
Novel Acetamide-Based HO-1 Inhibitor Counteracts Glioblastoma Progression by Interfering with the Hypoxic–Angiogenic Pathway


 ,
,  ,
,  , , ,
, , ,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
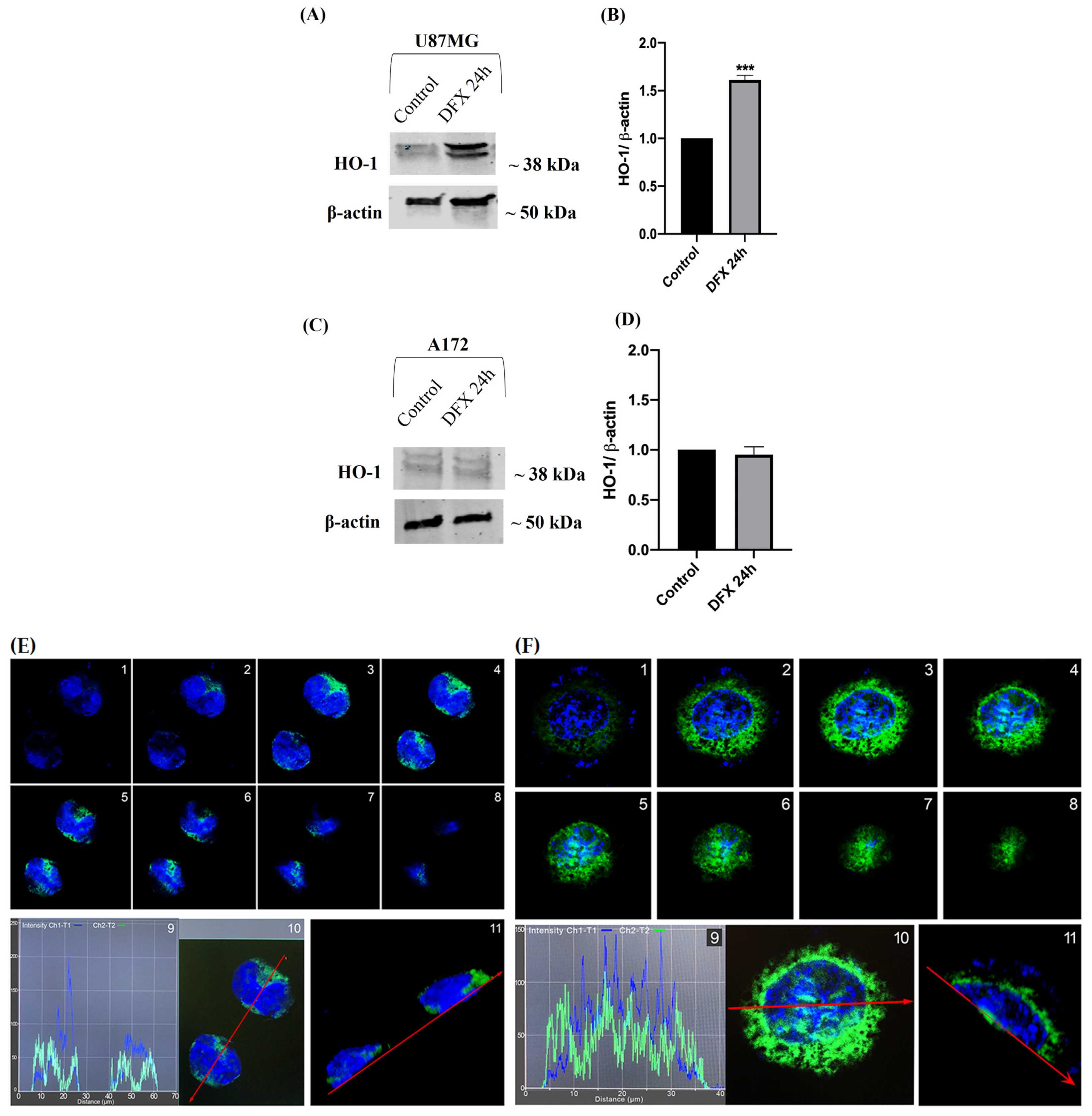
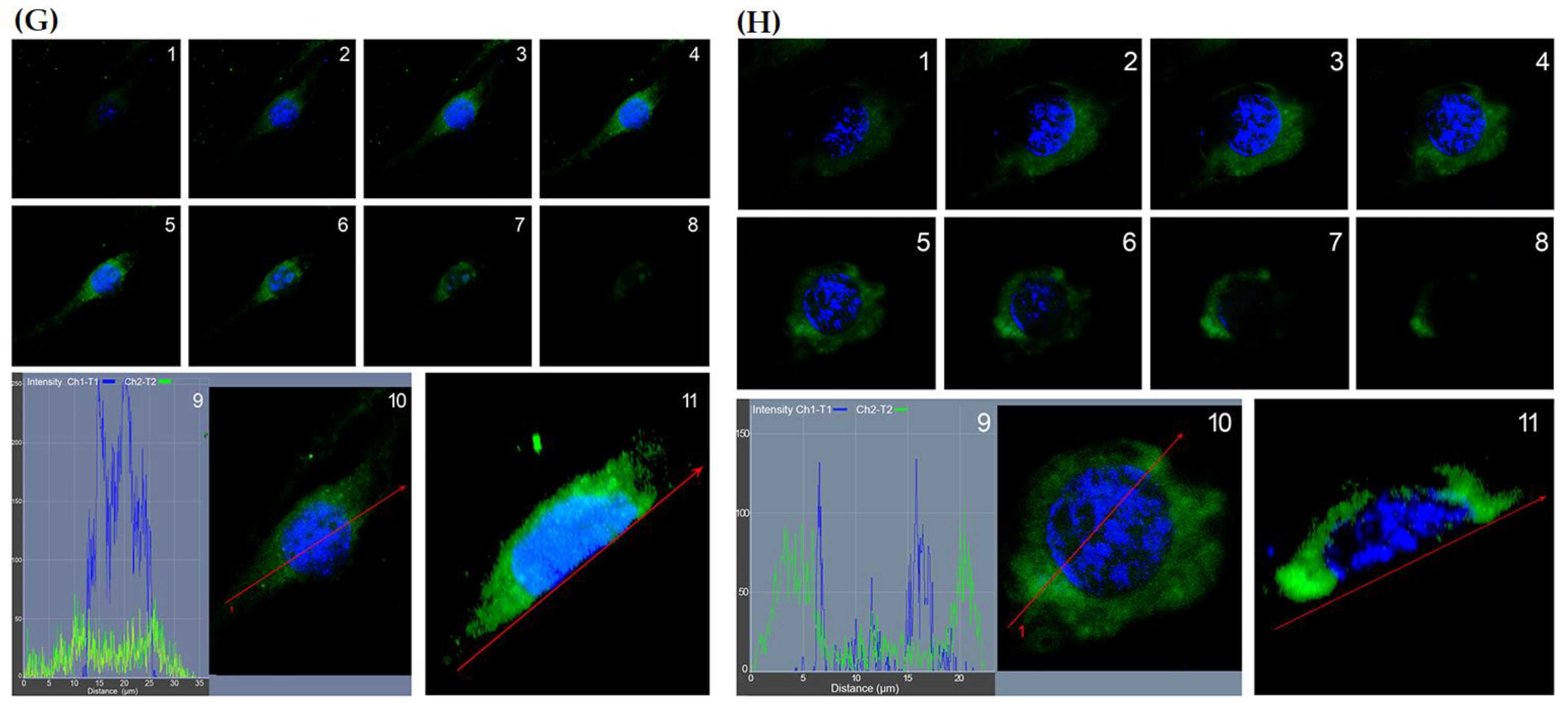
2.1. HO-1 Is Overexpressed in Human GBM Glioblastoma Cells Exposed to Hypoxia
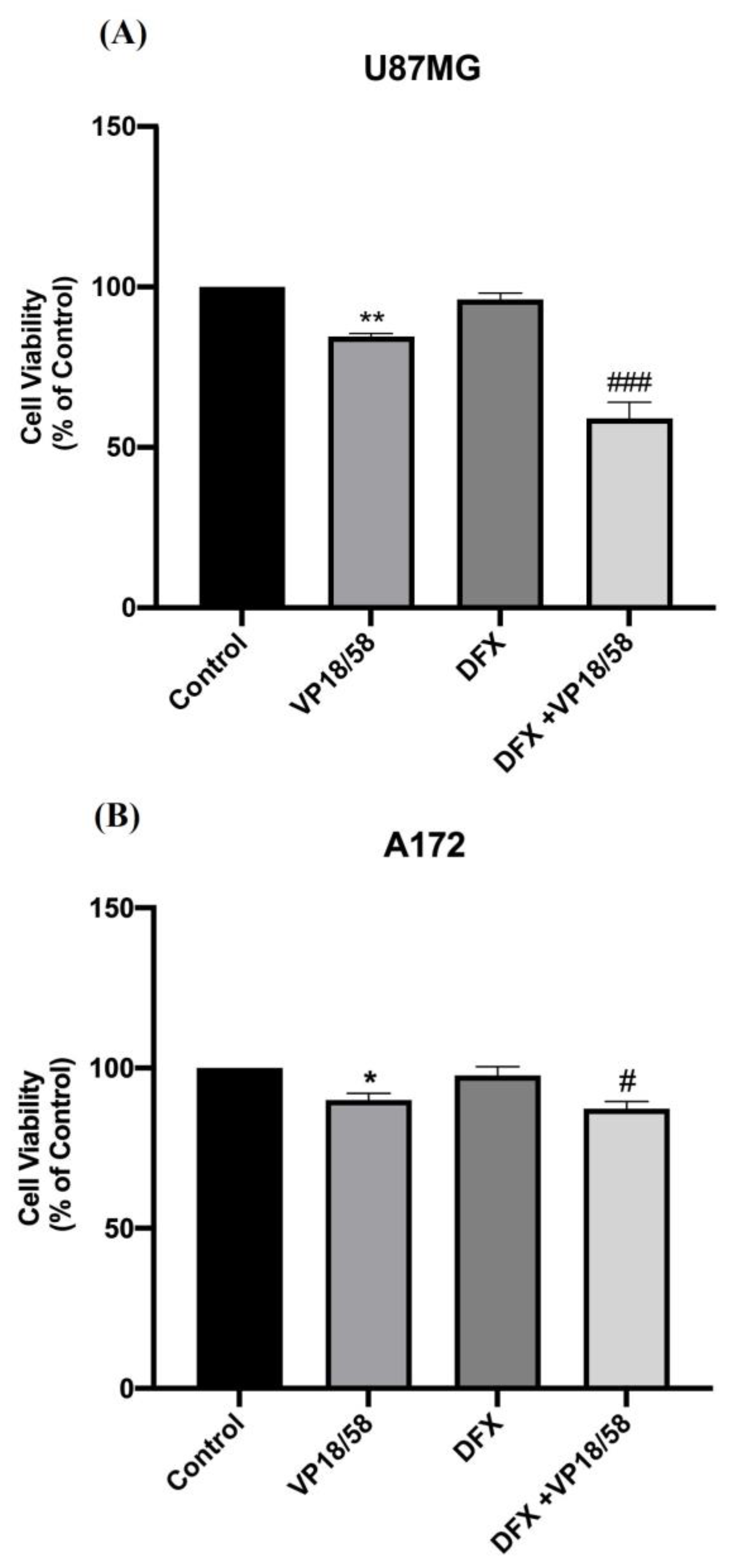
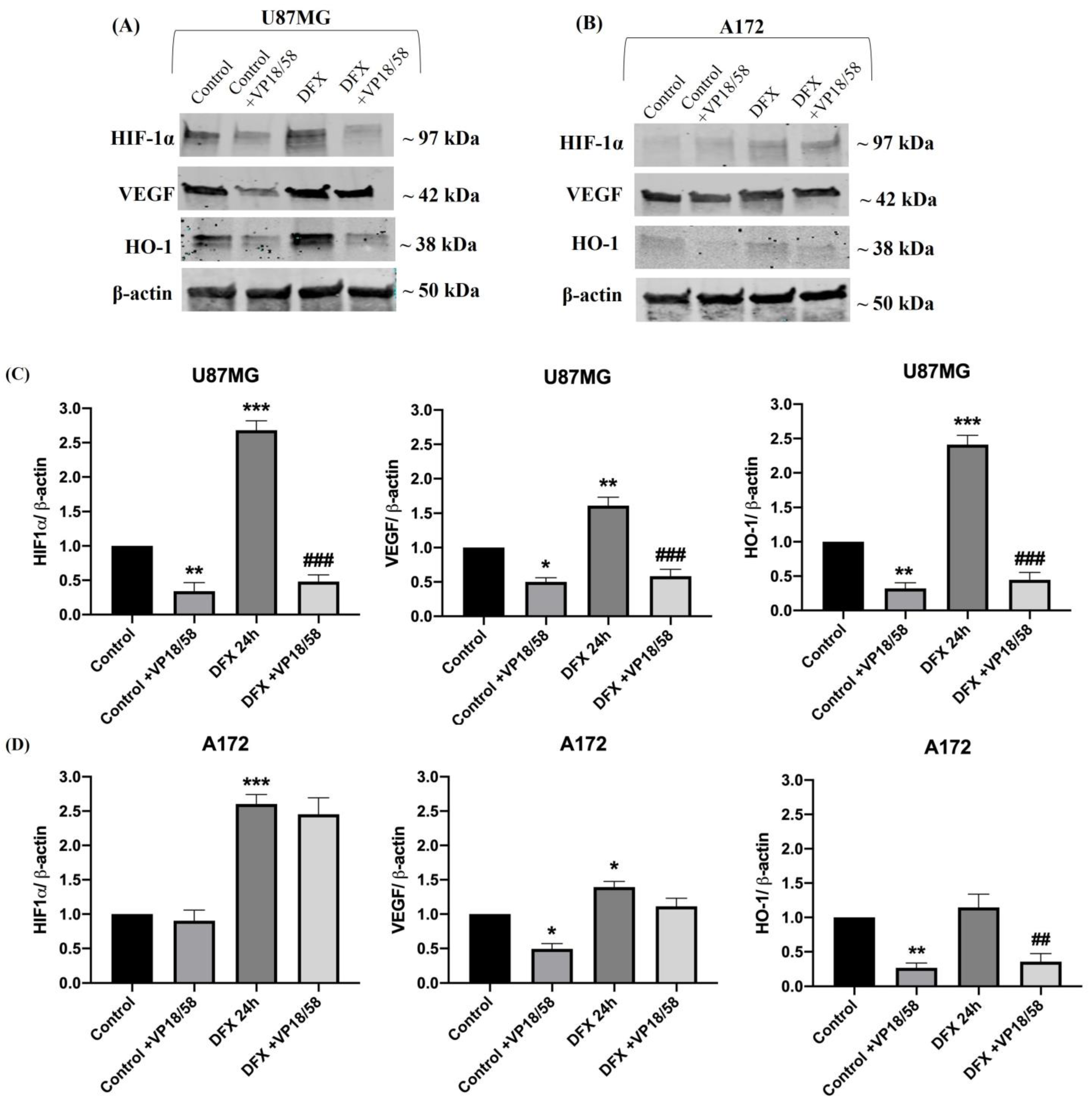
2.2. HO-1 Inhibition Affected GBM Cell Viability and Migration by Interfering with the Hypoxia-Driven Signaling Cascade
3. Discussion
4. Materials and Methods
4.1. Human GBM Cell Lines and Treatments
4.2. Cell Viability Assay
4.3. Wound-Healing Assay
4.4. Western Blot Analysis
4.5. Immunofluorescence Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- D‘Amico, A.G.; Maugeri, G.; Vanella, L.; Pittalà, V.; Reglodi, D.; D‘Agata, V. Multimodal Role of PACAP in Glioblastoma. Brain Sci. 2021, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Anjum, K.; Shagufta, B.I.; Abbas, S.Q.; Patel, S.; Khan, I.; Shah, S.A.A.; Akhter, N.; Hassan, S.S.U. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: A review. Biomed. Pharmacother. 2017, 92, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Marrinan, J.; Frishman, C.; Sampath, P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin. Dev. Immunol. 2012, 2012, 831090. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.L.; Meode, M.; Tan, M.; Maxwell, L.; Bering, E.A.; Pedersen, H.; Willms, J.; Liao, J.; Black, S.; Cairncross, J.G.; et al. PARP inhibition suppresses the emergence of temozolomide resistance in a model system. J. Neurooncol. 2020, 148, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef] [PubMed]
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-Balibrea, E.; Balañà, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.J.; Fu, X.L.; Guang, R.; To, S.T. Advances in the targeting of HIF-1α and future therapeutic strategies for glioblastoma multiforme (Review). Oncol. Rep. 2017, 37, 657–670. [Google Scholar] [CrossRef]
- Oliver, L.; Olivier, C.; Marhuenda, F.B.; Campone, M.; Vallette, F.M. Hypoxia and the malignant glioma microenvironment: Regulation and implications for therapy. Curr. Mol. Pharmacol. 2009, 2, 263–284. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D‘Amico, A.G.; Reitano, R.; Magro, G.; Cavallaro, S.; Salomone, S.; D‘Agata, V. PACAP and VIP Inhibit the Invasiveness of Glioblastoma Cells Exposed to Hypoxia through the Regulation of HIFs and EGFR Expression. Front. Pharmacol. 2016, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D‘Amico, A.G.; Rasà, D.M.; Saccone, S.; Federico, C.; Cavallaro, S.; D‘Agata, V. PACAP and VIP regulate hypoxia-inducible factors in neuroblastoma cells exposed to hypoxia. Neuropeptides 2018, 69, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ikeda, S.; Nara, M.; Kitadate, A.; Tagawa, H.; Takahashi, N. Hypoxia-induced oxidative stress promotes therapy resistance via upregulation of heme oxygenase-1 in multiple myeloma. Cancer Med. 2023, 12, 9709–9722. [Google Scholar] [CrossRef] [PubMed]
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front. Immunol. 2021, 12, 658315. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO system in tumor growth, angiogenesis and metabolism—Targeting HO-1 as an anti-tumor therapy. Vascul Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.I.; Choudhury, M.; da Silva, J.L.; Schwartzman, M.L.; Abraham, N.G. Overexpression of the heme oxygenase gene in renal cell carcinoma. Proc. Soc. Exp. Biol. Med. 1997, 214, 54–61. [Google Scholar] [CrossRef]
- Maines, M.D.; Abrahamsson, P.A. Expression of heme oxygenase-1 (HSP32) in human prostate: Normal, hyperplastic, and tumor tissue distribution. Urology 1996, 47, 727–733. [Google Scholar] [CrossRef]
- Sorrenti, V.; D‘Amico, A.G.; Barbagallo, I.; Consoli, V.; Grosso, S.; Vanella, L. Tin Mesoporphyrin Selectively Reduces Non-Small-Cell Lung Cancer Cell Line A549 Proliferation by Interfering with Heme Oxygenase and Glutathione Systems. Biomolecules 2021, 11, 917. [Google Scholar] [CrossRef] [PubMed]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.V.; Sapochnik, D.; Garcia Solá, M.; Coso, O. Regulation of the Expression of Heme Oxygenase-1: Signal Transduction, Gene Promoter Activation, and Beyond. Antioxid. Redox Signal. 2020, 32, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Lavrovsky, Y.; Schwartzman, M.L.; Levere, R.D.; Kappas, A.; Abraham, N.G. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5987–5991. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Ivaldo, C.; Traverso, N.; Furfaro, A.L. Clinical Significance of Heme Oxygenase 1 in Tumor Progression. Antioxidants 2021, 10, 789. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Lee, C.H.; Liang, J.A.; Yu, C.Y.; Shyu, W.C. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol. Rep. 2010, 24, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Alam, J.; Wiegand, G.W.; Choi, A.M. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 10393–10398. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Wang, T.; Liu, Y.; Irwin, M.G.; Ou, J.S.; Liao, X.L.; Gao, X.; Xu, Y.; Ng, K.F.; Vanhoutte, P.M.; et al. N-acetylcysteine and allopurinol confer synergy in attenuating myocardial ischemia injury via restoring HIF-1α/HO-1 signaling in diabetic rats. PLoS ONE 2013, 8, e68949. [Google Scholar] [CrossRef]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef]
- Ghosh, D.; Ulasov, I.V.; Chen, L.; Harkins, L.E.; Wallenborg, K.; Hothi, P.; Rostad, S.; Hood, L.; Cobbs, C.S. TGFβ-Responsive HMOX1 Expression Is Associated with Stemness and Invasion in Glioblastoma Multiforme. Stem Cells 2016, 34, 2276–2289. [Google Scholar] [CrossRef] [PubMed]
- Nishie, A.; Ono, M.; Shono, T.; Fukushi, J.; Otsubo, M.; Onoue, H.; Ito, Y.; Inamura, T.; Ikezaki, K.; Fukui, M.; et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin. Cancer Res. 1999, 5, 1107–1113. [Google Scholar] [PubMed]
- Deininger, M.H.; Meyermann, R.; Trautmann, K.; Duffner, F.; Grote, E.H.; Wickboldt, J.; Schluesener, H.J. Heme oxygenase (HO)-1 expressing macrophages/microglial cells accumulate during oligodendroglioma progression. Brain Res. 2000, 882, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dulak, J.; Deshane, J.; Jozkowicz, A.; Agarwal, A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: Focus on angiogenesis. Circulation 2008, 117, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Sferrazzo, G.; Di Rosa, M.; Barone, E.; Li Volti, G.; Musso, N.; Tibullo, D.; Barbagallo, I. Heme Oxygenase-1 in Central Nervous System Malignancies. J. Clin. Med. 2020, 9, 1562. [Google Scholar] [CrossRef] [PubMed]
- Castruccio Castracani, C.; Longhitano, L.; Distefano, A.; Di Rosa, M.; Pittalà, V.; Lupo, G.; Caruso, M.; Corona, D.; Tibullo, D.; Li Volti, G. Heme Oxygenase-1 and Carbon Monoxide Regulate Growth and Progression in Glioblastoma Cells. Mol. Neurobiol. 2020, 57, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Floresta, G.; Ciaffaglione, V.; Gentile, D.; Margani, F.; Turnaturi, R.; Rescifina, A.; Pittalà, V. Progress in the development of selective heme oxygenase-1 inhibitors and their potential therapeutic application. Eur. J. Med. Chem. 2019, 167, 439–453. [Google Scholar] [CrossRef]
- Barbagallo, I.; Giallongo, C.; Volti, G.L.; Distefano, A.; Camiolo, G.; Raffaele, M.; Salerno, L.; Pittalà, V.; Sorrenti, V.; Avola, R.; et al. Heme Oxygenase Inhibition Sensitizes Neuroblastoma Cells to Carfilzomib. Mol. Neurobiol. 2019, 56, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, M.; Pittalà, V.; Zingales, V.; Barbagallo, I.; Salerno, L.; Li Volti, G.; Romeo, G.; Carota, G.; Sorrenti, V.; Vanella, L. Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death. Int. J. Mol. Sci. 2019, 20, 2593. [Google Scholar] [CrossRef]
- Fang, J.; Sawa, T.; Akaike, T.; Akuta, T.; Sahoo, S.K.; Khaled, G.; Hamada, A.; Maeda, H. In vivo antitumor activity of pegylated zinc protoporphyrin: Targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003, 63, 3567–3574. [Google Scholar]
- Zhang, Y.; Zhang, X.; Zhou, B. Zinc Protoporphyrin-9 Potentiates the Anticancer Activity of Dihydroartemisinin. Antioxidants 2023, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Sasahira, T.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int. J. Cancer 2007, 120, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Sass, G.; Leukel, P.; Schmitz, V.; Raskopf, E.; Ocker, M.; Neureiter, D.; Meissnitzer, M.; Tasika, E.; Tannapfel, A.; Tiegs, G. Inhibition of heme oxygenase 1 expression by small interfering RNA decreases orthotopic tumor growth in livers of mice. Int. J. Cancer 2008, 123, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Fallica, A.N.; Sorrenti, V.; D‘Amico, A.G.; Salerno, L.; Romeo, G.; Intagliata, S.; Consoli, V.; Floresta, G.; Rescifina, A.; D‘Agata, V.; et al. Discovery of Novel Acetamide-Based Heme Oxygenase-1 Inhibitors with Potent In Vitro Antiproliferative Activity. J. Med. Chem. 2021, 64, 13373–13393. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Mechta-Grigoriou, F. Redox regulation of the hypoxia-inducible factor. Biol. Chem. 2006, 387, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zheng, A.; Chen, L.; Dang, F.; Liu, X.; Gao, J. Benzo(a)pyrene affects proliferation with reference to metabolic genes and ROS/HIF-1α/HO-1 signaling in A549 and MCF-7 cancer cells. Drug Chem. Toxicol. 2022, 45, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Belot, N.; Rorive, S.; Doyen, I.; Lefranc, F.; Bruyneel, E.; Dedecker, R.; Micik, S.; Brotchi, J.; Decaestecker, C.; Salmon, I.; et al. Molecular characterization of cell substratum attachments in human glial tumors relates to prognostic features. Glia 2001, 36, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, M.; Mori, K.; Sakaeda, Y.; Shimada, J.; Ohmori, Y. STAT1 represses hypoxia-inducible factor-1-mediated transcription. Biochem. Biophys. Res. Commun. 2009, 387, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Gil, L.; Federico, C.; Pinedo, F.; Bruno, F.; Rebolledo, A.B.; Montoya, J.J.; Olazabal, I.M.; Ferrer, I.; Saccone, S. Aging dependent effect of nuclear tau. Brain Res. 2017, 1677, 129–137. [Google Scholar] [CrossRef]
- D‘Amico, A.G.; Maugeri, G.; Magrì, B.; Giunta, S.; Saccone, S.; Federico, C.; Pricoco, E.; Broggi, G.; Caltabiano, R.; Musumeci, G.; et al. Modulatory activity of ADNP on the hypoxia-induced angiogenic process in glioblastoma. Int. J. Oncol. 2023, 62, 14. [Google Scholar] [CrossRef]
- Sadeghi, M.; Fathi, M.; Gholizadeh Navashenaq, J.; Mohammadi, H.; Yousefi, M.; Hojjat-Farsangi, M.; Namdar, A.; Movasaghpour Akbari, A.A.; Jadidi-Niaragh, F. The prognostic and therapeutic potential of HO-1 in leukemia and MDS. Cell Commun. Signal. 2023, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, M.; Sitasawad, S. miR-140-5p Attenuates Hypoxia-Induced Breast Cancer Progression by Targeting Nrf2/HO-1 Axis in a Keap1-Independent Mechanism. Cells 2021, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Tracey, N.; Creedon, H.; Kemp, A.J.; Culley, J.; Muir, M.; Klinowska, T.; Brunton, V.G. HO-1 drives autophagy as a mechanism of resistance against HER2-targeted therapies. Breast Cancer Res. Treat. 2020, 179, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C.; Guan, S.S.; Yang, H.J.; Chang, C.C.; Luo, T.Y.; Chang, J.; Ho, A.S. Blocking heme oxygenase-1 by zinc protoporphyrin reduces tumor hypoxia-mediated VEGF release and inhibits tumor angiogenesis as a potential therapeutic agent against colorectal cancer. J. Biomed. Sci. 2016, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Namba, F.; Go, H.; Murphy, J.A.; La, P.; Yang, G.; Sengupta, S.; Fernando, A.P.; Yohannes, M.; Biswas, C.; Wehrli, S.L.; et al. Expression level and subcellular localization of heme oxygenase-1 modulates its cytoprotective properties in response to lung injury: A mouse model. PLoS ONE 2014, 9, e90936. [Google Scholar] [CrossRef] [PubMed]
- Gandini, N.A.; Fermento, M.E.; Salomón, D.G.; Blasco, J.; Patel, V.; Gutkind, J.S.; Molinolo, A.A.; Facchinetti, M.M.; Curino, A.C. Nuclear localization of heme oxygenase-1 is associated with tumor progression of head and neck squamous cell carcinomas. Exp. Mol. Pathol. 2012, 93, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Lavrovsky, Y.; Schwartzman, M.L.; Abraham, N.G. Novel regulatory sites of the human heme oxygenase-1 promoter region. Biochem. Biophys. Res. Commun. 1993, 196, 336–341. [Google Scholar] [CrossRef]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Stern, A.; Chiu, D.T. G6PD: A hub for metabolic reprogramming and redox signaling in cancer. Biomed. J. 2021, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1α. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, A.; Hosea, R.; Wu, S.; Kasim, V. The Emerging Roles of the Metabolic Regulator G6PD in Human Cancers. Int. J. Mol. Sci. 2023, 24, 17238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, Z.; Ni, Y.; Bai, H.; Han, Q.; Yi, Z.; Yi, X.; Agbana, Y.L.; Kuang, Y.; Zhu, Y. NF-κB and pSTAT3 synergistically drive G6PD overexpression and facilitate sensitivity to G6PD inhibition in ccRCC. Cancer Cell Int. 2020, 20, 483. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.R.; Wang, H.M.; Liu, P.L.; Chen, Y.H.; Yang, M.C.; Chou, S.H.; Cheng, Y.J.; Yin, W.H.; Hwang, J.J.; Chong, I.W. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell Oncol. 2012, 35, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Sunamura, M.; Duda, D.G.; Ghattas, M.H.; Lozonschi, L.; Motoi, F.; Yamauchi, J.; Matsuno, S.; Shibahara, S.; Abraham, N.G. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 2003, 6, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Yang, S.F.; Tsai, C.H.; Chou, M.C.; Chou, M.Y.; Chang, Y.C. Upregulation of heme oxygenase-1 expression in areca-quid-chewing-associated oral squamous cell carcinoma. J. Formos. Med. Assoc. 2008, 107, 355–363. [Google Scholar] [CrossRef]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittalà, V. Heme Oxygenase Database (HemeOxDB) and QSAR Analysis of Isoform 1 Inhibitors. ChemMedChem 2017, 12, 1873–1881. [Google Scholar] [CrossRef]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittalà, V. Comprehensive data on a 2D-QSAR model for Heme Oxygenase isoform 1 inhibitors. Data Brief. 2017, 15, 281–299. [Google Scholar] [CrossRef]
- Kozakowska, M.; Dobrowolska-Glazar, B.; Okoń, K.; Józkowicz, A.; Dobrowolski, Z.; Dulak, J. Preliminary Analysis of the Expression of Selected Proangiogenic and Antioxidant Genes and MicroRNAs in Patients with Non-Muscle-Invasive Bladder Cancer. J. Clin. Med. 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D‘Amico, A.G.; Saccone, S.; Federico, C.; Rasà, D.M.; Caltabiano, R.; Broggi, G.; Giunta, S.; Musumeci, G.; D‘Agata, V. Effect of PACAP on Hypoxia-Induced Angiogenesis and Epithelial-Mesenchymal Transition in Glioblastoma. Biomedicines 2021, 9, 965. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D‘Amico, A.G.; Rasà, D.M.; Saccone, S.; Federico, C.; Magro, G.; Cavallaro, S.; D‘Agata, V. Caffeine Effect on HIFs/VEGF Pathway in Human Glioblastoma Cells Exposed to Hypoxia. Anticancer. Agents Med. Chem. 2018, 18, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Bach, F.H. Heme oxygenase-1: From biology to therapeutic potential. Trends Mol. Med. 2009, 15, 50–58. [Google Scholar] [CrossRef]
- Seo, G.S.; Jiang, W.Y.; Chi, J.H.; Jin, H.; Park, W.C.; Sohn, D.H.; Park, P.H.; Lee, S.H. Heme oxygenase-1 promotes tumor progression and metastasis of colorectal carcinoma cells by inhibiting antitumor immunity. Oncotarget 2015, 6, 19792–19806. [Google Scholar] [CrossRef]
- Mucha, O.; Podkalicka, P.; Czarnek, M.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Stepniewski, J.; Jozkowicz, A.; Dulak, J.; Loboda, A. Pharmacological versus genetic inhibition of heme oxygenase-1—The comparison of metalloporphyrins, shRNA and CRISPR/Cas9 system. Acta Biochim. Pol. 2018, 65, 277–286. [Google Scholar] [CrossRef]
- Bonaventura, G.; Iemmolo, R.; D‘Amico, A.G.; La Cognata, V.; Costanzo, E.; Zappia, M.; D‘Agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. PACAP and PAC1R are differentially expressed in motor cortex of amyotrophic lateral sclerosis patients and support survival of iPSC-derived motor neurons. J. Cell Physiol. 2018, 233, 3343–3351. [Google Scholar] [CrossRef]
- Maugeri, G.; Longo, A.; D‘Amico, A.G.; Rasà, D.M.; Reibaldi, M.; Russo, A.; Bonfiglio, V.; Avitabile, T.; D‘Agata, V. Trophic effect of PACAP on human corneal endothelium. Peptides 2018, 99, 20–26. [Google Scholar] [CrossRef]
- D‘Amico, A.G.; Maugeri, G.; Magro, G.; Salvatorelli, L.; Drago, F.; D‘Agata, V. Expression pattern of parkin isoforms in lung adenocarcinomas. Tumour Biol. 2015, 36, 5133–5141. [Google Scholar] [CrossRef]
- D‘Amico, A.G.; Maugeri, G.; Saccone, S.; Federico, C.; Cavallaro, S.; Reglodi, D.; D‘Agata, V. PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2943. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amico, A.G.; Maugeri, G.; Vanella, L.; Consoli, V.; Sorrenti, V.; Bruno, F.; Federico, C.; Fallica, A.N.; Pittalà, V.; D’Agata, V. Novel Acetamide-Based HO-1 Inhibitor Counteracts Glioblastoma Progression by Interfering with the Hypoxic–Angiogenic Pathway. Int. J. Mol. Sci. 2024, 25, 5389. https://doi.org/10.3390/ijms25105389
D’Amico AG, Maugeri G, Vanella L, Consoli V, Sorrenti V, Bruno F, Federico C, Fallica AN, Pittalà V, D’Agata V. Novel Acetamide-Based HO-1 Inhibitor Counteracts Glioblastoma Progression by Interfering with the Hypoxic–Angiogenic Pathway. International Journal of Molecular Sciences. 2024; 25(10):5389. https://doi.org/10.3390/ijms25105389
Chicago/Turabian StyleD’Amico, Agata Grazia, Grazia Maugeri, Luca Vanella, Valeria Consoli, Valeria Sorrenti, Francesca Bruno, Concetta Federico, Antonino Nicolò Fallica, Valeria Pittalà, and Velia D’Agata. 2024. "Novel Acetamide-Based HO-1 Inhibitor Counteracts Glioblastoma Progression by Interfering with the Hypoxic–Angiogenic Pathway" International Journal of Molecular Sciences 25, no. 10: 5389. https://doi.org/10.3390/ijms25105389
APA StyleD’Amico, A. G., Maugeri, G., Vanella, L., Consoli, V., Sorrenti, V., Bruno, F., Federico, C., Fallica, A. N., Pittalà, V., & D’Agata, V. (2024). Novel Acetamide-Based HO-1 Inhibitor Counteracts Glioblastoma Progression by Interfering with the Hypoxic–Angiogenic Pathway. International Journal of Molecular Sciences, 25(10), 5389. https://doi.org/10.3390/ijms25105389