
Unlocking Glioblastoma Vulnerabilities with CRISPR-Based Genetic Screening

Abstract
:1. Introduction
2. Therapy Resistance in GBM
3. CRISPR/Cas9-Based Genome Engineering Tools
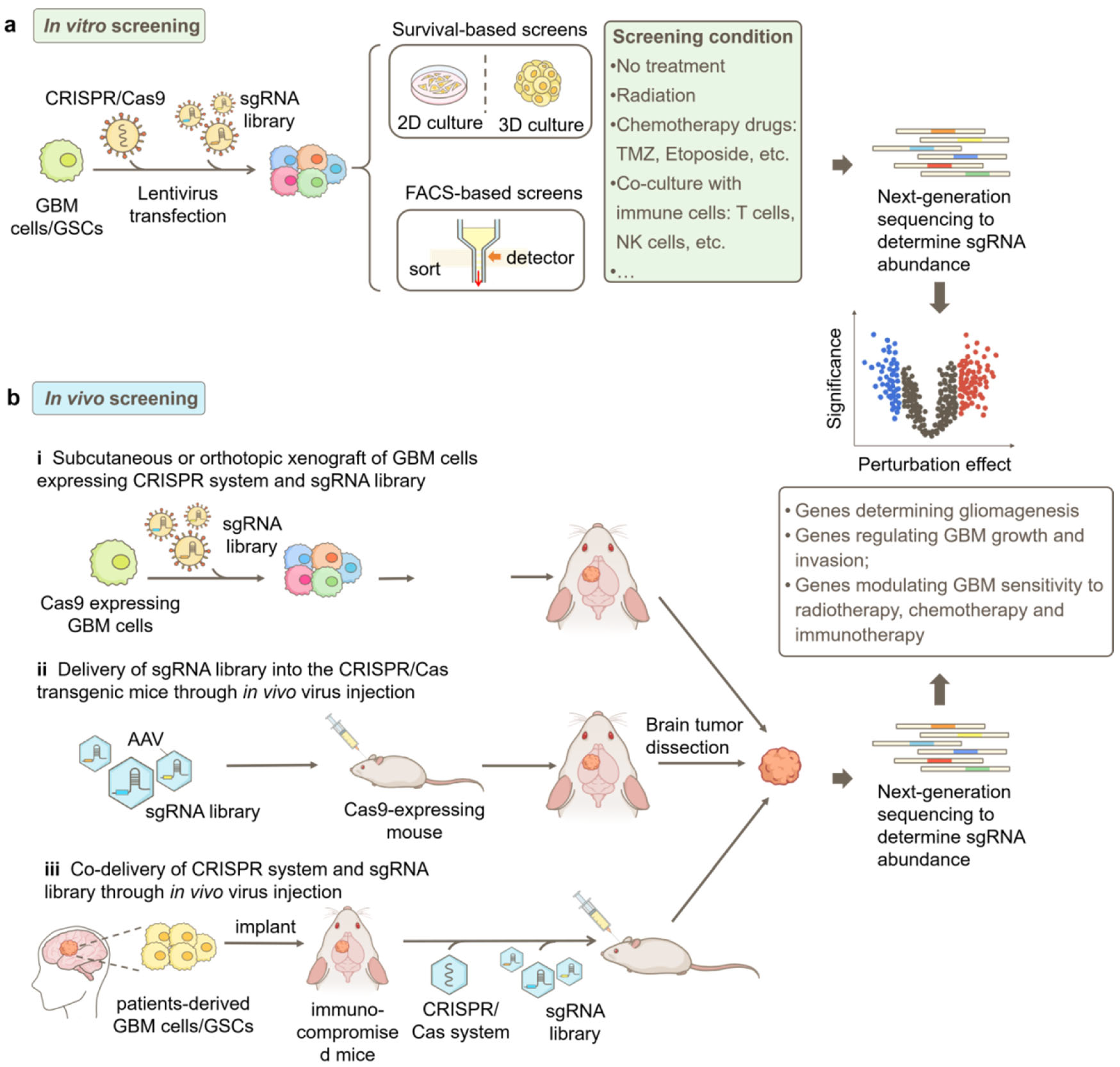
4. CRISPR/Cas9-Based Functional Genomics Screening Strategies in GBM
5. CRISPR/Cas9-Based Genetic Screening in GBM
5.1. GBM Progression
5.2. Responsiveness to Radiotherapy
5.3. Responsiveness to Chemotherapy
5.4. Responsiveness to Immunotherapy
6. Discussion and Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| CAR T | chimeric antigen receptor T |
| Chek2 | checkpoint kinase 2 |
| CRiNCL | CRISPRi Non-Coding Library |
| CRISPR | Regularly Interspaced Short Palindromic Repeats |
| CRISPRa | CRISPR activation |
| CRISPRi | CRISPR interference |
| CTH | cystathionine gamma-lyase |
| dCas9 | inactive Cas9, dead Cas9 |
| DDR | DNA damage response |
| DSBs | double-strand breaks |
| EpiDoKOL | Epigenetic Domain-specific Knockout Library |
| FACS | fluorescence-activated cell sorting |
| GA-amide | gambogic amide |
| GBM | Glioblastoma, Glioblastoma multiforme |
| GeCKO | genome-scale CRISPR-Cas9 knockout |
| GSCs | Glioblastoma stem cells, GBM stem-like cells |
| HR | homologous recombination |
| KD | knockdown |
| KO | knockout |
| lncGRS | lncRNA Glioma Radiation Sensitizers |
| lncRNAs | long non-coding RNAs |
| MGMT | methyl guanine methyl transferase |
| mTSG | mouse homolog tumor suppressor gene |
| nCas9 | Cas9 nickase |
| NGS | Next-generation sequencing |
| NHEJ | non-homologous end joining |
| NSCs | neural stem cells |
| PDX | patient-derived xenograft |
| pegRNA | prime editing guide RNA |
| RNAi | RNA interference |
| saRNASmall | activating RNA |
| sgRNA | single guide RNA |
| shRNA | short hairpin RNA |
| siRNA | small interfering RNA |
| TERT | telomerase reverse transcriptase |
| TMZ | temozolomide |
| TPMs | telomerase reverse transcriptase promoter mutations |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
| WDR1 | WD repeat domain 1 |
References
- Yabo, Y.A.; Niclou, S.P.; Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro Oncol. 2022, 24, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, C.; Ge, H.; Lin, Y.; Kang, D. Glioblastoma vaccine tumor therapy research progress. Chin. Neurosurg. J. 2022, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, S.; Sahu, A.K.; Baghel, P.; Saha, S. Current promising treatment strategy for glioblastoma multiform: A review. Oncol. Rev. 2019, 13, 417. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.Z.; Kim, C.Y.; Lim, D.H. The Overview of Practical Guidelines for Gliomas by KSNO, NCCN, and EANO. Brain Tumor Res. Treat. 2022, 10, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Immanuel, S.R.C.; Ghanate, A.D.; Parmar, D.S.; Yadav, R.; Uthup, R.; Panchagnula, V.; Raghunathan, A. Integrated genetic and metabolic landscapes predict vulnerabilities of temozolomide resistant glioblastoma cells. NPJ Syst. Biol. Appl. 2021, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Przybyla, L.; Gilbert, L.A. A new era in functional genomics screens. Nat. Rev. Genet. 2022, 23, 89–103. [Google Scholar] [CrossRef]
- Kulkarni, S.; Goel-Bhattacharya, S.; Sengupta, S.; Cochran, B.H. A Large-Scale RNAi Screen Identifies SGK1 as a Key Survival Kinase for GBM Stem Cells. Mol. Cancer Res. 2018, 16, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Sa, J.K.; Yoon, Y.; Kim, M.; Kim, Y.; Cho, H.J.; Lee, J.K.; Kim, G.S.; Han, S.; Kim, W.J.; Shin, Y.J.; et al. In vivo RNAi screen identifies NLK as a negative regulator of mesenchymal activity in glioblastoma. Oncotarget 2015, 6, 20145–20159. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Mao, A.; Xu, M.; Weng, Q.; Mao, J.; Ji, J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019, 447, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Annovazzi, L.; Mellai, M.; Schiffer, D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers 2017, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.; Kroon, P.; Zhang, Y.; Galan-Ganga, M.; Bao, Z.; Wang, Z.; Liu, H.; Sa, J.K.; et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 2020, 11, 3883. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.-H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro-Oncol. 2019, 21, 167–178. [Google Scholar] [CrossRef]
- Immanuel, S.R.C.; Ghanate, A.D.; Parmar, D.S.; Marriage, F.; Panchagnula, V.; Day, P.J.; Raghunathan, A. Integrative analysis of rewired central metabolism in temozolomide resistant cells. Biochem. Biophys. Res. Commun. 2018, 495, 2010–2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Prager, B.C.; Gimple, R.C.; Aguilar, B.; Alizadeh, D.; Tang, H.; Lv, D.; Starr, R.; Brito, A.; Wu, Q.; et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021, 11, 1192–1211. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhao, L.; Shen, J.Z.; Liang, Z.; Wu, Q.; Yang, K.; Min, L.; Gimple, R.C.; Yang, Q.; Bhargava, S.; et al. Transcription Elongation Machinery Is a Druggable Dependency and Potentiates Immunotherapy in Glioblastoma Stem Cells. Cancer Discov. 2022, 12, 502–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Prager, B.C.; Wu, Q.L.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.L.; Morton, A.R.; Zhou, W.C.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qi, L.S. A CRISPR-dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Braun, C.J.; Saur, D.; Rad, R. In vivo functional screening for systems-level integrative cancer genomics. Nat. Rev. Cancer 2020, 20, 573–593. [Google Scholar] [CrossRef]
- Bak, R.O.; Gomez-Ospina, N.; Porteus, M.H. Gene Editing on Center Stage. Trends Genet. 2018, 34, 600–611. [Google Scholar] [CrossRef]
- Makarova, K.S.; Zhang, F.; Koonin, E.V. SnapShot: Class 2 CRISPR-Cas Systems. Cell 2017, 168, 328.e1. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Ouyang, M.; Zhan, J.; Tian, R. CRISPR-based functional genomics screening in human-pluripotent-stem-cell-derived cell types. Cell Genom. 2023, 3, 100300. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPR-based functional genomics for neurological disease. Nat. Rev. Neurol. 2020, 16, 465–480. [Google Scholar] [CrossRef]
- Tian, R.; Gachechiladze, M.A.; Ludwig, C.H.; Laurie, M.T.; Hong, J.Y.; Nathaniel, D.; Prabhu, A.V.; Fernandopulle, M.S.; Patel, R.; Abshari, M.; et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 2019, 104, 239–255.e12. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Malatesta, M.; Lien, B.V.; Saha, P.; Thombare, S.S.; Hong, S.J.; Pedraza, L.; Koontz, M.; Seo, K.; Horlbeck, M.A.; et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 2020, 21, 83. [Google Scholar] [CrossRef] [PubMed]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Wang, X.; Liu, Y.; Liu, N.; Li, Y.; Luo, J.; Ma, Q.; Wu, D.; Li, J.; Xu, C.; et al. Programmable A-to-Y base editing by fusing an adenine base editor with an N-methylpurine DNA glycosylase. Nat. Biotechnol. 2023, 41, 1080–1084. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kasinski, A.L. In Vivo Cancer-Based Functional Genomics. Trends Cancer 2020, 6, 1002–1017. [Google Scholar] [CrossRef]
- Chow, R.D.; Chen, S. Cancer CRISPR Screens In Vivo. Trends Cancer 2018, 4, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.D.; Guzman, C.D.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.A.; Zhang, S.; et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017, 20, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Xie, Q.; Gimple, R.C.; Zhong, Z.; Tam, T.; Tian, J.; Kidwell, R.L.; Wu, Q.; Prager, B.C.; Qiu, Z.; et al. Three-dimensional bioprinted glioblastoma microenvironments model cellular dependencies and immune interactions. Cell Res. 2020, 30, 833–853. [Google Scholar] [CrossRef]
- Ozyerli-Goknar, E.; Kala, E.Y.; Aksu, A.C.; Bulut, I.; Cingoz, A.; Nizamuddin, S.; Biniossek, M.; Seker-Polat, F.; Morova, T.; Aztekin, C.; et al. Epigenetic-focused CRISPR/Cas9 screen identifies (absent, small, or homeotic)2-like protein (ASH2L) as a regulator of glioblastoma cell survival. Cell Commun. Signal 2023, 21, 328. [Google Scholar] [CrossRef]
- Humphreys, L.M.; Smith, P.; Chen, Z.; Fouad, S.; D’Angiolella, V. The role of E3 ubiquitin ligases in the development and progression of glioblastoma. Cell Death Differ. 2021, 28, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Attenello, F.J.; Tsung, K.; Bishara, I.; Loh, Y.H.E.; Chen, T.C. In vivo CRISPR screening for novel noncoding RNA functional targets in glioblastoma models. J. Neurosci. Res. 2021, 99, 2029–2045. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wei, Y.; Zhang, Q.; Sun, M.; Wang, Y.; Hou, J.; Zhang, P.; Lv, X.; Su, D.; Jiang, Y.; et al. Multiomics analyses reveal DARS1-AS1/YBX1–controlled posttranscriptional circuits promoting glioblastoma tumorigenesis/radioresistance. Sci. Adv. 2023, 9, eadf3984. [Google Scholar] [CrossRef]
- Verdugo, E.; Puerto, I.; Medina, M.A. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun. 2022, 42, 1083–1111. [Google Scholar] [CrossRef]
- Prolo, L.M.; Li, A.; Owen, S.F.; Parker, J.J.; Foshay, K.; Nitta, R.T.; Morgens, D.W.; Bolin, S.; Wilson, C.M.; Vega L, J.C.M.; et al. Targeted genomic CRISPR-Cas9 screen identifies MAP4K4 as essential for glioblastoma invasion. Sci. Rep. 2019, 9, 14020. [Google Scholar] [CrossRef]
- Garcia, J.H.; Akins, E.A.; Jain, S.; Wolf, K.J.; Zhang, J.; Choudhary, N.; Lad, M.; Shukla, P.; Rios, J.; Seo, K.; et al. Multiomic screening of invasive GBM cells reveals targetable transsulfuration pathway alterations. J. Clin. Investig. 2023, 134, e170397. [Google Scholar] [CrossRef]
- Dresser, L.; Wlodarski, R.; Rezania, K.; Soliven, B. Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations. J. Clin. Med. 2021, 10, 2235. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.J.; Stewart, C.E.; Hendrickson, P.G.; Regal, J.A.; Kim, S.Y.; Ashley, D.M.; Waitkus, M.S.; Reitman, Z.J. Pooled genetic screens to identify vulnerabilities in TERT-promoter-mutant glioblastoma. Oncogene 2023, 42, 3274–3286. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef]
- Zhu, G.D.; Yu, J.; Sun, Z.Y.; Chen, Y.; Zheng, H.M.; Lin, M.L.; Ou-Yang, S.; Liu, G.L.; Zhang, J.W.; Shao, F.M. Genome-wide CRISPR/Cas9 screening identifies CARHSP1 responsible for radiation resistance in glioblastoma. Cell Death Dis. 2021, 12, 724. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Reily Rocha, A.; Molina Silva, M.; Rodrigues Gomes, L.; Teatin Latancia, M.; Andrade Tomaz, M.; de Souza, I.; Karolynne Seregni Monteiro, L.; Menck, C.F.M. Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells 2020, 9, 2573. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liu, X.; Li, Y.; Wang, Q.; Zhou, J.; Wang, Y.; Dong, F.; Yang, C.; Sun, Z.; Fang, C.; et al. Genome-Wide CRISPR-Cas9 Screening Identifies NF-kappaB/E2F6 Responsible for EGFRvIII-Associated Temozolomide Resistance in Glioblastoma. Adv. Sci. 2019, 6, 1900782. [Google Scholar] [CrossRef]
- Tong, F.; Zhao, J.-X.; Fang, Z.-Y.; Cui, X.-T.; Su, D.-Y.; Liu, X.; Zhou, J.-H.; Wang, G.-X.; Qiu, Z.-J.; Liu, S.-Z.; et al. MUC1 promotes glioblastoma progression and TMZ resistance by stabilizing EGFRvIII. Pharmacol. Res. 2023, 187, 106606. [Google Scholar] [CrossRef]
- Cheng, X.; An, J.; Lou, J.; Gu, Q.; Ding, W.; Droby, G.N.; Wang, Y.; Wang, C.; Gao, Y.; Anand, J.R.; et al. Trans-lesion synthesis and mismatch repair pathway crosstalk defines chemoresistance and hypermutation mechanisms in glioblastoma. Nat. Commun. 2024, 15, 1957. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Awah, C.U.; Chen, L.; Bansal, M.; Mahajan, A.; Winter, J.; Lad, M.; Warnke, L.; Gonzalez-Buendia, E.; Park, C.; Zhang, D.; et al. Ribosomal protein S11 influences glioma response to TOP2 poisons. Oncogene 2020, 39, 5068–5081. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Liu, X.; Zhang, W.; Zhang, K.; Pan, L.; Zhu, M.; Qin, H.; Zou, C.; Wang, W.; Zhang, C.; et al. Biomimetic Macrophage Membrane-Camouflaged Nanoparticles Induce Ferroptosis by Promoting Mitochondrial Damage in Glioblastoma. ACS Nano 2023, 17, 23746–23760. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Qiu, B.; Zhang, Y.; Hu, Y.; Wang, Z.; Guan, Z.; Qin, Y.; Sui, T.; Wu, F.; Li, B.; et al. The tumor-enriched small molecule gambogic amide suppresses glioma by targeting WDR1-dependent cytoskeleton remodeling. Signal Transduct. Target. Ther. 2023, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Kann, M.C.; Bailey, S.R.; Haradhvala, N.J.; Llopis, P.M.; Bouffard, A.A.; Scarfó, I.; Leick, M.B.; Grauwet, K.; Berger, T.R.; et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature 2022, 604, 563–570. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Bernareggi, D.; Xie, Q.; Prager, B.C.; Yun, J.; Cruz, L.S.; Pham, T.V.; Kim, W.; Lee, X.; Coffey, M.; Zalfa, C.; et al. CHMP2A regulates tumor sensitivity to natural killer cell-mediated cytotoxicity. Nat. Commun. 2022, 13, 1899. [Google Scholar] [CrossRef]
- Mittrücker, H.W.; Visekruna, A.; Huber, M. Heterogeneity in the differentiation and function of CD8+ T cells. Arch. Immunol. Ther. Exp. 2014, 62, 449–458. [Google Scholar] [CrossRef]
- Dmello, C.; Zhao, J.; Chen, L.; Gould, A.; Castro, B.; Arrieta, V.A.; Zhang, D.Y.; Kim, K.-S.; Kanojia, D.; Zhang, P.; et al. Checkpoint kinase 1/2 inhibition potentiates anti-tumoral immune response and sensitizes gliomas to immune checkpoint blockade. Nat. Commun. 2023, 14, 1566. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, A.; Maus, M.V. Making CAR T Cells a Solid Option for Solid Tumors. Front. Immunol. 2018, 9, 2593. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, S.; Zhang, W.; Liang, Z.; Fang, Y.; Sun, T.; Wan, Y.; Ma, X.; Zhang, S.; Xu, Y.; et al. Identification of genetic modifiers enhancing B7-H3-targeting CAR T cell therapy against glioblastoma through large-scale CRISPRi screening. J. Exp. Clin. Cancer Res. 2024, 43, 95. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Shen, S.-H.; Lu, F.; Zheng, P.; Wu, S.; Liao, J.; Jiang, X.; Zeng, G.; Wei, D. CRISPR screening of E3 ubiquitin ligases reveals Ring Finger Protein 185 as a novel tumor suppressor in glioblastoma repressed by promoter hypermethylation and miR-587. J. Transl. Med. 2022, 20, 96. [Google Scholar] [CrossRef]
- Serra, R.; Mangraviti, A.; Gorelick, N.L.; Shapira-Furman, T.; Alomari, S.; Cecia, A.; Darjee, N.; Brem, H.; Rottenberg, Y.; Domb, A.J.; et al. Combined intracranial Acriflavine, temozolomide and radiation extends survival in a rat glioma model. Eur. J. Pharm. Biopharm. 2022, 170, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Phenotype | Cell Model | CRISPR System | Screening Condition | sgRNA Library | Main Findings | Ref. |
|---|---|---|---|---|---|---|
| GBM tumorigenesis | astrocytes | CRISPRn | AAV delivery of sgRNA library into the brain of LSL-Cas9 transgenic mice | mTSG library: 286 sgRNAs targeting 56 tumor suppressor genes | determined mutational profiles in GBM tumorigenesis; identified co-occurring driver combinations like B2m-Nf1 and Zc3h13-Rb1 | [42] |
| GBM growth | U87 | CRISPRn | normal condition, without treatment | Custom library: 557 sgRNA targeting 557 E3 ligase genes | identified RNF185 as a tumor suppressor regulated by miR-587 | [75] |
| U87 | CRISPRi | implanted in the brain of NU/J mice | CRinCL—Unique to U87: 23,317 sgRNAs targeting 2307 lncRNAs | identified 17 lncRNA essential for GBM growth in vivo | [48] | |
| U87, U251 | CRISPRi | normal condition, without treatment | Custom library: 9083 sgRNAs targeting 1209 lncRNAs dysregulated in GBM cells | DARS1-AS1 promotes GBM growth through interaction with YBX1 | [49] | |
| T98G-TERT-ON GBM cells | CRISPRn | normal condition, without treatment | Custom Library: sgRNAs targeting AAVS1, TERT and GABPB1L | did not detect genetic vulnerabilities specific to GBM carrying TERT promoter mutations (TPMs) | [54] | |
| T98G, U373 | CRISPRn | normal condition, without treatment | EpiDoKOL: 1628 sgRNA targeting 251 chromatin modifiers | ASH2L is essential for GBM cell survival by regulating cell cycle, transcription, and histone methylation through interactions with histone methyltransferases | [46] | |
| patient-derived GSCs, NSCs | CRISPRn | normal condition, without treatment | GeCKO library: 64,751 sgRNAs targeting 18,080 genes | knockout of PKMYT1 specifically inhibits the growth of GSCs by impairing cell division | [43] | |
| patient-derived GSCs | CRISPRn | normal condition, without treatment | Custom library: targeting 160 chromatin regulator genes from ChIP-seq profiling | knockout of YY1 inhibits the proliferation and self-renewal of GSCs by controlling transcription and m6A modification | [24] | |
| patient-derived GSCs, NSCs | CRISPRn | normal condition, without treatment | TKOv1/TKOv3 library: 70,948 sgRNAs targeting 18,053 genes | identified transcription factors SOX2 and SOX9, histone methyltransferase DOT1L, and cytokine signaling suppressor SOCS3 as important regulators of GSC stemness and fitness | [44] | |
| patient-derived GSCs | CRISPRn | cultured as spheres or in the 3D bioprinted tissue model | Brunello library: 76,441 sgRNAs targeting 19,114 genes | identified PAG1, ZNF830, ATP5H, RNF19A as essential genes of GSCs | [45] | |
| GBM invasion | U138 | CRISPRn | cultured in a transwell system | Custom library: 45,740 sgRNAs targeting 4574 genes relative to cell motility and drug targets | knockout of MAP4K4 reduces invasion and inhibits mesenchymal transition of GBM cells | [51] |
| patient-derived GSCs | CRISPRn | cultured in 3D hydrogel invasion devices | Custom library: 29,790 sgRNAs targeting 2981 metabolic genes | knockdown or inhibition of CTH impaired GBM invasion in vitro and in vivo and caused cysteine deficiency and ROS accumulation | [52] | |
| GBM responsiveness to radiotherapy | U87 | CRISPRi | treated with radiation | CRiNC-U87 and HEK293T and CRiNCL-Unique to U87:38,011 sgRNAs targeting 3750 lncRNAs | knockdown of lncGRS-1(CTC-338 M12.4) selectively inhibits GBM growth and enhances GBM sensitivity to radiation | [36] |
| U87, U251 | CRISPRa | treated with radiation | SAM library: 70,290 sgRNAs targeting 23,430 genes | CARHSP1 enhances radiation resistance in GBM via TNF-α/NF-kβ pathway | [56] | |
| GBM responsiveness to chemotherapy | U138 | CRISPRn | treated with TMZ | GeCKO v2 library: 123,411 sgRNAs targeting 19,050 genes | knockout of MSH2, MSH6, CLCA2 or PTCH2 enhances TMZ resistance | [57] |
| U138 | CRISPRa | treated with TMZ | SAM library: 70,290 sgRNAs targeting 23,430 genes | NRF2 enhances TMZ resistance by controlling the expression of enzymes in GSH synthesis | [57] | |
| RAD18+/+ and RAD18−/− U373 cells | CIRSPRn | treated with TMZ | DDR-CRISPR lentivirus library: 5040 sgRNAs targeting 504 DDR genes | knockout of POLD3 leads to greater TMZ sensitivity in RAD18-deficient GBM cells | [60] | |
| WT and EGFRvIII U87 cells | CRISPRn | treated with TMZ | GeCKO v2: library123,411 sgRNAs targeting 19,050 genes | E2F6 enhances TMZ resistance by promoting DNA repair | [58] | |
| WT and EGFRvIII U87 cells | CRISPRn | treated with TMZ | GeCKO v2 library: 123,411 sgRNAs targeting 19,050 genes | MUC1 enhances TMZ resistance by regulating DSB repair and autophagy | [59] | |
| patient-derived GSCs | CRISPRn | treated with TMZ | TKOv1/TKOv3 library: 70,948 sgRNAs targeting 18,053 genes | Knockout of genes in the MMR pathway, including MLH1, MSH2, MSH6, and PMS2, leads to TMZ resistance in GSCs; knockout of genes in the FA and HR pathways (such as FANCA, MCM8, and MCM9) sensitizes GSCs to TMZ | [44] | |
| LN229 | CRISPRn | treated with RSL3 | GeCKO v2 library: 123,411 sgRNAs targeting 19,050 genes | identified ALOX15 as an essential driver of ferroptosis in GBM | [63] | |
| SNB19, U251, patient-derived GBM cells | CRISPRn | treated with etoposide | Brunello Library: 76,441 sgRNAs targeting 19,114 genes | knockout of RPS11 reduces GBM responsiveness to etoposide by impairing the induction of the pro-apoptotic gene APAF1 | [62] | |
| patient-derived GSCs | CRISPRn | treated with Gambogic amide (GA-amide) | Brunello Library: 76,441 sgRNAs targeting 19,114 genes | WDR1 is the direct binding target of GA-amide, a potential new chemotherapy drug for GBM | [64] | |
| GBM responsiveness to T cell cytotoxicity | U87 | CRISPRn | co-cultured with EGFR-targeting CAR T cells | Brunello library: 76,441 sgRNAs targeting 19,114 genes | knockout of genes in the IFNγ signaling pathway, including IFNGR1, JAK1 and JAK2, induces GBM resistance to CAR T cell cytotoxicity | [66] |
| GBM responsiveness to T cell cytotoxicity | patient-derived GSCs | CRISPRn | co-cultured with IL13Rα2-targeting CAR T cells | Brunello library: 76,441 sgRNAs targeting 19,114 genes | knockout of RELA or NPLOC4 sensitizes GBM to CAR T cell-mediated killing | [23] |
| U87, U251 and T98G | CRISPRi | co-cultured with B7-H3 targeting CAR T cells | H1 library: 13,025 sgRNAs targeting 2318 genes of kinases, phosphatases, and drug target | knockdown of ARPC4 or NDUFV1 in GBM cells enhances their killing by CAR T cells by activating TNFSF15-mediated cytokine signaling pathways. | [73] | |
| GL261 | CRISPRn | implanted in WT and CD8 KO mice | Brie kinome KO library: 2856 sgRNA targeting 714 kinases | identified Chek2 as the most important kinase mediating GBM resistance to CD8+ T cell killing | [71] | |
| GBM responsiveness to NK cell cytotoxicity | patient-derived GSCs | CRISPRn | co-cultured with NK cells | Brunello library: 76,441 sgRNAs targeting 19,114 genes | knockout of CHMP2A in GSCs sensitizes them to NK cells by activating NF-κB signaling and increasing the secretion of chemokines like CXCL10 and CXCL12 | [69] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Y.; Li, X.; Tian, R. Unlocking Glioblastoma Vulnerabilities with CRISPR-Based Genetic Screening. Int. J. Mol. Sci. 2024, 25, 5702. https://doi.org/10.3390/ijms25115702
Fang Y, Li X, Tian R. Unlocking Glioblastoma Vulnerabilities with CRISPR-Based Genetic Screening. International Journal of Molecular Sciences. 2024; 25(11):5702. https://doi.org/10.3390/ijms25115702
Chicago/Turabian StyleFang, Yitong, Xing Li, and Ruilin Tian. 2024. "Unlocking Glioblastoma Vulnerabilities with CRISPR-Based Genetic Screening" International Journal of Molecular Sciences 25, no. 11: 5702. https://doi.org/10.3390/ijms25115702