Schmelzle, T.; Lewis, I.; Kallen, J.; Chène, P. Structure-Based Design of Potent Linear Peptide Inhibitors of the YAP-TEAD Protein-Protein Interaction Derived from the YAP Omega-Loop Sequence. Bioorg. Med. Chem. Lett. 2019, 29, 2316–2319. [Google Scholar] [CrossRef] [PubMed]
- Mesrouze, Y.; Gubler, H.; Villard, F.; Boesch, R.; Ottl, J.; Kallen, J.; Reid, P.C.; Scheufler, C.; Marzinzik, A.L.; Chène, P. Biochemical and Structural Characterization of a Peptidic Inhibitor of the YAP:TEAD Interaction That Binds to the α-Helix Pocket on TEAD. ACS Chem. Biol. 2023, 18, 643–651. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and Pharmacological Disruption of the TEAD–YAP Complex Suppresses the Oncogenic Activity of YAP. Genes. Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Kaan, H.Y.K.; Sim, A.Y.L.; Tan, S.K.J.; Verma, C.; Song, H. Targeting YAP/TAZ-TEAD Protein-Protein Interactions Using Fragment-Based and Computational Modeling Approaches. PLoS ONE 2017, 12, e0178381. [Google Scholar] [CrossRef]
- Kim, J.; Lim, H.; Moon, S.; Cho, S.Y.; Kim, M.; Park, J.H.; Park, H.W.; No, K.T. Hot Spot Analysis of YAP-TEAD Protein-Protein Interaction Using the Fragment Molecular Orbital Method and Its Application for Inhibitor Discovery. Cancers 2021, 13, 4246. [Google Scholar] [CrossRef]
- Furet, P.; Bordas, V.; Douget, M.L.; Salem, B.; Mesrouze, Y.; Imbach-Weese, P.; Sellner, H.; Voegtle, M.; Soldermann, N.; Chapeau, E.; et al. The First Class of Small Molecules Potently Disrupting the YAP-TEAD Interaction by Direct Competition. ChemMedChem 2022, 17, e202200303. [Google Scholar] [CrossRef] [PubMed]
- Amidon, B.; Frosch, H.; Syed, S.; Cavanaugh, J.; Turcotte, C.; O’Callaghan, K.; Natarajan, P.; Ecsedy, J.; McGovern, K.; Castro, A. Abstract P216: IK-930 Mediated TEAD Inhibition Decreases and Delays Tumor Growth and Enhances Targeted Apoptosis in Lung and Colon Cancer Xenografts When Combined with MEK or EGFR Inhibitors. Mol. Cancer Ther. 2021, 20 (Suppl. S12), P216. [Google Scholar] [CrossRef]
- Tang, T.T.; Post, L. Abstract 5364: The TEAD Autopalmitoylation Inhibitor VT3989 Improves Efficacy and Increases Durability of Efficacy of Osimertinib in Preclinical EGFR Mutant Tumor Models. Cancer Res. 2022, 82 (Suppl. S12), 5364. [Google Scholar] [CrossRef]
- Lu, W.; Wang, J.; Li, Y.; Tao, H.; Xiong, H.; Lian, F.; Gao, J.; Ma, H.; Lu, T.; Zhang, D.; et al. Discovery and Biological Evaluation of Vinylsulfonamide Derivatives as Highly Potent, Covalent TEAD Autopalmitoylation Inhibitors. Eur. J. Med. Chem. 2019, 184, 111767. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Ning, C.; Yue, J.; Zhang, C.; He, X.; Wang, Y.; Liu, Z. Discovering Inhibitors of TEAD Palmitate Binding Pocket through Virtual Screening and Molecular Dynamics Simulation. Comput. Biol. Chem. 2022, 98, 107648. [Google Scholar] [CrossRef] [PubMed]
- Liberelle, M.; Toulotte, F.; Renault, N.; Gelin, M.; Allemand, F.; Melnyk, P.; Guichou, J.-F.; Cotelle, P. Toward the Design of Ligands Selective for the C-Terminal Domain of TEADs. J. Med. Chem. 2022, 65, 5926–5940. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, A.; Seike, T.; Danjo, T.; Nakajima, T.; Otsubo, N.; Yamaguchi, D.; Tsuji, Y.; Hamaguchi, K.; Yasunaga, M.; Nishiya, Y.; et al. The Novel Potent TEAD Inhibitor, K-975, Inhibits YAP1/TAZ-TEAD Protein-Protein Interactions and Exerts an Anti-Tumor Effect on Malignant Pleural Mesothelioma. Am. J. Cancer Res. 2020, 10, 4399–4415. [Google Scholar] [PubMed]
- Mesrouze, Y.; Meyerhofer, M.; Bokhovchuk, F.; Fontana, P.; Zimmermann, C.; Martin, T.; Delaunay, C.; Izaac, A.; Kallen, J.; Schmelzle, T.; et al. Effect of the Acylation of TEAD4 on Its Interaction with Co-activators YAP and TAZ. Protein Sci. 2017, 26, 2399–2409. [Google Scholar] [CrossRef]
- Fan, M.; Lu, W.; Che, J.; Kwiatkowski, N.; Gao, Y.; Seo, H.-S.; Ficarro, S.B.; Gokhale, P.C.; Liu, Y.; Geffken, E.A.; et al. Covalent Disruptor of YAP-TEAD Association Suppresses Defective Hippo Signaling. Elife 2022, 11, e78810. [Google Scholar] [CrossRef]
- Heinrich, T.; Peterson, C.; Schneider, R.; Garg, S.; Schwarz, D.; Gunera, J.; Seshire, A.; Kötzner, L.; Schlesiger, S.; Musil, D.; et al. Optimization of TEAD P-Site Binding Fragment Hit into In Vivo Active Lead MSC-4106. J. Med. Chem. 2022, 65, 9206–9229. [Google Scholar] [CrossRef]
- Hu, L.; Sun, Y.; Liu, S.; Erb, H.; Singh, A.; Mao, J.; Luo, X.; Wu, X. Discovery of a New Class of Reversible TEA Domain Transcription Factor Inhibitors with a Novel Binding Mode. eLife 2022, 11, e80210. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 Model: A next Generation Energy Model for High Resolution Protein Structure Modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Mejuch, T.; Chakraborty, S.; Karatas, H.; Bharath, S.R.; Guèret, S.M.; Goy, P.-A.; Hahne, G.; Pahl, A.; Sievers, S.; et al. Identification of Quinolinols as Activators of TEAD-Dependent Transcription. ACS Chem. Biol. 2019, 14, 2909–2921. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Ma, M.; Li, J.; Yu, F.-X.; Guan, K.-L.; Post, L. Small Molecule Inhibitors of TEAD Auto-Palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-Deficient Mesothelioma. Mol. Cancer Ther. 2021, 20, 986–998. [Google Scholar] [CrossRef]
- Kumar, B.K.; Faheem; Sekhar, K.V.G.C.; Ojha, R.; Prajapati, V.K.; Pai, A.; Murugesan, S. Pharmacophore Based Virtual Screening, Molecular Docking, Molecular Dynamics and MM-GBSA Approach for Identification of Prospective SARS-CoV-2 Inhibitor from Natural Product Databases. J. Biomol. Struct. Dyn. 2022, 40, 1363–1386. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.; Hidalgo, I.J. New Horizons in Predictive Drug Metabolism and Pharmacokinetics. In New Horizons in Predictive Drug Metabolism and Pharmacokinetics; The Royal Society of Chemistry: London, UK, 2016; pp. 27–52. ISBN 9781849738286. [Google Scholar]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Alexeev, Y.; Mazanetz, M.P.; Ichihara, O.; Fedorov, D.G. GAMESS As a Free Quantum-Mechanical Platform for Drug Research. Curr. Top. Med. Chem. 2012, 12, 2013–2033. [Google Scholar] [CrossRef]
- Nakano, T.; Kaminuma, T.; Sato, T.; Akiyama, Y.; Uebayasi, M.; Kitaura, K. Fragment Molecular Orbital Method: Application to Polypeptides. Chem. Phys. Lett. 2000, 318, 614–618. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Second Order Møller-Plesset Perturbation Theory Based upon the Fragment Molecular Orbital Method. J. Chem. Phys. 2004, 121, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, D.G.; Kitaura, K.; Li, H.; Jensen, J.H.; Gordon, M.S. The Polarizable Continuum Model (PCM) Interfaced with the Fragment Molecular Orbital Method (FMO). J. Comput. Chem. 2006, 27, 976–985. [Google Scholar] [CrossRef]
- Lim, H.; Hong, H.; Hwang, S.; Kim, S.J.; Seo, S.Y.; No, K.T. Identification of Novel Natural Product Inhibitors against Matrix Metalloproteinase 9 Using Quantum Mechanical Fragment Molecular Orbital-Based Virtual Screening Methods. Int. J. Mol. Sci. 2022, 23, 4438. [Google Scholar] [CrossRef]
- Sastry, G.M.; Dixon, S.L.; Sherman, W. Rapid Shape-Based Ligand Alignment and Virtual Screening Method Based on Atom/Feature-Pair Similarities and Volume Overlap Scoring. J. Chem. Inf. Model. 2011, 51, 2455–2466. [Google Scholar] [CrossRef]
- Lim, H.; Jin, X.; Kim, J.; Hwang, S.; Shin, K.B.; Choi, J.; Nam, K.-Y.; No, K.T. Investigation of Hot Spot Region in XIAP Inhibitor Binding Site by Fragment Molecular Orbital Method. Comput. Struct. Biotechnol. J. 2019, 17, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Munnaluri, R.; Sivan, S.K.; Manga, V. Molecular Docking and MM/GBSA Integrated Protocol for Designing Small Molecule Inhibitors against HIV-1 Gp41. Med. Chem. Res. 2015, 24, 829–841. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
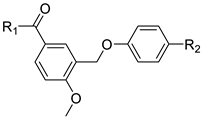 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (nM) | MMGBSA dG (kcal/mol) |
| BC-006 | ─NH─NH2 | H | 1515 | −63.38 |
| BC-007 | ─NH2 | H | 5608 | −58.98 |
| BC-008 | ─OH | H | >10,000 | −52.85 |
| BC-009 | ─OCH3 | H | 2607 | −60.81 |
| BC-010 | ─NH─NH2 | ─CF3 | 768.2 | −66.51 |
| BC-011 | ─NH2 | ─CF3 | 72.43 | −68.86 |
| BC-012 | ─OH | ─CF3 | 8904 | −56.77 |
| BC-013 | ─OCH3 | ─CF3 | 416.9 | −62.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Jin, H.; Kim, J.; Cho, S.Y.; Moon, S.; Wang, J.; Mao, J.; No, K.T. Leveraging the Fragment Molecular Orbital and MM-GBSA Methods in Virtual Screening for the Discovery of Novel Non-Covalent Inhibitors Targeting the TEAD Lipid Binding Pocket. Int. J. Mol. Sci. 2024, 25, 5358. https://doi.org/10.3390/ijms25105358
Kim J, Jin H, Kim J, Cho SY, Moon S, Wang J, Mao J, No KT. Leveraging the Fragment Molecular Orbital and MM-GBSA Methods in Virtual Screening for the Discovery of Novel Non-Covalent Inhibitors Targeting the TEAD Lipid Binding Pocket. International Journal of Molecular Sciences. 2024; 25(10):5358. https://doi.org/10.3390/ijms25105358
Chicago/Turabian StyleKim, Jongwan, Haiyan Jin, Jinhyuk Kim, Seon Yeon Cho, Sungho Moon, Jianmin Wang, Jiashun Mao, and Kyoung Tai No. 2024. "Leveraging the Fragment Molecular Orbital and MM-GBSA Methods in Virtual Screening for the Discovery of Novel Non-Covalent Inhibitors Targeting the TEAD Lipid Binding Pocket" International Journal of Molecular Sciences 25, no. 10: 5358. https://doi.org/10.3390/ijms25105358
APA StyleKim, J., Jin, H., Kim, J., Cho, S. Y., Moon, S., Wang, J., Mao, J., & No, K. T. (2024). Leveraging the Fragment Molecular Orbital and MM-GBSA Methods in Virtual Screening for the Discovery of Novel Non-Covalent Inhibitors Targeting the TEAD Lipid Binding Pocket. International Journal of Molecular Sciences, 25(10), 5358. https://doi.org/10.3390/ijms25105358