

Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
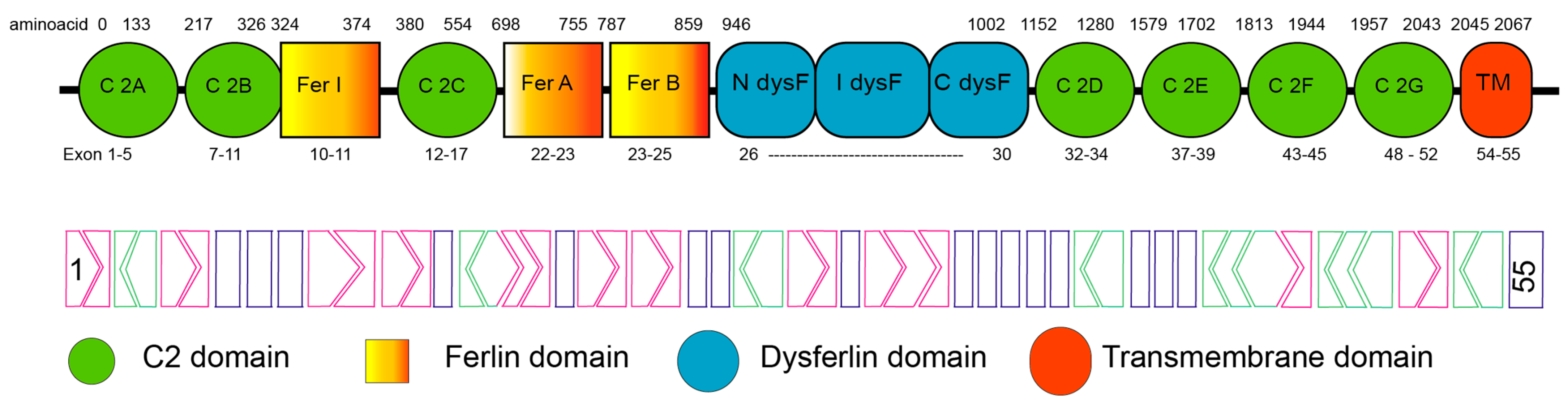
1.1. Dysferlin
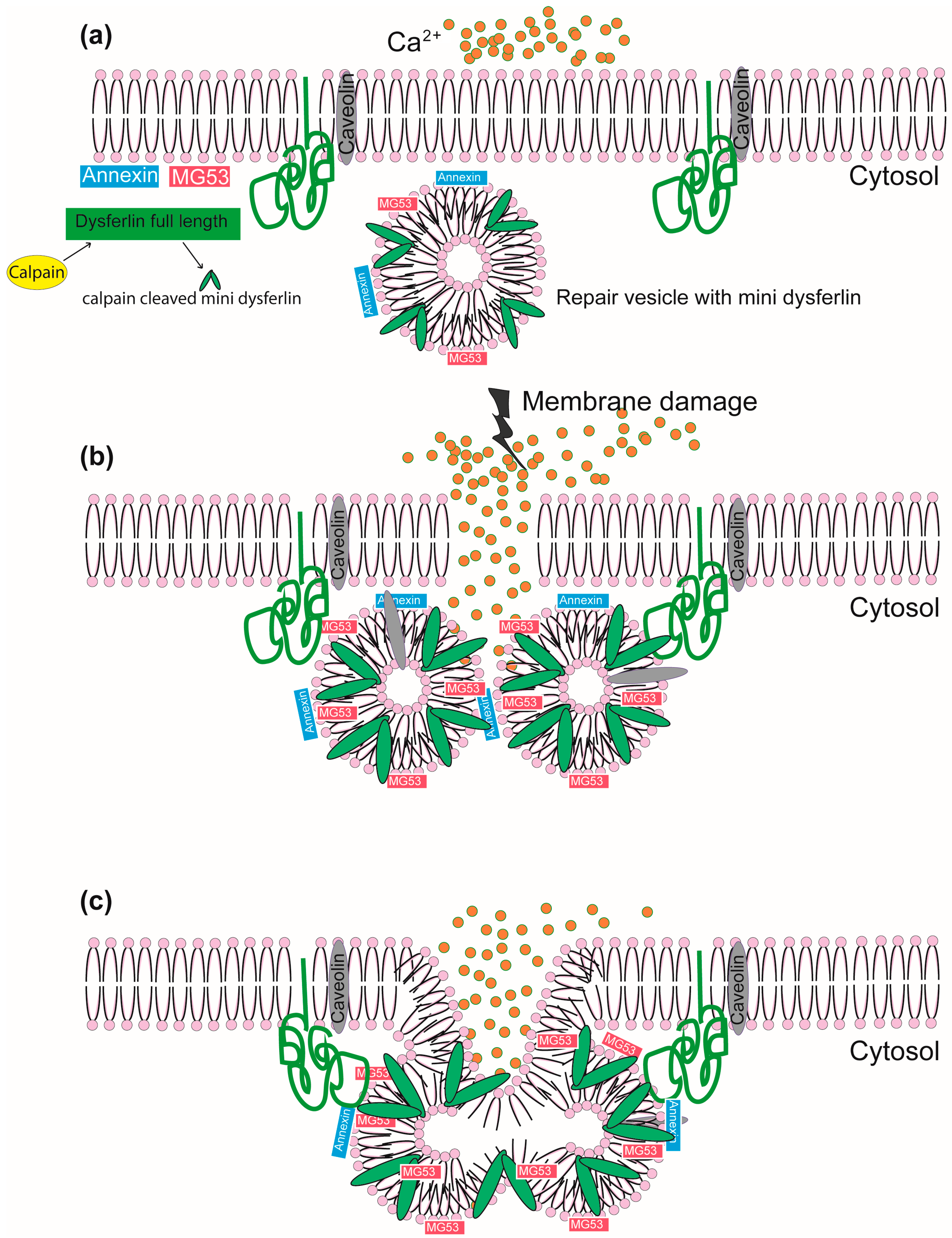
1.2. The Role of Dysferlin in Membrane Repair and the Intracellular Vesicular System
1.3. Mutations in Dysferlin and LGMD2B
1.4. Mouse Models to Study Dysferlinopathies
Stem Cells as a Model
2. LGMD2B Disease Symptoms and Diagnosis
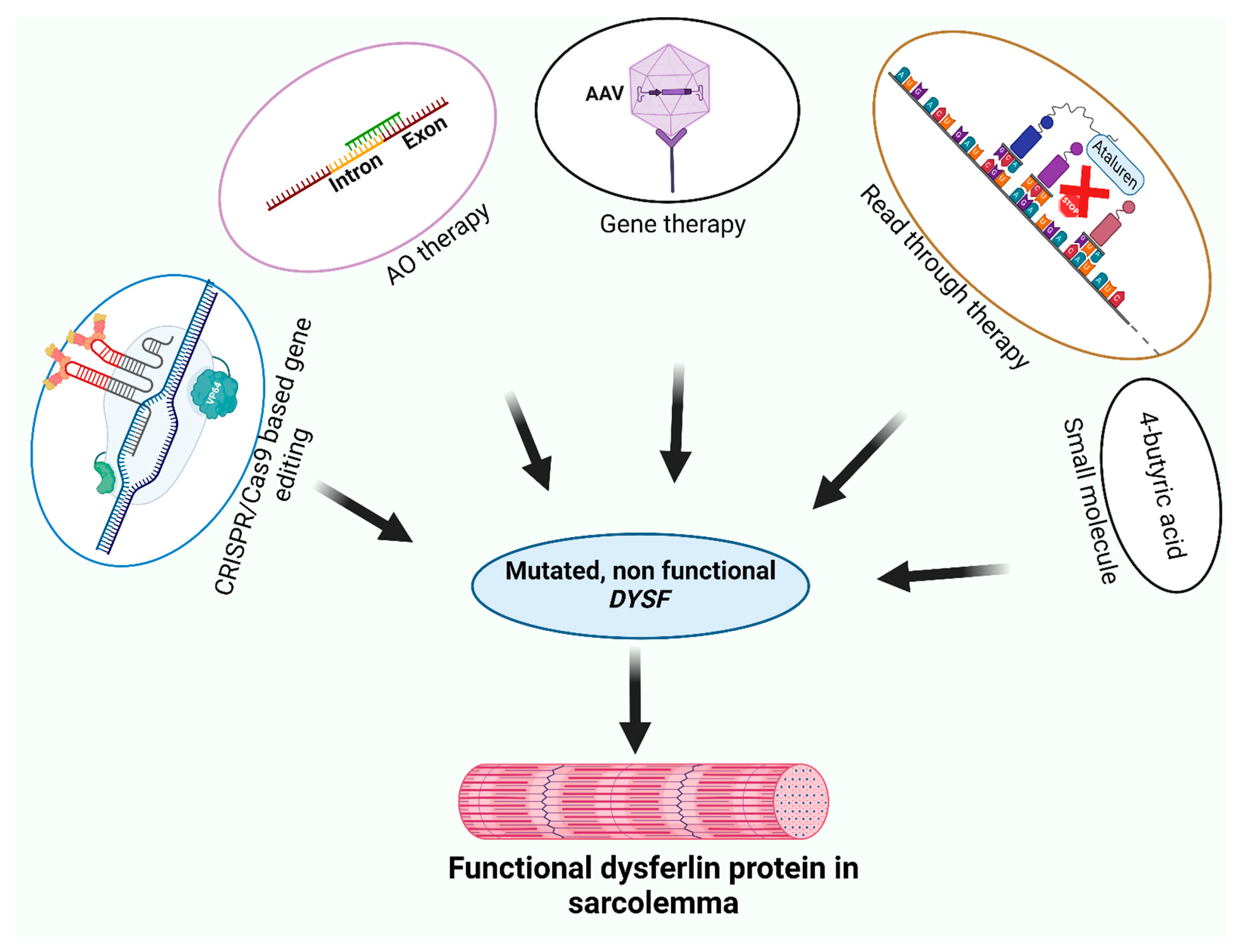
3. Therapeutic Strategies for Dysferlinopathies
3.1. Adeno-Associated Virus-Mediated Gene Therapy
3.2. CRISPR/Cas9-Mediated Precise Correction for Pathogenic DYSF Mutations
3.3. Readthrough of Nonsense Mutations to Treat Dysferlinopathies
3.4. Small Molecule Restoration of Membrane Repair Function
3.5. Antisense Oligonucleotide-Mediated Therapies
3.5.1. Antisense Oligonucleotide Mediated Strategies to Address Dysferlinopathy
3.5.2. Overcoming Limitations and Advancing Delivery Strategies of AO
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin interacts with Annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [PubMed]
- Britton, S.; Freeman, T.; Vafiadaki, E.; Keers, S.; Harrison, R.; Bushby, K.; Bashir, R. The third human FER-1-like protein is highly similar to dysferlin. Genomics 2000, 68, 313–321. [Google Scholar] [CrossRef]
- Bulankina, A.V.; Thoms, S. Functions of Vertebrate Ferlins. Cells 2020, 9, 534. [Google Scholar] [CrossRef]
- karishma Dhuri, C.B.; Quijano, E.; Pham, H.; Gupta, A.; Bikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. Clin. Med. 2020, 9, 2004. [Google Scholar]
- Aung-Htut, M.T.; Ham, K.A.; Tchan, M.; Johnsen, R.; Schnell, F.J.; Fletcher, S.; Wilton, S.D. Splice modulating antisense oligonucleotides restore some acid-alpha-glucosidase activity in cells derived from patients with late-onset Pompe disease. Sci. Rep. 2020, 10, 6702. [Google Scholar] [CrossRef]
- Pegoraro, E.; Hoffman, E.P. Limb-Girdle Muscular Dystrophy Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Blandin, G.; Beroud, C.; Labelle, V.; Nguyen, K.; Wein, N.; Hamroun, D.; Williams, B.; Monnier, N.; Rufibach, L.E.; Urtizberea, J.A.; et al. UMD-DYSF, a novel locus specific database for the compilation and interactive analysis of mutations in the dysferlin gene. Hum. Mutat. 2012, 33, E2317–E2331. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Takahashi, T. Dysferlinopathy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Salani, S.; Lucchiari, S.; Fortunato, F.; Crimi, M.; Corti, S.; Locatelli, F.; Bossolasco, P.; Bresolin, N.; Comi, G.P. Developmental and tissue-specific regulation of a novel dysferlin isoform. Muscle Nerve 2004, 30, 366–374. [Google Scholar] [CrossRef]
- Pramono, Z.A.; Lai, P.S.; Tan, C.L.; Takeda, S.; Yee, W.C. Identification and characterization of a novel human dysferlin transcript: Dysferlin_v1. Hum. Genet. 2006, 120, 410–419. [Google Scholar] [CrossRef]
- Aoki, M.; Liu, J.; Richard, I.; Bashir, R.; Britton, S.; Keers, S.M.; Oeltjen, J.; Brown, H.E.; Marchand, S.; Bourg, N.; et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology 2001, 57, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.V.B.; Davison, K.; Moss, J.A.; Young, C.; Cullen, M.J.; Walsh, J.; Johnson, M.A.; Bashir, R.; Britton, S.; Keers, S.; et al. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum. Mol. Genet. 1999, 8, 855–861. [Google Scholar] [CrossRef]
- Gallardo, E.; de Luna, N.; Diaz-Manera, J.; Rojas-Garcia, R.; Gonzalez-Quereda, L.; Flix, B.; de Morree, A.; van der Maarel, S.; Illa, I. Comparison of Dysferlin Expression in Human Skeletal Muscle with That in Monocytes for the Diagnosis of Dysferlin Myopathy. PLoS ONE 2011, 6, e0029061. [Google Scholar] [CrossRef] [PubMed]
- Harsini, F.M.; Chebrolu, S.; Fuson, K.L.; White, M.A.; Rice, A.M.; Sutton, R.B. FerA is a Membrane-Associating Four-Helix Bundle Domain in the Ferlin Family of Membrane-Fusion Proteins. Sci. Rep. 2018, 8, 10949. [Google Scholar] [CrossRef]
- Abdullah, N.; Padmanarayana, M.; Marty, N.J.; Johnson, C.P. Quantitation of the calcium and membrane binding properties of the C2 domains of dysferlin. Biophys. J. 2014, 106, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Davletov, B.A.; Sutton, R.B.; Sudhof, T.C.; Rizo, J. Bipartite Ca2+-binding motif in C2 domains of synaptotagmin and protein kinase C. Science 1996, 273, 248–251. [Google Scholar] [CrossRef]
- Marty, N.J.; Holman, C.L.; Abdullah, N.; Johnson, C.P. The C2 Domains of Otoferlin, Dysferlin, and Myoferlin Alter the Packing of Lipid Bilayers. Biochemistry 2013, 52, 5585–5592. [Google Scholar] [CrossRef]
- Sula, A.; Cole, A.R.; Yeats, C.; Orengo, C.; Keep, N.H. Crystal structures of the human Dysferlin inner DysF domain. BMC Struct. Biol. 2014, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Campbell, K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004, 14, 206–213. [Google Scholar] [CrossRef]
- Glover, L.; Brown, R.H. Dysferlin in membrane trafficking and patch repair. Traffic 2007, 8, 785–794. [Google Scholar] [CrossRef]
- Sharma, A.; Yu, C.; Leung, C.; Trane, A.; Lau, M.; Utokaparch, S.; Shaheen, F.; Sheibani, N.; Bernatchez, P. A new role for the muscle repair protein dysferlin in endothelial cell adhesion and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Lek, A.; Evesson, F.J.; Lemckert, F.A.; Redpath, G.M.; Lueders, A.K.; Turnbull, L.; Whitchurch, C.B.; North, K.N.; Cooper, S.T. Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. J. Neurosci. 2013, 33, 5085–5094. [Google Scholar] [CrossRef] [PubMed]
- Han, R. Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet. Muscle 2011, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.P.; Ziman, A.P.; Mueller, A.L.; Muriel, J.M.; Kleinhans-Welte, E.; Gumerson, J.D.; Vogel, S.S.; Ward, C.W.; Roche, J.A.; Bloch, R.J. Dysferlin stabilizes stress-induced Ca2+ signaling in the transverse tubule membrane. Proc. Natl. Acad. Sci. USA 2013, 110, 20831–20836. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.P.; Ward, C.W.; Bloch, R.J. Dysferlin at transverse tubules regulates Ca2+ homeostasis in skeletal muscle. Front. Physiol. 2014, 5, 77998. [Google Scholar] [CrossRef] [PubMed]
- Muriel, J.; Lukyanenko, V.; Kwiatkowski, T.; Bhattacharya, S.; Garman, D.; Weisleder, N.; Bloch, R.J. The C2 domains of dysferlin: Roles in membrane localization, Ca(2+) signalling and sarcolemmal repair. J. Physiol. 2022, 600, 1953–1968. [Google Scholar] [CrossRef]
- Wang, Y.; Tadayon, R.; Santamaria, L.; Mercier, P.; Forristal, C.J.; Shaw, G.S. Calcium binds and rigidifies the dysferlin C2A domain in a tightly coupled manner. Biochem. J. 2021, 478, 197–215. [Google Scholar] [CrossRef]
- Vincent, A.E.; Rosa, H.S.; Alston, C.L.; Grady, J.P.; Rygiel, K.A.; Rocha, M.C.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Dysferlin mutations and mitochondrial dysfunction. Neuromuscular Disord. 2016, 26, 782–788. [Google Scholar] [CrossRef]
- Codding, S.J.; Marty, N.; Abdullah, N.; Johnson, C.P. Dysferlin Binds SNAREs (Soluble N-Ethylmaleimide-sensitive Factor (NSF) Attachment Protein Receptors) and Stimulates Membrane Fusion in a Calcium-sensitive Manner. J. Biol. Chem. 2016, 291, 14575–14584. [Google Scholar] [CrossRef]
- Hofhuis, J.; Bersch, K.; Bussenschutt, R.; Drzymalski, M.; Liebetanz, D.; Nikolaev, V.; Wagner, S.; Maier, L.S.; Gartner, J.; Klinge, L.; et al. Dysferlin mediates membrane tubulation and links T-tubule biogenesis to muscular dystrophy. J. Cell Sci. 2017, 130, 841–852. [Google Scholar] [CrossRef]
- Hofhuis, J.; Bersch, K.; Wagner, S.; Molina, C.; Fakuade, F.E.; Iyer, L.M.; Streckfuss-Bomeke, K.; Toischer, K.; Zelarayan, L.C.; Voigt, N.; et al. Dysferlin links excitation-contraction coupling to structure and maintenance of the cardiac transverse-axial tubule system. Europace 2020, 22, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Rossi, A.E.; Alvarez, M.G.; Swanson, K.E.; Deveaux, H.K.; Earley, J.U.; Hadhazy, M.; Vohra, R.; Walter, G.A.; Pytel, P.; et al. Dysferlin and myoferlin regulate transverse tubule formation and glycerol sensitivity. Am. J. Pathol. 2014, 184, 248–259. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Allen, M.V.; Warner, J.L.; Barefield, D.Y.; Krishnan, S.; Swanson, K.E.; Earley, J.U.; McNally, E.M. Enhanced Muscular Dystrophy from Loss of Dysferlin Is Accompanied by Impaired Annexin A6 Translocation after Sarcolemmal Disruption. Am. J. Pathol. 2016, 186, 1610–1622. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef]
- Rezvanpour, A.; Shaw, G.S. Unique S100 target protein interactions. Gen. Physiol. Biophys. 2009, 28, F39–F46. [Google Scholar]
- Ampong, B.N.; Imamura, M.; Matsumiya, T.; Yoshida, M.; Takeda, S. Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol. 2005, 24, 134–144. [Google Scholar]
- Sinnreich, M.; Therrien, C.; Karpati, G. Lariat branch point mutation in the dysferlin gene with mild limb-girdle muscular dystrophy. Neurology 2006, 66, 1114–1116. [Google Scholar] [CrossRef] [PubMed]
- Cacciottolo, M.; Numitone, G.; Aurino, S.; Caserta, I.R.; Fanin, M.; Politano, L.; Minetti, C.; Ricci, E.; Piluso, G.; Angelini, C.; et al. Muscular dystrophy with marked Dysferlin deficiency is consistently caused by primary dysferlin gene mutations. Eur. J. Hum. Genet. 2011, 19, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Krahn, M. The UMD-DYSF Locus-Specific Database. [Web Page] 2011 06/26/2015. Available online: http://www.umd.be/DYSF/ (accessed on 15 September 2018).
- Izumi, R.; Takahashi, T.; Suzuki, N.; Niihori, T.; Ono, H.; Nakamura, N.; Katada, S.; Kato, M.; Warita, H.; Tateyama, M.; et al. The genetic profile of dysferlinopathy in a cohort of 209 cases: Genotype-phenotype relationship and a hotspot on the inner DysF domain. Hum. Mutat. 2020, 41, 1540–1554. [Google Scholar] [CrossRef]
- Krahn, M.; Beroud, C.; Labelle, V.; Nguyen, K.; Bernard, R.; Bassez, G.; Figarella-Branger, D.; Fernandez, C.; Bouvenot, J.; Richard, I.; et al. Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum. Mutat. 2009, 30, E345–E375. [Google Scholar] [CrossRef]
- Zhong, H.; Yu, M.; Lin, P.; Zhao, Z.; Zheng, X.; Xi, J.; Zhu, W.; Zheng, Y.; Zhang, W.; Lv, H.; et al. Molecular landscape of DYSF mutations in dysferlinopathy: From a Chinese multicenter analysis to a worldwide perspective. Hum. Mutat. 2021, 42, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.Q.; Yu, M.; Zhang, W.; Lyu, H.; Yuan, Y.; Wang, Z.X. Dysferlin Gene Mutation Spectrum in a Large Cohort of Chinese Patients with Dysferlinopathy. Chinese Med. J.-Peking. 2016, 129, 2287. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.Y.; Lian, Y.J.; Xu, H.L.; Zheng, Y.K.; Li, C.F.; Zhang, J.W.; Yan, S.P. Novel, de novo dysferlin gene mutations in a patient with Miyoshi myopathy. Neurosci. Lett. 2018, 664, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Izawa, T.; Kuwamura, M.; Yamate, J. Dysferlin and animal models for dysferlinopathy. J. Toxicol. Pathol. 2012, 25, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Rayavarapu, S.; Van der Meulen, J.H.; Gordish-Dressman, H.; Hoffman, E.P.; Nagaraju, K.; Knoblach, S.M. Characterization of dysferlin deficient SJL/J mice to assess preclinical drug efficacy: Fasudil exacerbates muscle disease phenotype. PLoS ONE 2010, 5, e12981. [Google Scholar] [CrossRef]
- Vafiadaki, E.; Reis, A.; Keers, S.; Harrison, R.; Anderson, L.V.; Raffelsberger, T.; Ivanova, S.; Hoger, H.; Bittner, R.E.; Bushby, K.; et al. Cloning of the mouse dysferlin gene and genomic characterization of the SJL-Dysf mutation. Neuroreport 2001, 12, 625–629. [Google Scholar] [CrossRef]
- The Jackson Laboratory. Mouse Strain Datasheet. Available online: https://www.jax.org (accessed on 11 August 2018).
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Malcher, J.; Heidt, L.; Goyenvalle, A.; Escobar, H.; Marg, A.; Beley, C.; Benchaouir, R.; Bader, M.; Spuler, S.; Garcia, L.; et al. Exon Skipping in a Dysf-Missense Mutant Mouse Model. Mol. Ther. Nucleic Acids 2018, 13, 198–207. [Google Scholar] [CrossRef]
- Kokubu, Y.; Nagino, T.; Sasa, K.; Oikawa, T.; Miyake, K.; Kume, A.; Fukuda, M.; Fuse, H.; Tozawa, R.; Sakurai, H. Phenotypic Drug Screening for Dysferlinopathy Using Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2019, 8, 1017–1029. [Google Scholar] [CrossRef]
- Kesari, A.; Fukuda, M.; Knoblach, S.; Bashir, R.; Nader, G.A.; Rao, D.; Nagaraju, K.; Hoffman, E.P. Dysferlin deficiency shows compensatory induction of Rab27A/Slp2a that may contribute to inflammatory onset. Am. J. Pathol. 2008, 173, 1476–1487. [Google Scholar] [CrossRef]
- Nagaraju, K.; Rawat, R.; Veszelovszky, E.; Thapliyal, R.; Kesari, A.; Sparks, S.; Raben, N.; Plotz, P.; Hoffman, E.P. Dysferlin deficiency enhances monocyte phagocytosis: A model for the inflammatory onset of limb-girdle muscular dystrophy 2B. Am. J. Pathol. 2008, 172, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Bassez, G.; Bernard, R.; Krahn, M.; Labelle, V.; Figarella-Branger, D.; Pouget, J.; Hammouda el, H.; Beroud, C.; Urtizberea, A.; et al. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum. Mutat. 2005, 26, 165. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Angelini, C. Muscle pathology in dysferlin deficiency. Neuropath Appl. Neuro 2002, 28, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.; Bladen, C.L.; Mayhew, A.; James, M.; Bettinson, K.; Moore, U.; Smith, F.E.; Rufibach, L.; Cnaan, A.; Bharucha-Goebel, D.X.; et al. The Clinical Outcome Study for dysferlinopathy: An international multicenter study. Neurol. Genet. 2016, 2, e89. [Google Scholar] [CrossRef] [PubMed]
- Dastur, R.S.; Gaitonde, P.S.; Kachwala, M.; Nallamilli, B.R.R.; Ankala, A.; Khadilkar, S.V.; Atchayaram, N.; Gayathri, N.; Meena, A.K.; Rufibach, L.; et al. Detection of Dysferlin Gene Pathogenic Variants in the Indian Population in Patients Predicted to have a Dysferlinopathy Using a Blood-based Monocyte Assay and Clinical Algorithm: A Model for Accurate and Cost-effective Diagnosis. Ann. Indian. Acad. Neur 2017, 20, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Aoki , M.; Takahashi, T. Mutational and clinical features of Japanese patients with dysferlinopathy (Miyoshi myopathy and limb girdle muscular dystrophy type 2B). Rinsho Shinkeigaku 2005, 45, 938–942. [Google Scholar] [PubMed]
- Fanin, M.; Angelini, C. Progress and challenges in diagnosis of dysferlinopathy. Muscle Nerve 2016, 54, 821–835. [Google Scholar] [CrossRef]
- Anderson, L.V.; Harrison, R.M.; Pogue, R.; Vafiadaki, E.; Pollitt, C.; Davison, K.; Moss, J.A.; Keers, S.; Pyle, A.; Shaw, P.J.; et al. Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies). Neuromuscul. Disord. 2000, 10, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Therrien, C.; Dodig, D.; Karpati, G.; Sinnreich, M. Mutation impact on dysferlin inferred from database analysis and computer-based structural predictions. J. Neurol. Sci. 2006, 250, 71–78. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. rAAVrh74.MHCK7.DYSF.DV for Treatment of Dysferlinopathies. The Proposed Clinical Trial is a Double-Blind, Randomized Controlled Study with Direct Intramuscular Injection of rAAVrh.74.MHCK7.DYSF.DV Gene Vector to the Extensor Digitorum Brevis Muscle (EDB). Two Cohorts of Subjects with Dysferlin Deficiency, Each with Proven Mutations will Undergo Gene Transfer. A Minimum of Three Subjects will be Enrolled into Each Cohort.]. 2021. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02710500 (accessed on 7 March 2024).
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Allocca, M.; Doria, M.; Petrillo, M.; Colella, P.; Garcia-Hoyos, M.; Gibbs, D.; Kim, S.R.; Maguire, A.; Rex, T.S.; Di Vicino, U.; et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J. Clin. Investig. 2008, 118, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Lostal, W.; Bartoli, M.; Bourg, N.; Roudaut, C.; Bentaïb, A.; Miyake, K.; Guerchet, N.; Fougerousse, F.; McNeil, P.; Richard, I. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum. Mol. Genet. 2010, 19, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, P.C.; Griffin, D.A.; Pozsgai, E.R.; Johnson, R.W.; Grose, W.E.; Heller, K.N.; Shontz, K.M.; Montgomery, C.L.; Liu, J.; Clark, K.R.; et al. AAV.Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models. Ann. Clin. Transl. Neurol. 2015, 2, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Potter, R.A.; Griffin, D.A.; Sondergaard, P.C.; Johnson, R.W.; Pozsgai, E.R.; Heller, K.N.; Peterson, E.L.; Lehtimaki, K.K.; Windish, H.P.; Mittal, P.J.; et al. Systemic Delivery of Dysferlin Overlap Vectors Provides Long-Term Gene Expression and Functional Improvement for Dysferlinopathy. Hum. Gene Ther. 2018, 29, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Grose, W.E.; Clark, K.R.; Griffin, D.; Malik, V.; Shontz, K.M.; Montgomery, C.L.; Lewis, S.; Brown, R.H.; Janssen, P.M.L.; Mendell, J.R.; et al. Homologous Recombination Mediates Functional Recovery of Dysferlin Deficiency following AAV5 Gene Transfer. PLoS ONE 2012, 7, e0039233. [Google Scholar] [CrossRef]
- Pryadkina, M.; Lostal, W.; Bourg, N.; Charton, K.; Roudaut, C.; Hirsch, M.L.; Richard, I. A comparison of AAV strategies distinguishes overlapping vectors for efficient systemic delivery of the 6.2 kb Dysferlin coding sequence. Mol. Ther.-Meth Clin. D 2015, 2, 15009. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. A Gene Transfer Study to Evaluate the Safety, Tolerability and Efficacy of SRP-6004 in Ambulatory Participants with Limb Girdle Muscular Dystrophy, Type 2B/R2 (LGMD2B/R2, Dysferlin [DYSF] Related). 2023. Available online: https://clinicaltrials.gov/study/NCT05906251#publications (accessed on 7 March 2024).
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C. Record number of gene-therapy trials, despite setbacks. Nat. Med. 2021, 27, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Venditti, C.P. Safety questions for AAV gene therapy. Nat. Biotechnol. 2021, 39, 24–26. [Google Scholar] [CrossRef]
- Wills, C.A.; Drago, D.; Pietrusko, R.G. Clinical holds for cell and gene therapy trials: Risks, impact, and lessons learned. Mol. Ther. Methods Clin. Dev. 2023, 31, 101125. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Horodecka, K.; Duchler, M. CRISPR/Cas9: Principle, Applications, and Delivery through Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 6072. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Turan, S.; Farruggio, A.P.; Srifa, W.; Day, J.W.; Calos, M.P. Precise Correction of Disease Mutations in Induced Pluripotent Stem Cells Derived From Patients With Limb Girdle Muscular Dystrophy. Mol. Ther. 2016, 24, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Mou, H.; Smith, J.L.; Peng, L.; Yin, H.; Moore, J.; Zhang, X.O.; Song, C.Q.; Sheel, A.; Wu, Q.; Ozata, D.M.; et al. CRISPR/Cas9-mediated genome editing induces exon skipping by alternative splicing or exon deletion. Genome Biol. 2017, 18, 108. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Li, M.; Berger, S.; Meilak, M.; Rientjes, J.; Currie, P.D. Effect of Ataluren on dystrophin mutations. J. Cell Mol. Med. 2020, 24, 6680–6689. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Sacristan-Reviriego, A.; Perdigao, P.R.L.; Sai, H.; Georgiou, M.; Kalitzeos, A.; Carr, A.F.; Coffey, P.J.; Michaelides, M.; Bainbridge, J.; et al. Investigation of PTC124-mediated translational readthrough in a retinal organoid model of AIPL1-associated Leber congenital amaurosis. Stem Cell Rep. 2022, 17, 2187–2202. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I.; Boeck, K.D.; Casimir, G.J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 1262–1272. [Google Scholar] [CrossRef]
- Samanta, A.; Stingl, K.; Kohl, S.; Ries, J.; Linnert, J.; Nagel-Wolfrum, K. Ataluren for the Treatment of Usher Syndrome 2A Caused by Nonsense Mutations. Int. J. Mol. Sci. 2019, 20, 6274. [Google Scholar] [CrossRef]
- Huang, S.; Bhattacharya, A.; Ghelfi, M.D.; Li, H.; Fritsch, C.; Chenoweth, D.M.; Goldman, Y.E.; Cooperman, B.S. Ataluren binds to multiple protein synthesis apparatus sites and competitively inhibits release factor-dependent termination. Nat. Commun. 2022, 13, 2413. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Kim, E.K.; Choi, J.; Kim, D.S.; Shin, J.H. Functional recovery of a novel knockin mouse model of dysferlinopathy by readthrough of nonsense mutation. Mol. Ther. Methods Clin. Dev. 2021, 21, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Tominaga, N.; Williams, E.O.; Rufibach, L.; Schowel, V.; Spuler, S.; Viswanathan, M.; Guarente, L.P. 4-Phenylbutyrate restores localization and membrane repair to human dysferlin mutations. iScience 2022, 25, 103667. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.; Mastaglia, F.L.; Fletcher, S.; Wilton, S.D. Precision Medicine through Antisense Oligonucleotide-Mediated Exon Skipping. Trends Pharmacol. Sci. 2018, 39, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Flynn, L.L.; Mitrpant, C.; Adams, A.; Pitout, I.L.; Stirnweiss, A.; Fletcher, S.; Wilton, S.D. Targeted SMN Exon Skipping: A Useful Control to Assess In Vitro and In Vivo Splice-Switching Studies. Biomedicines 2021, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Zaw, K.; Greer, K.; Aung-Htut, M.T.; Mitrpant, C.; Veedu, R.N.; Fletcher, S.; Wilton, S.D. Consequences of Making the Inactive Active Through Changes in Antisense Oligonucleotide Chemistries. Front. Genet. 2019, 10, 1249. [Google Scholar] [CrossRef]
- Hall, J. Future directions for medicinal chemistry in the field of oligonucleotide therapeutics. RNA 2023, 29, 423–433. [Google Scholar] [CrossRef]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Wilton-Clark, H.; Yokota, T. Casimersen for Duchenne muscular dystrophy. Drugs Today Barc 2021, 57, 707–717. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid Ther 2017, 27, 67–69. [Google Scholar] [CrossRef]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 1756285618754459. [Google Scholar] [CrossRef]
- Pfaff, A.L.; Singleton, L.M.; Koks, S. Mechanisms of disease-associated SINE-VNTR-Alus. Exp. Biol. Med. Maywood 2022, 247, 756–764. [Google Scholar] [CrossRef]
- Barthelemy, F.; Blouin, C.; Wein, N.; Mouly, V.; Courrier, S.; Dionnet, E.; Kergourlay, V.; Mathieu, Y.; Garcia, L.; Butler-Browne, G.; et al. Exon 32 Skipping of Dysferlin Rescues Membrane Repair in Patients’ Cells. J. Neuromuscul. Dis. 2015, 2, 281–290. [Google Scholar] [CrossRef]
- Dominov, J.A.; Uyan, O.; Sapp, P.C.; McKenna-Yasek, D.; Nallamilli, B.R.; Hegde, M.; Brown, R.H., Jr. A novel dysferlin mutant pseudoexon bypassed with antisense oligonucleotides. Ann. Clin. Transl. Neurol. 2014, 1, 703–720. [Google Scholar] [CrossRef]
- Dominov, J.A.; Uyan, O.; McKenna-Yasek, D.; Nallamilli, B.R.R.; Kergourlay, V.; Bartoli, M.; Levy, N.; Hudson, J.; Evangelista, T.; Lochmuller, H.; et al. Correction of pseudoexon splicing caused by a novel intronic dysferlin mutation. Ann. Clin. Transl. Neur 2019, 6, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.; Aartsma-Rus, A. Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Mir, F.; Yokota, T. Enhancing the Effectiveness of Oligonucleotide Therapeutics Using Cell-Penetrating Peptide Conjugation, Chemical Modification, and Carrier-Based Delivery Strategies. Pharmaceutics 2023, 15, 1130. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S. The Evolution of Antisense Oligonucleotide Chemistry-A Personal Journey. Biomedicines 2021, 9, 503. [Google Scholar] [CrossRef]
- Iribe, H.; Miyamoto, K.; Takahashi, T.; Kobayashi, Y.; Leo, J.; Aida, M.; Ui-Tei, K. Chemical Modification of the siRNA Seed Region Suppresses Off-Target Effects by Steric Hindrance to Base-Pairing with Targets. ACS Omega 2017, 2, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Terada, C.; Oh, K.; Tsubaki, R.; Chan, B.; Aibara, N.; Ohyama, K.; Shibata, M.A.; Wada, T.; Harada-Shiba, M.; Yamayoshi, A.; et al. Dynamic and static control of the off-target interactions of antisense oligonucleotides using toehold chemistry. Nat. Commun. 2023, 14, 7972. [Google Scholar] [CrossRef]
- Maksudov, F.; Kliuchnikov, E.; Pierson, D.; Ujwal, M.L.; Marx, K.A.; Chanda, A.; Barsegov, V. Therapeutic phosphorodiamidate morpholino oligonucleotides: Physical properties, solution structures, and folding thermodynamics. Mol. Ther. Nucleic Acids 2023, 31, 631–647. [Google Scholar] [CrossRef]
- Langner, H.K.; Jastrzebska, K.; Caruthers, M.H. Synthesis and Characterization of Thiophosphoramidate Morpholino Oligonucleotides and Chimeras. J. Am. Chem. Soc. 2020, 142, 16240–16253. [Google Scholar] [CrossRef]
- Le, B.T.; Paul, S.; Jastrzebska, K.; Langer, H.; Caruthers, M.H.; Veedu, R.N. Thiomorpholino oligonucleotides as a robust class of next generation platforms for alternate mRNA splicing. Proc. Natl. Acad. Sci. USA 2022, 119, e2207956119. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Caruthers, M.H. Synthesis of Backbone-Modified Morpholino Oligonucleotides Using Phosphoramidite Chemistry. Molecules 2023, 28, 5380. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.; Weldemichael, T.; Liu, Z.; Li, H.Y. Delivery of Oligonucleotides: Efficiency with Lipid Conjugation and Clinical Outcome. Pharmaceutics 2022, 14, 342. [Google Scholar] [CrossRef] [PubMed]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.R.; Chiou, S.H.; Huang, Y.W.; Lee, H.J. Bio-Membrane Internalization Mechanisms of Arginine-Rich Cell-Penetrating Peptides in Various Species. Membranes 2022, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Ait Benichou, S.; Jauvin, D.; De Serres-Berard, T.; Bennett, F.; Rigo, F.; Gourdon, G.; Boutjdir, M.; Chahine, M.; Puymirat, J. Enhanced Delivery of Ligand-Conjugated Antisense Oligonucleotides (C16-HA-ASO) Targeting Dystrophia Myotonica Protein Kinase Transcripts for the Treatment of Myotonic Dystrophy Type 1. Hum. Gene Ther. 2022, 33, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Jearawiriyapaisarn, N.; Moulton, H.M.; Buckley, B.; Roberts, J.; Sazani, P.; Fucharoen, S.; Iversen, P.L.; Kole, R. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 2008, 16, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, O.; Yokota, T. Pharmacology and toxicology of eteplirsen and SRP-5051 for DMD exon 51 skipping: An update. Arch. Toxicol. 2021, 96, 1–9. [Google Scholar] [CrossRef]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef]
- Hald Albertsen, C.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef] [PubMed]
- Calero, M.; Moleiro, L.H.; Sayd, A.; Dorca, Y.; Miquel-Rio, L.; Paz, V.; Robledo-Montana, J.; Enciso, E.; Accion, F.; Herraez-Aguilar, D.; et al. Lipid nanoparticles for antisense oligonucleotide gene interference into brain border-associated macrophages. Front. Mol. Biosci. 2022, 9, 887678. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, W.; Lin, L.; Chen, A.; Feng, J.; Qu, X.; Zhang, H.; Yue, J. Delivery of therapeutic oligonucleotides in nanoscale. Bioact. Mater. 2022, 7, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood-Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem. Int. Ed. Engl. 2020, 59, 8173–8180. [Google Scholar] [CrossRef]
- Chu, M.L.; Moran, E. The Limb-Girdle Muscular Dystrophies: Is Treatment on the Horizon? Neurotherapeutics 2018, 15, 849–862. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poudel, B.H.; Fletcher, S.; Wilton, S.D.; Aung-Htut, M. Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities. Int. J. Mol. Sci. 2024, 25, 5572. https://doi.org/10.3390/ijms25115572
Poudel BH, Fletcher S, Wilton SD, Aung-Htut M. Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities. International Journal of Molecular Sciences. 2024; 25(11):5572. https://doi.org/10.3390/ijms25115572
Chicago/Turabian StylePoudel, Bal Hari, Sue Fletcher, Steve D. Wilton, and May Aung-Htut. 2024. "Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities" International Journal of Molecular Sciences 25, no. 11: 5572. https://doi.org/10.3390/ijms25115572
APA StylePoudel, B. H., Fletcher, S., Wilton, S. D., & Aung-Htut, M. (2024). Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities. International Journal of Molecular Sciences, 25(11), 5572. https://doi.org/10.3390/ijms25115572