
Strong Bases and beyond: The Prominent Contribution of Neutral Push–Pull Organic Molecules towards Superbases in the Gas Phase


Abstract
:1. Introduction
2. When Do Exceptionally Strong Organic Bases Become Superbases?
3. Thermodynamic Acid–Base Parameters in the Gas Phase
4. Experimental Methods of PA/GB Determinations: Superbase History and Recent Advances
5. Procedures for PA/GB Theoretical Estimations
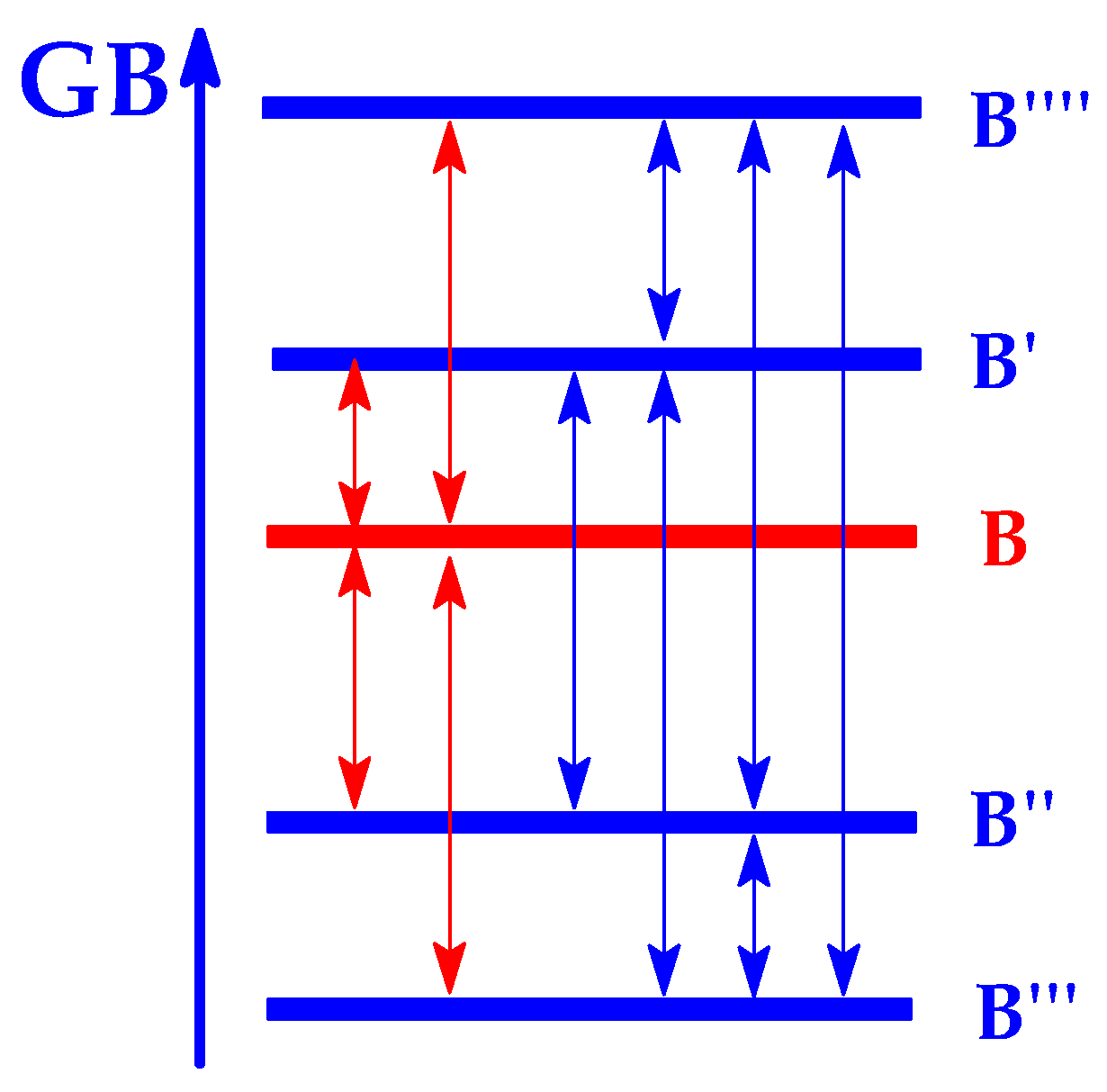
6. Theoretical Treatment of Isomeric Bases
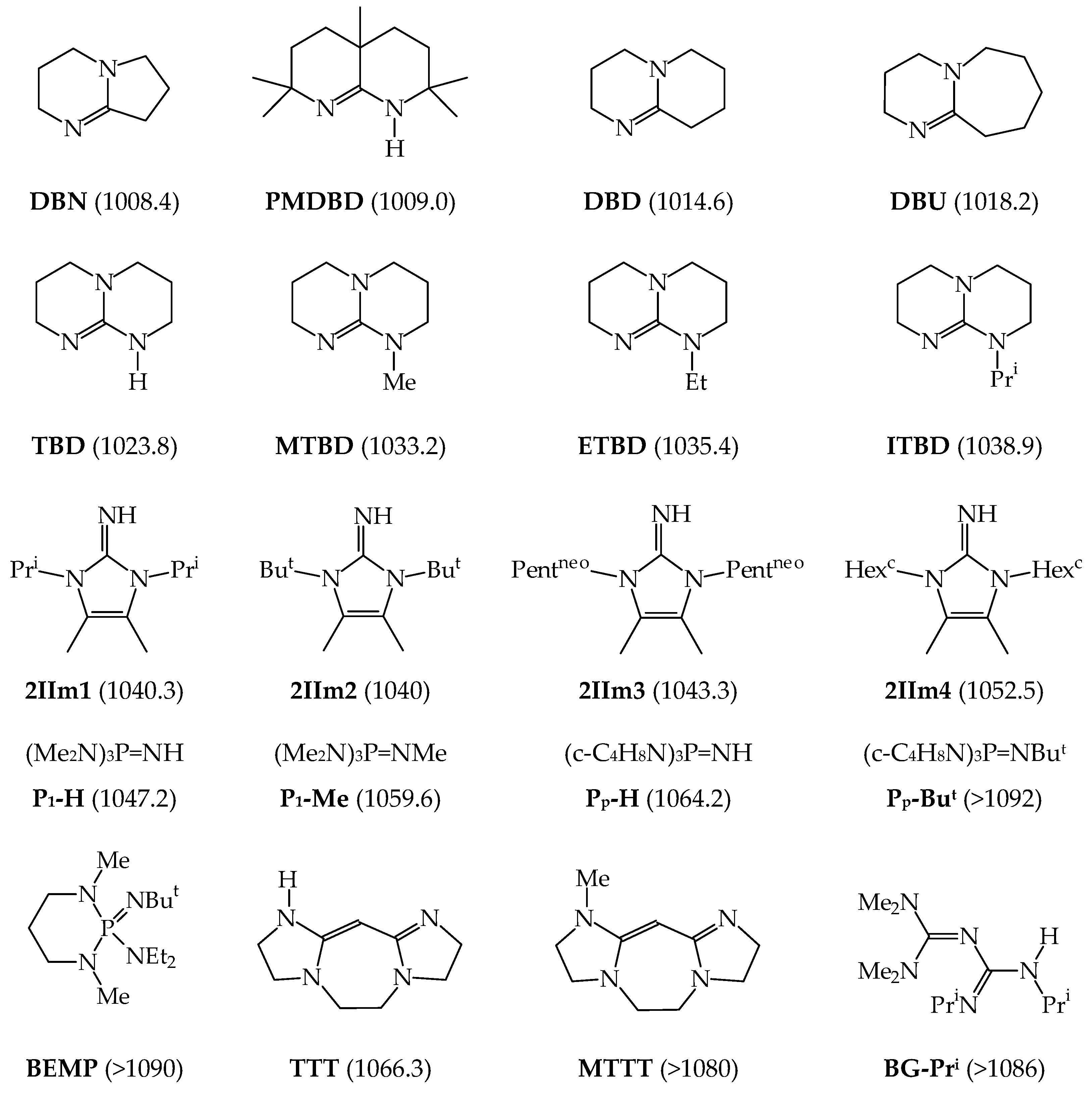
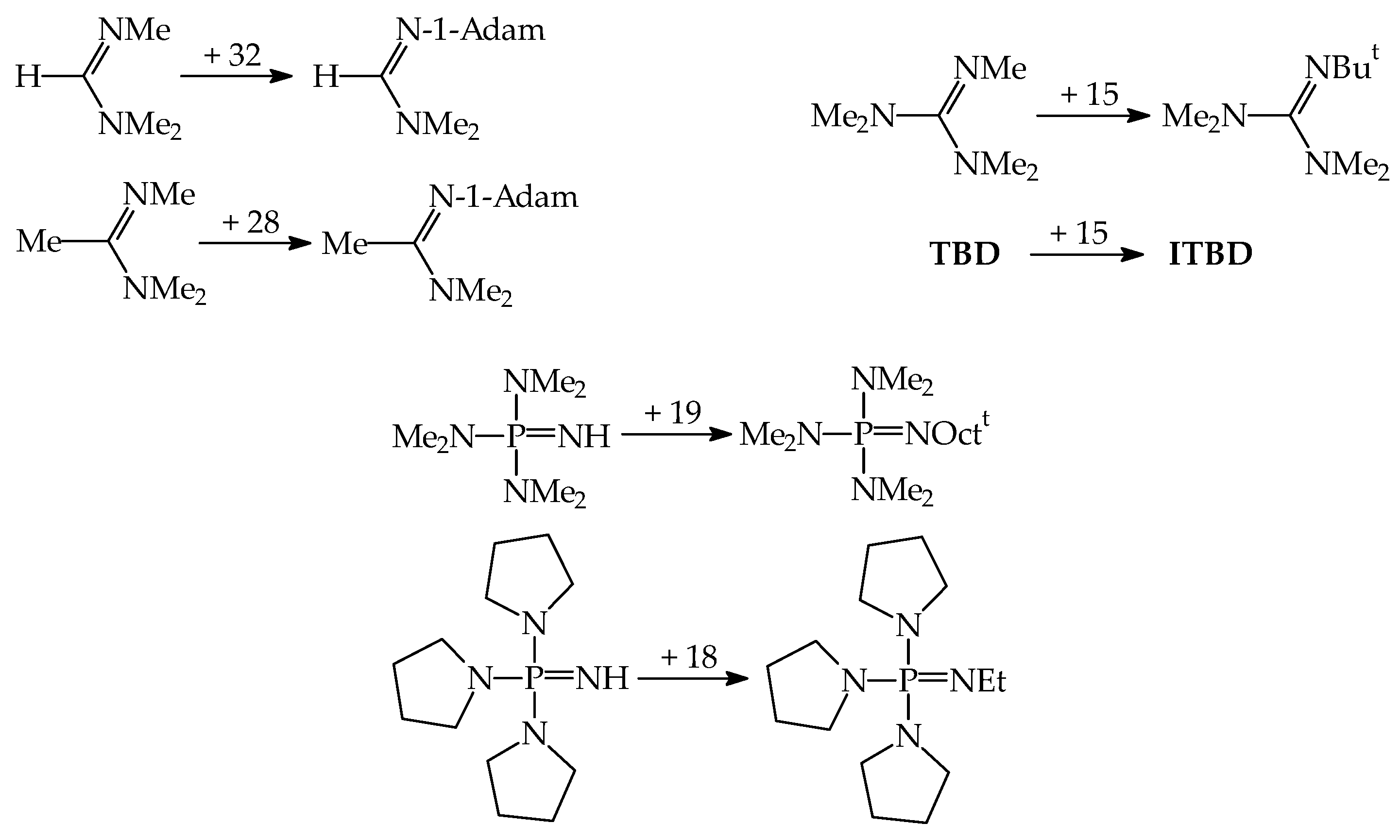
7. Series of Brønsted Organic Nitrogen Superbases with Known Experimental GBs
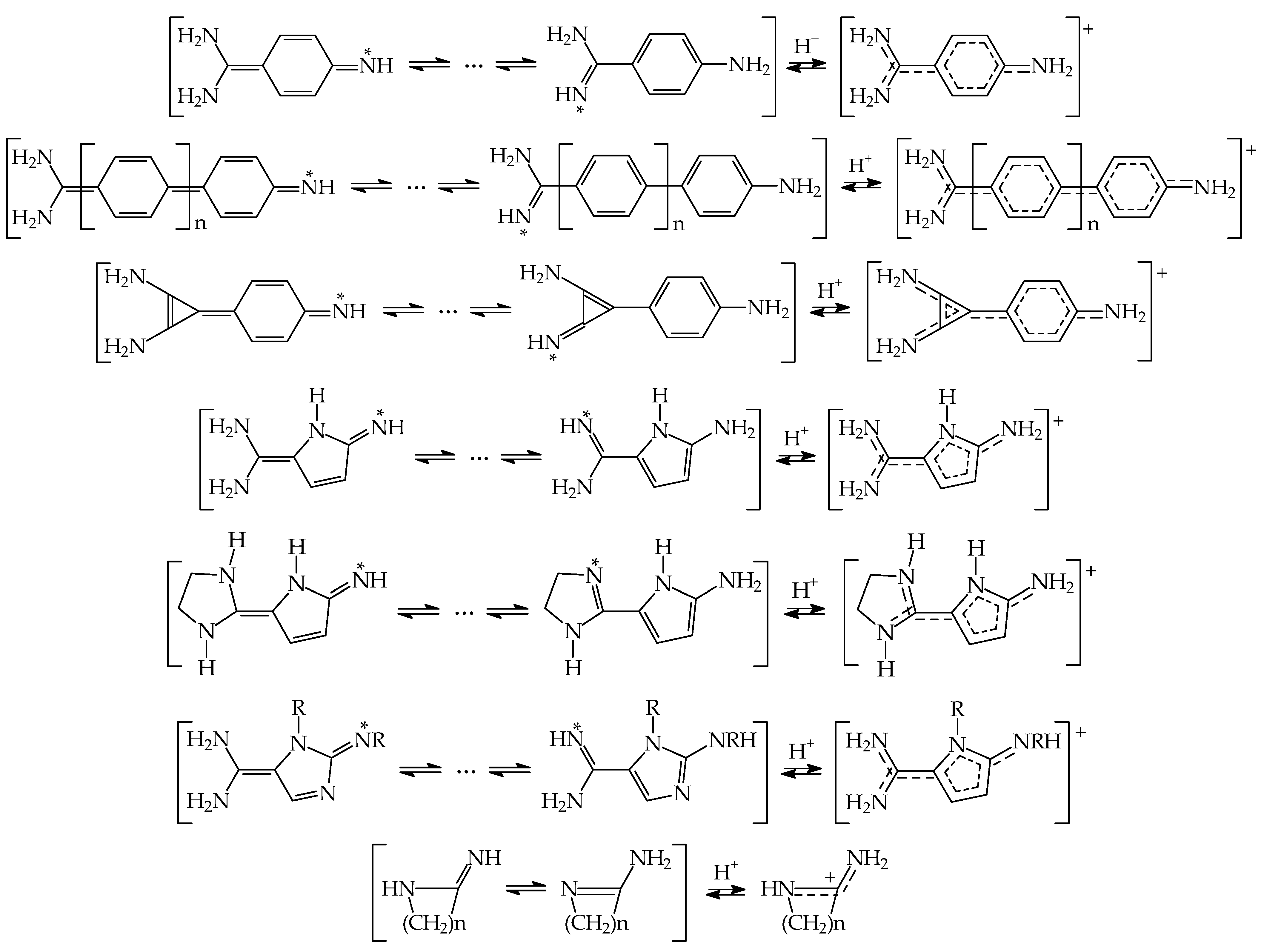
8. Substituent Effects and Intramolecular Interactions on Superbasicity of N Bases
9. Selected Methods for Organic Nitrogen Superbases Preparation





10. Overview and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A | acceptor |
| A− | conjugate base of AH |
| 1-Adam | 1-adamantyl group |
| AEP | 2-(β-aminoethyl)pyridine |
| AH | neutral acid |
| Arg | arginine |
| Agm | agmatine (N-(4-aminobutyl)guanidine) |
| B | neutral base |
| Bun | n-butyl group |
| But | tert-butyl group |
| BEMP | 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosporine |
| BH+ | conjugate acid of B |
| BG | biguanide (guanidinoamidine) |
| Cad | cadaverine (1,5-diaminopentan) |
| CCSD(T) | coupled cluster method including single-, double- and triple-excitation |
| D | donor |
| DBD | 1,5-diazabicyclo[4.4.0]dec-5-ene |
| DBN | 1,5-diazabicyclo[4.3.0]non-5-ene |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DE | deprotonation enthalpy |
| DFT | density functional theory |
| DMAN | 1,8-bis(dimethylamino)naphthalene |
| DMAP | N,N-dimethyl-4-aminopyridine |
| E | energy |
| Eprot | protonation energy |
| Et | ethyl group |
| ETBD | 7-ethyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene |
| FDMEP | N1,N1-dimethyl-N2-β-(2-pyridylethyl)formamidine |
| FT-ICR | Fourier transform mode of ICR |
| G | Gibbs energy |
| G2 | Gaussian-2 theory |
| G2MP2 | MP2 variant of Gaussian-2 theory |
| G3MP2B3 | Gaussian-3 theory with B3LYP structures and frequencies, and MP2 energy correction |
| G4MP2 | MP2 variant of Gaussian-4 theory |
| Gn | Gaussian-n theory |
| GA | gas-phase acidity |
| GB | gas-phase basicity |
| H | enthalpy |
| fH | enthalpy of formation |
| HA | histamine |
| HF | Hartree–Fock self-consistent field |
| His | histidine |
| HMPN | 1,8-bis(hexamethylaminophosphazeno)naphthalene |
| HPMS | high pressure mass spectrometry |
| I | intensity |
| I | imeglimin |
| ICR | ion cyclotron resonance |
| 2IIm1 | 1,3-di(iso-propyl)-4,5-dimethyl-2-iminoimidazoline |
| 2IIm2 | 1,3-di(tert-butyl)- 4,5-dimethyl-2-iminoimidazoline |
| 2IIm3 | 1,3-di(neo-pentyl)- 4,5-dimethyl-2-iminoimidazoline |
| 2IIm4 | 1,3-dicyclohexyl-4,5-dimethyl-2-iminoimidazoline |
| ITBD | 7-iso-propyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene |
| IUPAC | International Union of Pure and Applied Chemistry |
| K | equilibrium constant |
| Lys | lysine |
| M | metformin |
| Me | methyl group |
| MP2 | second-order Møller–Plesset (perturbation theory) |
| MPn | n-order Møller–Plesset (perturbation theory) |
| MTBD | 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-7,9-ene |
| MTTT | 11-methyl-1,4,7,11-tetraazatricyclo[8.3.0.04.8]tridec-7,9-diene |
| MW | microwave |
| NIST | National Institute of Standards and Technology |
| Octt | tert-octyl group |
| p | pressure |
| P1-H | tris(dimethylamino)iminophosphorane |
| P1-Me | tris(dimethylamino)(methylimino)phosphorane |
| Pp-H | tris(pyrrolidin-1-yl)iminophosphorane |
| Pp-But | tris(pyrrolidin-1-yl)(tert-butylimino)phosphorane |
| PA | proton affinity |
| Ph | phenyl group |
| pKa | negative logarithm of acidity dissociation constant |
| PMDBD | 3,3,6,9,9-pentamethyl-2,10-diazabicyclo[4.4.0]dec-1-ene |
| Prn | n-propyl group |
| Pii | iso-propyl group |
| QCISD(T) | quadratic configuration interaction method including single-, double- and triple-excitation |
| R | molar gas constant |
| S | entropy |
| σα | polarizability substituent constant |
| σF | field/inductive substituent constant |
| σR | resonance substituent constant |
| T | temperature |
| T | transmitter |
| TBD | 1,5,7-triazabicyclo[4.4.0]dec-7,9-ene |
| TBPN | 1,8-bis(tri-n-butylphosphoimino)naphthalene |
| TcyPPN | 1,8-bis(tri-cyclopentylphosphoimino)naphthalene |
| TiPrPN | 1,8-bis(tri-iso-propylmethylphosphoimino)naphthalene |
| TMG | N1,N1,N3,N3-tetramethylguanidine |
| TMGN | 1,8-bis(tetramethylguanidino)naphthalene |
| TMPN | 1,8-bis(trimethylphosphoimino)naphthalene |
| TTT | 1,4,7,11-tetraazatricyclo[8.3.0.04.8]tridec-7,9-diene |
| V | volume |
| W1 | Weizmann-1 theory |
| W2 | Weizmann-2 theory |
| x | molar fraction of B isomer |
| y | molar fraction of BH+ isomer |
| ZPE | zero-point energy |
References
- Trofimov, B.A.; Schmidt, E.Y. Superbases in Organic Synthesis. Chem. Probl. 2022, 4, 325–339. [Google Scholar] [CrossRef]
- Brown, C.A. The Remarkable Fast Reaction of Potassium Hydride with Amines and Other Feeble Organic Acids. A Convenient Rapid Route to Elusive New Superbases. J. Am. Chem. Soc. 1973, 95, 982–983. [Google Scholar] [CrossRef]
- Caubère, P. Unimetal Super Bases. Chem. Rev. 1993, 93, 2317–2334. [Google Scholar] [CrossRef]
- Armentrout, P.; Hodges, R.; Beauchamp, J.L. Metal Atoms as Superbases: The Gas Phase Proton Affinity of Uranium. J. Am. Chem. Soc. 1977, 99, 3162–3163. [Google Scholar] [CrossRef]
- Taft, R.W.; Gal, J.-F.; Geribaldi, S.; Maria, P.-C. Unique Basicity Properties of Conjugated Amino Cyclohexenone Derivatives. The Effects of Molecular Structure on the Disparate Basicities Toward Protonic Acids. J. Am. Chem. Soc. 1986, 108, 861–863. [Google Scholar] [CrossRef]
- Perrin, C.L.; Agranat, I.; Bagno, A.; Braslavsky, S.E.; Fernandes, P.A.; Gal, J.-F.; Lloyd-Jones, G.C.; Mayr, H.; Murdoch, J.R.; Nudelman, N.S.; et al. Glossary of Terms Used in Physical Organic Chemistry (IUPAC Recommendations 2021). Pure Appl. Chem. 2022, 94, 353–534. [Google Scholar] [CrossRef]
- Ishikawa, T. Superbases for Organic Synthesis: Guanidines, Amidines and Phosphazenes and Related Organocatalysts; Wiley: Chichester, UK, 2009. [Google Scholar]
- Weitkamp, R.F.; Neumann, B.; Stammler, H.-G.; Hoge, B. Phosphorus-Containing Superbases: Recent Progress in the Chemistry of Electron-Abundant Phosphines and Phosphazenes. Chem. Eur. J. 2021, 27, 10807–10825. [Google Scholar] [CrossRef] [PubMed]
- Hunter, E.P.L.; Lias, S.G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]
- Linstrom, P.J.; Mallard, W.G. (Eds.) NIST Chemistry WebBook, NIST Standard Reference Database No. 69; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2014. Available online: http://webbook.nist.gov/chemistry (accessed on 15 March 2024).
- Jena, P.; Sun, Q. (Eds.) Superatoms: Principles, Synthesis and Applications; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2022. [Google Scholar] [CrossRef]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2022. [Google Scholar]
- Olah, G.A.; Prakash, G.K.S.; Sommer, J. Superacids; John Wiley & Sons, Inc.: New York, NY, USA, 1985. [Google Scholar]
- Olah, G.A.; Prakash, G.K.S.; Molnár, Á.; Sommer, J. Superacid Chemistry, 2nd ed.; Wiley-Blackwell: New York, NY, USA, 2009. [Google Scholar]
- Alder, R.W.; Bowman, P.S.; Steele, W.R.S.; Winterman, D.R. The Remarkable Basicity of 1,8-Bis(dimethylamino)naphthalene. Chem. Commun. 1968, 723–724. [Google Scholar] [CrossRef]
- Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I.A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. [Google Scholar] [CrossRef]
- Koppel, I.A.; Schwesinger, R.; Breuer, T.; Burk, P.; Herodes, K.; Koppel, I.; Leito, I.; Mishima, M. Intrinsic Basicities of Phosphorus Imines and Ylides: A Theoretical Study. J. Phys. Chem. A 2001, 105, 9575–9586. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C. Enhanced Basicity of Push-Pull Nitrogen Bases in the Gas Phase. Chem. Rev. 2016, 116, 13454–13511. [Google Scholar] [CrossRef] [PubMed]
- Leito, I.; Koppel, I.A.; Koppel, I.; Kaupmees, K.; Tshepelevitsh, S.; Saame, J. Basicity Limits of Neutral Organic Superbases. Angew. Chem. Int. Ed. 2015, 54, 9262–9265. [Google Scholar] [CrossRef]
- Decouzon, M.; Gal, J.-F.; Maria, P.-C.; Raczyńska, E.D. Superbases in the Gas Phase: Amidine and Guanidine Derivatives with Proton Affinities Larger than 1000 kJ mol−1. Rapid Commun. Mass Spectrom. 1993, 7, 599–602. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Maria, P.-C.; Gal, J.-F.; Decouzon, M. Superbases in the Gas-Phase. Part II. Further Extension of the Basicity Scale Using Acyclic and Cyclic Guanidines. J. Phys. Org. Chem. 1994, 7, 725–733. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Decouzon, M.; Gal, J.-F.; Maria, P.-C.; Woźniak, K.; Kurg, R.; Carins, S.N. Superbases and Superacids in the Gas Phase. Trends Org. Chem. 1998, 7, 95–103. [Google Scholar]
- Raczyńska, E.D.; Decouzon, M.; Gal, J.-F.; Maria, P.-C.; Gelbard, G.; Vielfaure-Joly, F. Gas-Phase Structural (Internal) Effects in Strong Organic Nitrogen Bases. J. Phys. Org. Chem. 2001, 14, 25–34. [Google Scholar] [CrossRef]
- Kaljurand, I.; Koppel, I.A.; Kütt, A.; Rõõm, E.-I.; Rodima, T.; Koppel, I.; Mishima, M.; Leito, I. Experimental Gas-Phase Basicity Scale of Superbasic Phosphazenes. J. Phys. Chem. A 2007, 111, 1245–1250. [Google Scholar] [CrossRef]
- Kaljurand, I.; Saame, J.; Rodima, T.; Koppel, I.; Koppel, I.A.; Kögel, J.F.; Sundermeyer, J.; Köhn, U.; Coles, M.P.; Leito, I. Experimental Basicities of Phosphazene, Guanidinophosphazene and Proton Sponge Superbases in the Gas Phase and Solution. J. Phys. Chem. A 2016, 120, 2591–2604. [Google Scholar] [CrossRef] [PubMed]
- Glasovac, Z.; Štrukil, V.; Eckert-Maksić, M.; Schröder, D.; Kaczorowska, M.; Schwarz, H. Gas-Phase Proton Affinities of Guanidines with Heteroalkyl Side Chains. Int. J. Mass Spectrom. 2008, 270, 39–46. [Google Scholar] [CrossRef]
- Glasovac, Z.; Pavošević, F.; Štrukil, V.; Eckert-Maksić, M.; Schlangen, M.; Kretschmer, R. Toward Extension of the Gas-Phase Basicity Scale by Novel Pyridine Containing Guanidines. Int. J. Mass Spectrom. 2013, 354, 113–122. [Google Scholar] [CrossRef]
- Glasovac, Z.; Eckert-Maksić, M.; Kaljurand, I.; Saame, J.; Leito, I. Gas Phase Basicity of Biguanides—Comparison of the Equilibrium and the Kinetic Methods. Int. J. Mass Spectrom. 2019, 435, 61–68. [Google Scholar] [CrossRef]
- Despotović, I.; Kovacević, B.; Maksić, Z.B. Hyperstrong Neutral Organic Bases: Phosphazeno Azacalix [3](2,6)pyridines. Org. Lett. 2007, 9, 4709–4712. [Google Scholar] [CrossRef] [PubMed]
- Peran, N.; Maksić, Z.B. Polycyclic Croissant-Like Organic Compounds Are Powerful Superbases in the Gas Phase and Acetonitrile—A DFT Study. Chem. Comm. 2011, 47, 1327–1329. [Google Scholar] [CrossRef] [PubMed]
- Brønsted, J.N. Some Remarks on the Concept of Acids and Bases. Rec. Trav. Chim. Pays Bas 1923, 42, 718–728. [Google Scholar]
- Lowry, T.M. Co-Ordination and Acidity. J. Soc. Chem. Ind. Lond. 1923, 42, 1048–1052. [Google Scholar] [CrossRef]
- Gal, J.-F.; Maria, P.-M.; Raczyńska, E.D. Thermochemical Aspects of Proton Transfer in the Gas Phase. J. Mass Spectrom. 2001, 36, 699–716. [Google Scholar] [CrossRef] [PubMed]
- Ervin, K.M. Experimental Techniques in Gas-Phase Ion Thermochemistry. Chem. Rev. 2001, 101, 391–444. [Google Scholar] [CrossRef] [PubMed]
- Bouchoux, G. Gas-Phase Basicities of Polyfunctional Molecules Part 1 Theory and Methods. Mass Spectrom. Rev. 2007, 26, 775–835. [Google Scholar] [CrossRef]
- Marshall, A.G.; Hendrickson, C.L.; Jackson, G.S. Fourier Transform Ion Cyclotron Resonance Mass Spectrometry: A Primer. Mass Spectrom. Rev. 1998, 17, 1–35. [Google Scholar] [CrossRef]
- Cooks, R.G.; Patrick, J.S.; Kotiaho, T.; McLuckey, S.A. Thermochemical Determinations by the Kinetic Method. Mass Spectrom. Rev. 1994, 13, 287–339. [Google Scholar] [CrossRef]
- Cooks, R.G.; Wong, P.S.H. Kinetic Method of Making Thermochemical Determinations: Advances and Applications. Acc. Chem. Res. 1998, 31, 379–386. [Google Scholar] [CrossRef]
- Bouchoux, G. Evaluation of the Protonation Thermochemistry Obtained by the Extended Kinetic Method. J. Mass Spectrom. 2006, 41, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.K.; Saluja, P.P.S.; Kebarle, P.; Alder, R.W. Gas-Phase Basicities of N-Methyl Substituted 1,8-Diaminonaphthalenes and Related Compounds. J. Am. Chem. Soc. 1978, 100, 7328–7333. [Google Scholar] [CrossRef]
- Munson, M.S.B. Proton Affinities and the Methyl Inductive Effect. J. Am. Chem. Soc. 1965, 87, 2332–2336. [Google Scholar] [CrossRef]
- Lias, S.G.; Liebman, J.F.; Levin, R.D. Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules. J. Phys. Chem. Ref. Data 1984, 13, 695–808. [Google Scholar] [CrossRef]
- Smith, B.J.; Radom, L. Assigning Absolute Values to Proton Affinities: A Differentiation Between Competing Scales. J. Am. Chem. Soc. 1993, 115, 4885–4888. [Google Scholar] [CrossRef]
- Bowers, M.T.; Aue, D.H.; Webb, H.M.; McIver, R.T. Equilibrium Constants for Gas-Phase Ionic Reactions. Accurate Determination of Relative Proton Affinities. J. Am. Chem. Soc. 1971, 93, 4314–4315. [Google Scholar]
- Aue, D.H.; Webb, H.M.; Bowers, M.T. Quantitative Proton Affinities, Ionization Potentials, and Hydrogen Affinities of Alkylamines. J. Am. Chem. Soc. 1976, 98, 311–317. [Google Scholar] [CrossRef]
- Taft, R.W. Gas-Phase Proton-Transfer Equilibria; Caldin, E.F., Gold, V., Eds.; Springer Science: Dordrecht, The Netherlands, 1975; Chapter 2. [Google Scholar]
- Gal, J.-F.; Maria, P.-C.; Decouzon, M. The gas-phase acidity and bond dissociation energies of hydrogen telluride. Int. J. Mass Spectrom. Ion Proc. 1989, 93, 87–94. [Google Scholar] [CrossRef]
- Aue, D.H.; Webb, H.M.; Bowers, M.T. Quantitative Evaluation of Intramolecular Strong Hydrogen Bonding in the Gas Phase. J. Am. Chem. Soc. 1973, 95, 2699–2701. [Google Scholar] [CrossRef]
- Aue, D.H.; Bowers, M.T. Stabilities of Positive Ions From Equilibrium Gas-Phase Basicity Measurements. In Gas-Phase Ion Chemistry; Bowers, M.T., Ed.; Academic Press: New York, NY, USA, 1979; Volume 2, Chapter 9; pp. 1–51. [Google Scholar]
- Taft, R.W. Protonic Acidities and Basicities in the Gas Phase and in Solution: Substituent and Solvent Effects. Prog. Phys. Org. Chem. 1983, 14, 247–350. [Google Scholar]
- Borgarello, M.; Houriet, R.; Raczyńska, E.D.; Drapała, T. Gas-Phase Basicity of N1,N1-Dimethyl-N2-phenylformamidines. J. Org. Chem. 1990, 55, 38–42. [Google Scholar] [CrossRef]
- Taft, R.W.; Raczyńska, E.D.; Maria, P.-C.; Leito, I.; Gal, J.-F.; Decouzon, M.; Drapała, T.; Anvia, F. Experimental (FT-ICR) vs. Calculated (AM1) Gas-Phase Basicities of N1,N1-Dimethyl-N2-phenylformamidines. Substituent and Solvent Effects. Pol. J. Chem. 1995, 69, 41–53. [Google Scholar]
- Decouzon, M.; Gal, J.-F.; Maria, P.-C.; Raczyńska, E.D. Gas Phase Basicity of N1,N1-Dimethyl-N2-alkylformamidines. Polarizability Effects. J. Org. Chem. 1991, 56, 3669–3673. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Maria, P.-C.; Gal, J.-F.; Decouzon, M. Gas-Phase Basicity of N1,N1-Dimethylformamidines: Substituent Polarizability and Field Effects, and Comparison with Brönsted Basicity in Solution. J. Org. Chem. 1992, 57, 5730–5735. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Darowska, M.; Dąbkowska, I.; Decouzon, M.; Gal, J.-F.; Maria, P.-C.; Dubin Poliart, C. Experimental and Theoretical Evidence of Basic Site Preference in Polyfunctional Superbasic Amidinazine: N1,N1-Dimethyl-N2-beta-(2-pyridylethyl)formamidine. J. Org. Chem. 2004, 69, 4023–4030. [Google Scholar] [CrossRef] [PubMed]
- Bouchoux, G.; Eckert-Maksić, M. Gas Phase Basicities of Polyfunctional Molecules. Part 5: Non-Aromatic sp2 Nitrogen Containing Compounds. Mass Spectrom. Rev. 2016, 37, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Kaljurand, I.; Rodima, T.; Pihl, A.; Mäemets, V.; Leito, I.; Koppel, I.A.; Mishima, M. Acid-Base Equilibria in Nonpolar Media. 4. Extension of the Self-Consistent Basicity Scale in THF Medium. Gas-Phase Basicities of Phosphazenes. J. Org. Chem. 2003, 68, 9988–9993. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, R.S. Electronic Structures of Polyatomic Molecules and Valence. V. Molecules RXn. J. Chem. Phys. 1933, 1, 492–503. [Google Scholar] [CrossRef]
- Burk, P.; Koppel, I.A.; Koppel, I.; Leito, I.; Travnikova, O. Critical Test of Performance of B3LYP Functional for Prediction of Gas-Phase Acidities and Basicities. Chem. Phys. Lett. 2000, 323, 482–489. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Kovačević, B.; Vianello, R. Advances in Determining the Absolute Proton Affinities of Neutral Organic Molecules in the Gas Phase and Their Interpretation: A Theoretical Account. Chem. Rev. 2012, 112, 5240–5270. [Google Scholar] [CrossRef] [PubMed]
- Bouchoux, G. Gas Phase Basicities of Polyfunctional Molecules. Part 3: Amino Acids. Mass Spectrom. Rev. 2012, 31, 391–435. [Google Scholar] [CrossRef]
- Bartmess, J.E. Thermodynamics of the Electron and the Proton. J. Phys. Chem. 1994, 98, 6420–6424, Erratum in J. Phys. Chem. 1995, 98, 6755. [Google Scholar] [CrossRef]
- Fifen, J.J.; Dhaouadi, Z.; Nsangou, M. Revision of the Thermodynamics of the Proton in the Gas Phase. J. Phys. Chem. A 2014, 118, 11090–11097. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, Æ. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian, Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Wu, Z.; Fenselau, C. Proton Affinity of Arginine Measured by the Kinetic Approach. Rapid Commun. Mass Spectrom. 1992, 6, 403–405. [Google Scholar] [CrossRef]
- Bouchoux, G.; Salpin, J.-Y. Gas Phase Basicities of Polyfunctional Molecules. Part 2: Saturated Basic Sites. Mass Spectrom. Rev. 2012, 31, 353–390. [Google Scholar] [CrossRef]
- Meot-Ner (Mautner), M. The Ionic Hydrogen Bond. Chem. Rev. 2005, 105, 213–284. [Google Scholar] [CrossRef] [PubMed]
- Meot-Ner (Mautner), M. Update 1 of: Strong Ionic Hydrogen Bonds. Chem. Rev. 2012, 112, PR22–PR103. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Darowska, M.; Cyrański, M.K.; Makowski, M.; Rudka, T.; Gal, J.-F.; Maria, P.-C. Ab Initio Study of Tautomerism and of Basicity Center Preferences in Histamine, from Gas Phase to Solution—Comparison with Experimental Data (Gas Phase, Solution, Solid State). J. Phys. Org. Chem. 2003, 16, 783–796. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C. The Guanylated Bioamine Agmatine—A Theoretical Investigation of Its Structure and Exceptional High Basicity in the Gas Phase. Comput. Theoret. Chem. 2017, 1109, 10–18. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Rudka, T.; Darowska, M.; Dąbkowska, I.; Gal, J.-F.; Maria, P.-C. Experimental (FT-ICR) and Theoretical (DFT) Estimation of the Basic Site Preference for the Bidentate Molecule 2-(β-Aminoethyl)-pyridine: Similarity with Histamine. J. Phys. Org. Chem. 2005, 18, 856–863. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Michalec, P.; Zalewski, M. Exceptionally High Proton and Lithium Cation Gas-Phase Basicity of the Anti-Diabetic Drug Metformin. J. Phys. Chem. A 2017, 121, 8706–8718. [Google Scholar] [CrossRef] [PubMed]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Fontaine-Vive, F. Biguanide Antidiabetic Drugs: Imeglimin Exhibits Higher Proton Basicity but Smaller Lithium-Cation Basicity than Metformin in Vacuo. ACS Omega 2018, 3, 17842–17852. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Saeidian, H. Push–Pull Effect on the Gas-Phase Basicity of Nitriles: Transmission of the Resonance Effects by Methylenecyclopropene and Cyclopropenimine π-Systems Substituted by Two Identical Strong Electron Donors. Symmetry 2021, 13, 1554. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Sakhawat, G.S.; Fahim, M.Q.; Saeidian, H. Nitriles with High Gas-Phase Basicity—Part II Transmission of the Push–Pull Effect through Methylenecyclopropene and Cyclopropenimine Scaffolds Intercalated between Different Electron Donor(s) and the Cyano N-Protonation Site. Molecules 2022, 27, 4370. [Google Scholar] [CrossRef] [PubMed]
- González, A.I.; Mó, O.; Yáñez, M.; Léon, E.; Tortajada, J.; Morizur, J.P.; Leito, I.; Maria, P.-C.; Gal, J.F. Basicity of Acetamidine. Experimental and Theoretical Study. J. Phys. Chem. 1996, 100, 10490–10496. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Kovačević, B. Toward Organic Superbases: The Electronic Structure and the Absolute Proton Affinity of Quinodiimines and Some Related Compounds. J. Phys. Chem. A 1998, 102, 7324–7328. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Vianello, R. Quest for the Origin of Basicity: Initial vs Final State Effect in Neutral Nitrogen Bases. J. Phys. Chem. A 2002, 106, 419–430. [Google Scholar] [CrossRef]
- Despotović, I.; Maksić, Z.B.; Vianello, R. Computational Design of Brønsted Neutral Organic Superbases—[3]Iminoradia-lenes and Quinonimines Are Important Synthetic Targets. New J. Chem. 2007, 31, 52–62. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Glasovac, Z.; Kovačević, B. Predicted High Proton Affinity of Poly-2,5-dihydropyrrolimines—The Aromatic Domino Effect. J. Phys. Org. Chem. 2002, 15, 499–508. [Google Scholar] [CrossRef]
- Koneshlou, T.; Rouhani, M.; Saeidian, H.; Aliabad, J.M. Super/Hyperbasicity of Novel Diquinonimino Derivatives of Guanidine in Gas Phase. Chem. Phys. Lett. 2022, 804, 139915. [Google Scholar] [CrossRef]
- Khazali, M.; Rouhani, M.; Saeidian, H. Utilizing the Synergistic Effect between Imidazole Aromaticity and Guanidine Structure for the Computational Design of Novel Uncharged Organic Superbases. J. Mol. Struct. 2023, 1273, 134348. [Google Scholar] [CrossRef]
- Raczyńska, E.D. On Some Origins of Tautomeric Preferences in Neutral Creatinine in Vacuo: Search for Analogies and Differences in Cyclic Azoles and Azines. Symmetry 2024, 16, 98. [Google Scholar] [CrossRef]
- Kolomeitsev, A.A.; Koppel, I.A.; Rodima, T.; Barten, J.; Lork, E.; Röschenthaler, G.-V.; Kaljurand, I.; Kütt, A.; Koppel, I.; Mäemets, V.; et al. Guanidinophosphazenes: Design, Synthesis, and Basicity in THF and in the Gas Phase. J. Am. Chem. Soc. 2005, 127, 17656–17666. [Google Scholar] [CrossRef] [PubMed]
- Meot-Ner, M.; Hamlet, P.; Hunter, E.P.; Field, F.H. Internal and External Solvation of Polyfunctional Ions. J. Am. Chem. Soc. 1980, 102, 6393–6399. [Google Scholar] [CrossRef]
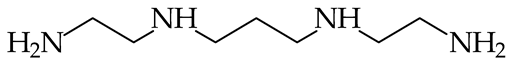
- Reyzer, M.L.; Brodbelt, J.S. Gas-Phase Basicities of Polyamines. J. Am. Soc. Mass Spectrom. 1998, 9, 1043–1048. [Google Scholar] [CrossRef]
- Tian, Z.; Fattahi, A.; Lis, L.; Kass, S.R. Neutral Intramolecular Hydrogen-Bond Bases. Croat. Chim. Acta 2009, 82, 41–45. [Google Scholar]
- Raczyńska, E.D.; Taft, R.W. Semiempirical (AM1) Studies of Possible and Preferred Site(s) of Protonation in the Gas Phase for Polyfunctional Nitrogen Bases: N1,N1-Dimethyl-N2-azinylformamidines. Bull. Chem. Soc. Jpn. 1997, 70, 1297–1305. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Decouzon, M.; Gal, J.-F.; Maria, P.-C.; Taft, R.W.; Anvia, F. Gas-Phase Basicity of Polyfunctional Amidinazines: Experimental Evidence of Preferred Site(s) of Protonation. J. Org. Chem. 2000, 65, 4635–4640. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C. Gas-Phase Basicity of Aromatic Azines: A Short Review on Structural Effects. Int. J. Mass Spectrom. 2017, 418, 130–139. [Google Scholar] [CrossRef]
- Despotović, I.; Vianello, R. Engineering Exceptionally Strong Oxygen Superbases with 1,8-Diazanaphthalene Di-N-oxides. Chem. Commun. 2014, 50, 10941–10944. [Google Scholar] [CrossRef] [PubMed]
- Schwesinger, R. Strong Uncharged Nitrogen Bases. Nachr. Chem. Tech. Lab. 1990, 38, 1214–1226. [Google Scholar] [CrossRef]
- Gelbard, G.; Vielfaure-Joly, F. Polynitrogen Strong Bases: 1-New Syntheses of Biguanides and Their Catalytic Properties in Transesterification Reactions. Tetrahedron Lett. 1998, 39, 2743–2746. [Google Scholar] [CrossRef]
- Polyakova, S.M.; Kunetskiy, R.A.; Schröder, D. Proton Affinities of 2-Iminoimidazolines with Bulky N-Alkyl-Substituents. Int. J. Mass Spectrom. 2012, 314, 13–17. [Google Scholar] [CrossRef]
- Kovačević, B.; Glasovac, Z.; Maksić, Z.B. The Intramolecular Hydrogen Bond and Intrinsic Proton Affinity of Neutral Organic Molecules: N,N′,N″-Tris(3-aminopropyl)guanidine and Some Related Systems. J. Phys. Org. Chem. 2002, 15, 765–774. [Google Scholar] [CrossRef]
- Barić, D.; Dragičević, I.; Kovačević, B. Design of Superbasic Guanidines: The Role of Multiple Intramolecular Hydrogen Bonds. J. Org. Chem. 2013, 78, 4075–4082. [Google Scholar] [CrossRef]
- Schwesinger, R.; Schlemper, H.; Hasenfratz, C.; Willaredt, J.; Dambacher, T.; Breuer, T.; Ottaway, C.; Fletschinger, M.; Boele, J.; Fritz, H.; et al. Extremely Strong, Uncharged Auxiliary Bases; Monomeric and Polymer-Supported Polyaminophosphazenes (P2–P5). Liebigs Ann. 1996, 1055–1081. [Google Scholar] [CrossRef]
- Berthelot, M.; Helbert, M.; Laurence, C.; Le Questel, J.-Y.; Anvia, F.; Taft, R.W. Super-Basic Nitriles. J. Chem. Soc. Perkin Trans. 2 1993, 625–627. [Google Scholar] [CrossRef]
- Makowski, M.; Raczyńska, E.D.; Chmurzyński, L. Ab Initio Study of Possible and Preferred Basic Site(s) in Polyfunctional N1,N1-Dimethyl-N2-cyanoformamidine. J. Phys. Chem. A 2001, 105, 869–874. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C. Exceptional Proton Affinities of Push-Pull Nitriles Substituted by the Guanidino and Phosphazeno Groups. RSC Adv. 2015, 5, 25513–25517. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Makowski, M.; Maria, P.-C.; Gal, J.-F. Can Nitriles Be Stronger Bases than Proton Sponges in the Gas Phase? A Computational Analysis. J. Phys. Chem. A 2015, 119, 8225–8236. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.W.; Topsom, R.D. The Nature and Analysis of Substituent Electronic Effects. Prog. Phys. Org. Chem. 1987, 16, 1–83. [Google Scholar]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Taagepera, M.; Henderson, W.G.; Brownlee, R.T.C.; Beauchamp, J.L.; Holtz, D.; Taft, R.W. Gas-Phase Basicities and Pyridine Substituent Effects. J. Am. Chem. Soc. 1972, 94, 1369–1370. [Google Scholar] [CrossRef]
- Abboud, J.-L.M.; Catalan, J.; Elguero, J.; Taft, R.W. Polarizability Effects on the Aqueous Solution Basicity of Substituted Pyridines. J. Org. Chem. 1988, 53, 1137–1140. [Google Scholar] [CrossRef]
- Kaljurand, I.; Lilleorg, R.; Murumaa, A.; Mishima, M.; Burk, P.; Koppel, I.; Koppel, I.A.; Leito, I. The basicity of substituted N,N-dimethylanilines in solution and in the gas phase. J. Phys. Org. Chem. 2013, 26, 171–181. [Google Scholar] [CrossRef]
- Häfelinger, G.; Kuske, F.K.H. General and Theoretical Aspects of Amidines and Related Compounds. In The Chemistry of Amidines and Imidates; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons Ltd.: New York, NY, USA, 1991; Volume 2, Chapter 1; pp. 1–100. [Google Scholar]
- Raczyńska, E.D.; Laurence, C.; Nicolet, P. Hydrogen Bonding Basicity of Amidines. J. Chem. Soc. Perkin Trans. 2 1988, 1491–1494. [Google Scholar] [CrossRef]
- Laurence, C.; Berthelot, M.; Raczyńska, E.; Le Questel, J.-Y.; Duguay, G.; Hudhomme, P. Hydrogen-Bond Basicity of Cyanamide, Amide, Thioamide, and Sulphonamide Iminologues. J. Chem. Res. (S) 1990, 250–251. [Google Scholar]
- Krygowski, T.M.; Anulewicz, R.; Raczyńska, E.D.; Laurence, C. Structural Studies of 1,3-Di(N,N-dimethylformamidyl)-2-cyanoguanidine. The Case of a Strongly Lewis Basic Nitrogen Atom in the Cyano Group VIII. Crystallographic Studies of Intramolecular and Intermolecular Interactions. J. Phys. Org. Chem. 1991, 4, 689–692. [Google Scholar] [CrossRef]
- Maria, P.C.; Gal, J.F.; Taft, R.W. Proton Affinities Versus Enthalpies of Complexation With Boron Trifluoride in Solution, in the Nitrile Series: Role of the Polarizability. New J. Chem. 1987, 11, 617–621. [Google Scholar]
- Peerboom, R.A.L.; Ingemann, S.; Nibbering, N.M.M.; Liebman, J.F. Proton Affinities and Heat of Formation of the Imines CH2=NH, CH2=NMe and PhCH=NH. J. Chem. Soc. Perkin Trans. 2 1990, 1825–1828. [Google Scholar] [CrossRef]
- Roithová, J.; Schröder, D.; Mišek, J.; Stará, I.G.; Starý, I. Chiral Superbases: The Proton Affinities of 1- and 2-Aza [6]helicene in the Gas Phase. J. Mass Spectrom. 2007, 42, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, A.; Sims, L.A.; Snead, R.; Gronert, S.; Maclagan, R.G.A.R.; Meot-Ner (Mautner), M. Protonation Energies of 1–5 Ring Polycyclic Aromatic Nitrogen Heterocycles: Comparing Experiment and Theory. J. Phys. Chem. A 2015, 119, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Maclagan, R.G.A.R.; Gronert, S.; Meot-Ner (Mautner), M. Protonated Polycyclic aromatic Nitrogen Heterocycles: Proton Affinities, Polarizabilities, and Atomic and Ring Charges of 1–5 Ring Ions. J. Phys. Chem. A 2015, 119, 127–139. [Google Scholar] [CrossRef]
- Valadbeigi, Y. Effects of Intramolecular Hydrogen Bond and Electron Delocalization on the Basicity of Proton Sponges and Superbases with Benzene, Pyridine, Pyrazine and Pyrimidine Scaffolds. Comput. Theor. Chem. 2020, 1188, 112947. [Google Scholar] [CrossRef]
- Gautier, J.-A.; Miocque, M.; Farnoux, C.C. Preparation and Synthetic Uses of Amidines. In The Chemistry of Amidines and Imidates; Patai, S., Ed.; Wiley: New York, NY, USA, 1975; Chapter 7; pp. 283–347. [Google Scholar]
- Boyd, G.V. Recent Advances in the Synthesis of Amidines. In The Chemistry of Amidines and Imidates; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons Ltd.: New York, NY, USA, 1991; Volume 2, Chapter 7; pp. 339–424. [Google Scholar]
- Yamamoto, Y.; Kojima, S. Synthesis and Chemistry of Guanidine Derivatives. In The Chemistry of Amidines and Imidates; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons Ltd.: New York, NY, USA, 1991; Volume 2, Chapter 10; pp. 485–526. [Google Scholar]
- Scoggins, M.W. A Rapid Gas Chromatographic Analysis of Diastereomeric Diamines. J. Chrom. Sci. 1975, 13, 146–148. [Google Scholar] [CrossRef]
- Bredereck, H.; Simchen, G.; Rebsdat, S.; Kantlehner, W.; Horn, P.; Wahl, R.; Hoffmann, H.; Grieshaber, P.; Säureamid-Reaktionen, L.; Orthoamide, I. Darstellung und Eigenschaften der Amidacetale und Aminalester. Chem. Ber. 1968, 101, 41–50. [Google Scholar] [CrossRef]
- Bredereck, H.; Bredereck, K. Säureamid-Reaktionen, XXVII. Umsetzungen mit Säureamid-Phosphoroxychlorid-Addukten und Amidchloriden. Chem. Ber. 1961, 94, 2278–2295. [Google Scholar] [CrossRef]
- Oszczapowicz, J.; Osek, J. Guanidines. 1. Influence of Substitution of Imino Nitrogen Atom on Basicity of Tetramethylguanidines. Pol. J. Chem. 1983, 57, 93–98. [Google Scholar]
- Pruszynski, P. Synthesis and Properties of Phenyl Substituted Derivatives of 2-Phenyl-1,1,3,3-tetramethylguanidines. Can. J. Chem. 1987, 65, 626–629. [Google Scholar] [CrossRef]
- Grytsai, O.; Ronco, C.; Benhida, R. Synthetic Accesses to Biguanide Compounds. Beilstein J. Org. Chem. 2021, 17, 1001–1040. [Google Scholar] [CrossRef]
- Glasovac, Z.; Trošelj, P.; Jušinski, I.; Margetić, D.; Eckert-Maksić, M. Synthesis of Highly Basic Hexasubstituted Biguanides by Environmentally Friendly Methods. Synlett 2013, 24, 2540–2544. [Google Scholar]
- Schwesinger, R. Extremely Strong, Non-Ionic Bases—Syntheses and Applications. Chimia 1985, 39, 269–272. [Google Scholar]
- Schwesinger, R.; Schlemper, H. Peralkylated Polyaminophosphazenes—Extremely Strong, Neutral Nitrogen Bases. Angew. Chem. Int. Ed. Engl. 1987, 26, 1167–1169. [Google Scholar] [CrossRef]
- Schwesinger, R.; Hasenfratz, C.; Schlemper, H.; Walz, L.; Peters, E.-M.; Peters, K.; von Schnering, H.G. How Strong and How Hindered Can Uncharged Phosphazene Bases Be? Angew. Chem. Int. Ed. Engl. 1993, 32, 1361–1363. [Google Scholar] [CrossRef]
- Schwesinger, R.; Willaredt, J.; Schlemper, H.; Keller, M.; Schmitt, D.; Fritz, H. Novel, Very Strong, Uncharged Auxiliary Bases; Design and Synthesis of Monomeric and Polymer-Bound Triaminoiminophosphorane Bases of Broadly Varied Steric Demand. Chem. Ber. 1994, 127, 2435–2454. [Google Scholar] [CrossRef]
- Schwesinger, R.; Dambacher, T. A Short Novel Synthesis of the Phosphazene Base Et-P2. Z. Naturforsch. 2006, 61, 1229–1233. [Google Scholar] [CrossRef]
- Schwesinger, R. Tricyclic 2,4-Diaminovinamidines—Readily Accessible, Very Strong CHN Bases. Angew. Chem. Int. Ed. Engl. 1987, 26, 1164–1165. [Google Scholar] [CrossRef]
- Schwesinger, R.; Miβfeldt, M.; Peters, K.; von Schnering, H.G. Novel, Very Strongly Basic, Pentacyclic “Proton Sponges” with Vinamidine Structure. Angew. Chem. Int. Ed. Engl. 1987, 26, 1165–1166. [Google Scholar] [CrossRef]
- Singh, A.; Ojha, A.K.; Jang, H.M. Strategic Design and Utilization of Molecular Flexibility for Straddling the Application of Organic Superbases: A DFT Study. ChemistrySelect 2018, 3, 837–842. [Google Scholar] [CrossRef]
- Barić, D. Utilizing the Azaazulene Scaffolds in the Design of New Organic Superbases. ACS Omega 2019, 4, 15197–15207. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Barić, D.; Xie, X.; Kovačević, B.; Sundermeyer, J. Basicity Enhancement by Multiple Intramolecular Hydrogen Bonding in Organic Superbase N,N′,N″,N′″-Tetrakis(3-(dimethylamino)propyl)triaminophosphazene. Org. Lett. 2019, 21, 9142–9146. [Google Scholar] [CrossRef] [PubMed]
- Saadat, K.; Shiri, A.; Kovačević, B. Step Forward to Stronger Neutral Step Forward to Stronger Neutral Organic Superbases: Fused Troponimines. J. Org. Chem. 2020, 85, 11375–11381. [Google Scholar] [CrossRef] [PubMed]
- Briš, A.; Glasovac, Z.; Margetić, D. Gas-Phase Basicity of Cyclic Guanidine Derivatives—A DFT Study. New J. Chem. 2021, 45, 2384–2392. [Google Scholar] [CrossRef]
- Koneshlou, T.; Rouhani, M.; Saeidian, H.; Aliabad, J.M. Biguanide-dihydropyrimidine Dual Scaffolds with Impressive Basicities According to DFT Calculations. Comput. Theor. Chem. 2023, 1225, 114178. [Google Scholar] [CrossRef]
- Valadbeigi, Y.; Taheri, R. Superbasicity of Imines with Bicyclo [5.1.0]octa-1,3,5,7-tetraene Scaffold due to Electron Delocalization in the Conjugated Acids. Comput. Theor. Chem. 2023, 1222, 114076. [Google Scholar] [CrossRef]
- Valadbeigi, Y.; Causon, T. Nitriles with Exceptionally High Proton Affinity Due to a C−N Bond Formation Upon Protonation. Comput. Theor. Chem. 2023, 1230, 114372. [Google Scholar] [CrossRef]
- Puleo, T.R.; Sujansky, S.J.; Wright, S.E.; Bandar, J.S. Organic Superbases in Recent Synthetic Methodology Research. Chem. Eur. J. 2021, 27, 4216–4229. [Google Scholar] [CrossRef] [PubMed]
- Vazdar, K.; Margetić, D.; Kovačević, B.; Sundermeyer, J.; Leito, I.; Jahn, U. Design of Novel Uncharged Organic Superbases: Merging Basicity and Functionality. Acc. Chem. Res. 2021, 54, 3108–3123. [Google Scholar] [CrossRef]
- Sujansky, S.; Hoteling, G.; Bandar, J. A Strategy for the Controllable Generation of Organic Superbases from Benchtop-Stable Salts. ChemRxiv 2024, 11, 2897–2904. [Google Scholar] [CrossRef]
- Zhang, Y.; Ning, L.; Zhu, T.; Xie, Z.; Dong, S.; Feng, X.; Liu, X. Chiral Guanidine Catalyzed Cyclization Reactions of 1,3-Enynes for Lactone Synthesis: Switchable H-Bond Catalysis. Org. Chem. Front. 2024. [Google Scholar] [CrossRef]
- Formica, M.; Rozsar, D.; Su, G.; Farley, A.J.M.; Dixon, D.J. Bifunctional Iminophosphorane Superbase Catalysis: Applications in Organic Synthesis. Acc. Chem. Res. 2020, 53, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Nam, H.; Lee, S.Y. Enantio- and Diastereoselective Variations on α-Iminonitriles: Harnessing Chiral Cyclopropenimine-Thiourea Organocatalysts. J. Am. Chem. Soc. 2024, 146, 3065–3074. [Google Scholar] [CrossRef]
- Duan, W.; Li, Z.; Yang, Z.; He, F.; Chen, S.; Qiao, C.; Yao, J.; Zhang, C.; Zhao, H.; Lia, M.; et al. Synthesis of Guanidinophosphazene Superbase for the Facile Preparation of High Molecular Weight Polysiloxane Under Mild Conditions. Polym. Chem. 2023, 14, 172–182. [Google Scholar] [CrossRef]
- Kim, J.G.; Lee, G.S.; Lee, A. Triazabicyclodecene: A versatile catalyst for polymer synthesis. J. Polym. Sci. 2024, 62, 42–91. [Google Scholar] [CrossRef]
- Zhou, L.; Reilly, L.T.; Shi, C.; Quinn, E.C.; Chen, E.Y.-X. Proton-Triggered Topological Transformation in Superbase-Mediated Selective Polymerization Enables Access to Ultrahigh-Molar-Mass Cyclic Polymers. Nat. Chem. 2024. [Google Scholar] [CrossRef]
- Wei, H.; Cheng, Z.; Wu, T.; Liu, Y.; Guo, J.; Chen, P.A.; Xia, J.; Xie, H.; Qiu, X.; Liu, T.; et al. Novel Organic Superbase Dopants for Ultraefficient N-Doping of Organic Semiconductors. Adv. Mater. 2023, 35, 2300084. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.-D.; Fabian Bauch, F.; Hub, Y.; Schumacher, S. Charge Transfer in Superbase n-Type Doping of PCBM Induced by Deprotonation. Phys. Chem. Chem. Phys. 2024, 26, 4194–4199. [Google Scholar] [CrossRef]
- Nishinaka, M.; Harada, I.; Akaike, K.; Wei, Q.; Koshiba, Y.; Horike, S.; Ishida, K. Electrochemical Charge-Carrier Modulation of Carbon Nanotubes Using Ionic Liquids Derived from Organic Superbases for Stable Thermoelectric Materials. Carbon 2024, 218, 118667. [Google Scholar] [CrossRef]
- Wilm, L.F.B.; Das, M.; Janssen-Müller, D.; Mück-Lichtenfeld, C.; Frank Glorius, F.; Dielmann, F. Photoswitchable Nitrogen Superbases: Using Light for Reversible Carbon Dioxide Capture. Angew. Chem. Int. Ed. 2022, 61, e202112344. [Google Scholar] [CrossRef]
- Gabriele, B.; Della Ca’, N.; Mancuso, R.; Veltri, L.; Ziccarelli, I. Amidine- and Guanidine-Based Synthetic Methods for CO2 Capture and Utilization. Curr. Opinion Green Sust. Chem. 2023, 41, 100793. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, R. A Critical Review on New and Efficient Adsorbents for CO2 Capture. Chem. Eng. J. 2024, 485, 149495. [Google Scholar] [CrossRef]
- Yue, W.; Han, W.; Yuan, M.; Zhou, X.; Hui Fu, H. Three-Component CO2 Binding Organic Liquids for Efficient and Reversible CO2 Capture: Effect of Molar Ratio of Component on Mechanism. J. Mol. Liquids 2024, 399, 124400. [Google Scholar] [CrossRef]
- Wu, D.; Martin, R.T.; Piña, J.; Kwon, J.; Crockett, M.P.; Thomas, A.A.; Gutierrez, O.; Park, N.H.; Hedrick, J.L.; Campos, L.M. Cyclopropenimine-Mediated CO2 Activation for the Synthesis of Polyurethanes and Small-Molecule Carbonates and Carbamates. Angew. Chem. Int. Ed. 2024, 136, e202401281. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Chen, Q.; Cong, S.; An, N.; Fan, J.; Song, F.; Zhang, X. Investigation of Effective CO2 Capture by Ternary Deep Eutectic Solvents Based on Superbase. J. Mol. Liquids 2024, 401, 124755. [Google Scholar] [CrossRef]
- Elliott, T.; Charbonneau, L.; Gazagnaire, E.; Kilpeläinen, I.; Kótai, B.; Laczko, G.; Papai, I.; Repo, T. Synergetic Effects on the Capture and Release of CO2 Using Guanidine and Amidine Superbases. RSC Sustain. 2024. [Google Scholar] [CrossRef]
- Graut, R.J. The Chemistry of Amidines and Imidates; Patai, S., Ed.; Wiley: New York, NY, USA, 1975; Chapter 6. [Google Scholar]
- Saczewski, F.; Balewski, Ł. Biological Activities of Guanidine Compounds. Expert Opin. Ther. Patents 2009, 19, 1417–1448. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M. N-(Hetero)aryl-2-imidazolines: An Emerging Privileged Motif for Contemporary Drug Design. Chem. Heterocycl. Comp. 2017, 53, 240–255. [Google Scholar] [CrossRef]
- Kim, S.-H.; Semenya, D.; Castagnolo, D. Antimicrobial Drugs Bearing Guanidine Moieties: A Review. Eur. J. Med. Chem. 2021, 216, 113293. [Google Scholar] [CrossRef]
- Kathuria, D.; Raul, A.D.; Wanjari, P.; Bharatam, P.V. Biguanides: Species with versatile therapeutic applications. Eur. J. Med. Chem. 2021, 219, 113378. [Google Scholar] [CrossRef]
- Zhao, H.; Swanson, K.D.; Zheng, B. Therapeutic Repurposing of Biguanides in Cancer. Trends Cancer 2021, 7, 714–730. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Romminger, S. The Chemistry and Biology of Guanidine Natural Products. Nat. Prod. Rep. 2016, 33, 456–490. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.R.; Carla, L.; Varela, C.L.; Ana, S.; Pires, A.S.; Tavares-da-Silva, E.J.; Roleira, F.M.F. Synthetic and Natural Guanidine Derivatives as Antitumor and Antimicrobial Agents: A Review. Bioorg. Chem. 2023, 138, 106600. [Google Scholar] [CrossRef]
- Sadanandan, A.M.; Khatri, P.K.; Saxena, R.C.; Jain, S.L. Guanidine Based Amino Acid Derived Task Specific Ionic Liquids as Noncorrosive Lubricant Additives for Tribological Performance. J. Mol. Liquids 2020, 313, 113527. [Google Scholar] [CrossRef]
- Lucio, A.J.; Efimov, I.; Efimov, O.N.; Zaleski, C.J.; Viles, S.; Ignatiuk, B.B.; Abbott, A.P.; Hillman, A.R.; Ryder, K.S. Amidine-Based Ionic Liquid Analogues with AlCl3: A Credible New Electrolyte for Rechargeable Al Batteries. Chem. Commun. 2021, 57, 9834–9837. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, S.; Hummel, M.; Sawada, D.; Guizani, C.; Rissanen, M.; Sixta, H. Superbase-Based Protic Ionic Liquids for Cellulose Filament Spinning. Cellulose 2021, 28, 533–547. [Google Scholar] [CrossRef]
- Gazagnaire, E.; Helminen, J.; King, A.W.T.; Almeida, T.G.; Kurten, T.; Kilpeläinen, I. Bicyclic Guanidine Superbase Carboxylate Salts for Cellulose Dissolution. RSC Adv. 2024, 14, 12119. [Google Scholar] [CrossRef]
- Kitos, A.A.; Mavragani, N.; Murugesu, M.; Brusso, J. A Chelate Like no Other: Exploring the Synthesis, Coordination Chemistry and Applications of Imidoyl Amidine Frameworks. Mater. Adv. 2020, 1, 2688–2706. [Google Scholar] [CrossRef]








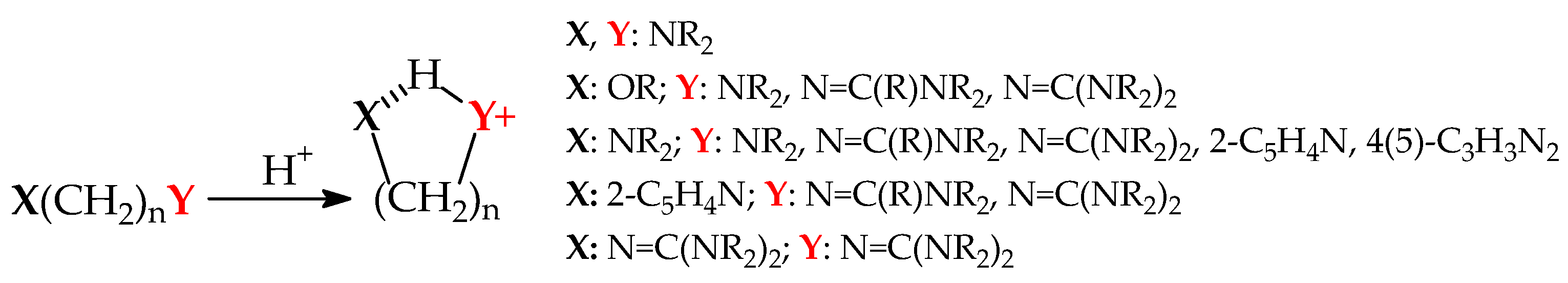



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | GB [Ref.] | Structure | GB [Ref.] |
|---|---|---|---|
 |  | 966–1021 [86] | |
| 995.8 [9] | 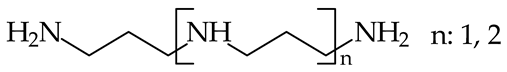 | 994–1012 [86] | |
| DMAN | |||
 |  | 1000 [86] | |
| 1004 [86] | 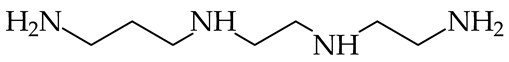 | 1004 [86] | |
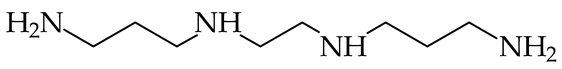 | 1004 [86] | ||
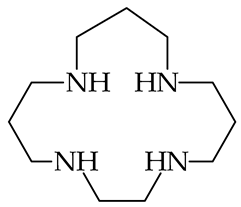 | 1021 [86] | 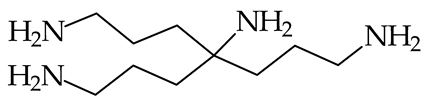 | 1019 b [87] |
| Derivative | GB | Derivative | GB |
|---|---|---|---|
| (PrnNH)2C=NPrn | 1024 | (PrnNH)2C=N(CH2)3NMe2 | 1054 |
| (PriNH)2C=N(CH2)3SMe | 1039 | (PriNH)2C=N(CH2)3NMe2 | 1055 |
| (Me2N)2C=N(CH2)3OMe | 1050 | (MeNH)2C=N(CH2)2-2-C5H4N | 1044 |
| [MeO(CH2)3NH]2C=NPrn | 1059 | (PriNH)2C=N(CH2)2-2-C5H4N | 1052 |
| (PrnNH)2C=N(CH2)3OMe | 1046 | [2-C5H4N-(CH2)2NH]2C=NPri | 1068 |
| [MeO(CH2)3NH]2C=N(CH2)3OMe | 1070 | [2-C5H4N-(CH2)2NH]2C=N(CH2)2-2-C5H4N | 1082 |
| [Me2N(CH2)3NH]2C=NPrn | 1072 | (PriNH)2C=N(CH2)3N=C(NHPri)2 | 1070 |
| Derivative | GBeq | GBkin |
|---|---|---|
| (Me2N)2C=NC(=N*Ph)NH(CH2)3NMe2 | - | 1062 |
| (Me2N)2C=NC(=N*4-C6H4OMe)NH(CH2)3NMe2 | - | 1071 |
| (Me2N)2C=NC(=N*Et)NH(CH2)3NMe2 | 1089 | 1085 |
| (Me2N)2C=NC(=N*Pri)NHPri | 1070 | 1064 |
| (Me2N)2C=NC(=N*c-C6H11)NHc-C6H11 | 1080 | 1071 |
| (Me2N)2C=NC{=N*(CH2)3OMe}NH(CH2)3OMe | 1095 | - |
| (Me2N)2C=NC{=N*(CH2)3NMe2}NH(CH2)3NMe2 | 1106 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C. Strong Bases and beyond: The Prominent Contribution of Neutral Push–Pull Organic Molecules towards Superbases in the Gas Phase. Int. J. Mol. Sci. 2024, 25, 5591. https://doi.org/10.3390/ijms25115591
Raczyńska ED, Gal J-F, Maria P-C. Strong Bases and beyond: The Prominent Contribution of Neutral Push–Pull Organic Molecules towards Superbases in the Gas Phase. International Journal of Molecular Sciences. 2024; 25(11):5591. https://doi.org/10.3390/ijms25115591
Chicago/Turabian StyleRaczyńska, Ewa Daniela, Jean-François Gal, and Pierre-Charles Maria. 2024. "Strong Bases and beyond: The Prominent Contribution of Neutral Push–Pull Organic Molecules towards Superbases in the Gas Phase" International Journal of Molecular Sciences 25, no. 11: 5591. https://doi.org/10.3390/ijms25115591