


From Immunity to Neurogenesis: Toll-like Receptors as Versatile Regulators in the Nervous System
 ,
,
Abstract
:1. Introduction
2. TLR Expression and Function in Glial Cells and Neurons
2.1. Microglia
2.2. Astrocytes
2.3. Oligodendrocytes
2.4. Neurons
3. Role of TLRs in the Blood–Brain Barrier
3.1. Brain Endothelial Cells
3.2. Pericytes
3.3. End-Feet of Perivascular Astrocytes
3.4. Other Cell Types of the NVU
4. Regulation of Neurogenesis by TLRs
5. Effects of TLR Misregulation on Behavior and Cognitive Function
6. Involvement of TLRs in Neuroinflammation and Neurodegeneration
6.1. TLRs in Bacterial and Viral Infections of the Nervous System
6.2. Neurodegenerative Diseases and TLRs
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Su, S.B.; Tao, L.; Deng, Z.P.; Chen, W.; Qin, S.Y.; Jiang, H.X. TLR10: Insights, Controversies and Potential Utility as a Therapeutic Target. Scand. J. Immunol. 2021, 93, e12988. [Google Scholar] [CrossRef] [PubMed]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like Receptors: Significance, Ligands, Signaling Pathways, and Functions in Mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Anthoney, N.; Foldi, I.; Hidalgo, A. Toll and Toll-like Receptor Signalling in Development. Development 2018, 145, 156018. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, S.; Tapping, R.I. Toll-like Receptor Signaling in Cell Proliferation and Survival. Cytokine 2010, 49, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Starkey, J.; Horstick, E.J.; Ackerman, S.D. Glial Regulation of Critical Period Plasticity. Front. Cell. Neurosci. 2023, 17, 1247335. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Ravid, R.; Gveric, D.; Van Noort, J.M. Broad Expression of Toll-like Receptors in the Human Central Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Mishra, M.K.; Rawji, K.S.; Keough, M.B.; Kappen, J.; Dowlatabadi, R.; Vogel, H.J.; Chopra, S.; Distéfano-Gagné, F.; Dufour, A.; Gosselin, D.; et al. Harnessing the Benefits of Neuroinflammation: Generation of Macrophages/Microglia with Prominent Remyelinating Properties. J. Neurosci. 2021, 41, 3366–3385. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Li, D.; Zhou, Q.; Hu, X. Mitochondria-Targeted TPP-MoS2 with Dual Enzyme Activity Provides Efficient Neuroprotection through M1/M2 Microglial Polarization in an Alzheimer’s Disease Model. Biomaterials 2020, 232, 119752. [Google Scholar] [CrossRef] [PubMed]
- Catania, G.; Rodella, G.; Vanvarenberg, K.; Préat, V.; Malfanti, A. Combination of Hyaluronic Acid Conjugates with Immunogenic Cell Death Inducer and CpG for Glioblastoma Local Chemo-Immunotherapy Elicits an Immune Response and Induces Long-Term Survival. Biomaterials 2023, 294, 122006. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Nam, H.; Kim, L.E.; Jeon, Y.; Min, H.; Ha, S.; Lee, Y.; Kim, S.Y.; Lee, S.J.; Kim, E.K.; et al. TLR4 (Toll-like Receptor 4) Activation Suppresses Autophagy through Inhibition of FOXO3 and Impairs Phagocytic Capacity of Microglia. Autophagy 2019, 15, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Ifuku, M.; Hinkelmann, L.; Kuhrt, L.D.; Efe, I.E.; Kumbol, V.; Buonfiglioli, A.; Krüger, C.; Jordan, P.; Fulde, M.; Noda, M.; et al. Activation of Toll-like Receptor 5 in Microglia Modulates Their Function and Triggers Neuronal Injury. Acta Neuropathol. Commun. 2020, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglioli, A.; Efe, I.E.; Guneykaya, D.; Ivanov, A.; Huang, Y.; Orlowski, E.; Krüger, C.; Deisz, R.A.; Markovic, D.; Flüh, C.; et al. Let-7 MicroRNAs Regulate Microglial Function and Suppress Glioma Growth through Toll-Like Receptor 7. Cell Rep. 2019, 29, 3460–3471.e7. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Kim, J.Y. 1,25-Dihydroxyvitamin D3 Facilitates M2 Polarization and Upregulates TLR10 Expression on Human Microglial Cells. Neuroimmunomodulation 2016, 23, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and Therapeutic Value of Astrocytes in Neurological Diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef] [PubMed]
- Rupareliya, V.P.; Singh, A.A.; Butt, A.M.; Hariharan, A.; Kumar, H. The “Molecular Soldiers” of the CNS: Astrocytes, a Comprehensive Review on Their Roles and Molecular Signatures. Eur. J. Pharmacol. 2023, 959, 176048. [Google Scholar] [CrossRef]
- Farina, C.; Krumbholz, M.; Giese, T.; Hartmann, G.; Aloisi, F.; Meinl, E. Preferential Expression and Function of Toll-like Receptor 3 in Human Astrocytes. J. Neuroimmunol. 2005, 159, 12–19. [Google Scholar] [CrossRef]
- Deng, S.; Chen, X.; Lei, Q.; Lu, W. AQP2 Promotes Astrocyte Activation by Modulating the TLR4/NFκB-P65 Pathway following Intracerebral Hemorrhage. Front. Immunol. 2022, 13, 847360. [Google Scholar] [CrossRef]
- Li, L.; Ni, L.; Eugenin, E.A.; Heary, R.F.; Elkabes, S. Toll-like Receptor 9 Antagonism Modulates Astrocyte Function and Preserves Proximal Axons following Spinal Cord Injury. Brain Behav. Immun. 2019, 80, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ni, L.; Heary, R.F.; Elkabes, S. Astroglial TLR9 Antagonism Promotes Chemotaxis and Alternative Activation of Macrophages via Modulation of Astrocyte-Derived Signals: Implications for Spinal Cord Injury. J. Neuroinflamm. 2020, 17, 73. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, B.; Baron-Van Evercooren, A. Schwann Cells: Rescuers of Central Demyelination. Glia 2020, 68, 1945–1956. [Google Scholar] [CrossRef]
- Sloane, J.A.; Batt, C.; Ma, Y.; Harris, Z.M.; Trapp, B.; Vartanian, T. Hyaluronan Blocks Oligodendrocyte Progenitor Maturation and Remyelination through TLR2. Proc. Natl. Acad. Sci. USA 2010, 107, 11555–11560. [Google Scholar] [CrossRef] [PubMed]
- Gyetvai, G.; Roe, C.; Heikal, L.; Ghezzi, P.; Mengozzi, M. Leukemia Inhibitory Factor Inhibits Erythropoietin-Induced Myelin Gene Expression in Oligodendrocytes. Mol. Med. 2018, 24, 51. [Google Scholar] [CrossRef] [PubMed]
- Esser, S.; Göpfrich, L.; Bihler, K.; Kress, E.; Nyamoya, S.; Tauber, S.C.; Clarner, T.; Stope, M.B.; Pufe, T.; Kipp, M.; et al. Toll-Like Receptor 2-Mediated Glial Cell Activation in a Mouse Model of Cuprizone-Induced Demyelination. Mol. Neurobiol. 2018, 55, 6237–6249. [Google Scholar] [CrossRef] [PubMed]
- Boccazzi, M.; Van Steenwinckel, J.; Schang, A.L.; Faivre, V.; Le Charpentier, T.; Bokobza, C.; Csaba, Z.; Verderio, C.; Fumagalli, M.; Mani, S.; et al. The Immune-Inflammatory Response of Oligodendrocytes in a Murine Model of Preterm White Matter Injury: The Role of TLR3 Activation. Cell Death Dis. 2021, 12, 166. [Google Scholar] [CrossRef]
- Göttle, P.; Schichel, K.; Reiche, L.; Werner, L.; Zink, A.; Prigione, A.; Küry, P. TLR4 Associated Signaling Disrupters as a New Means to Overcome HERV-W Envelope-Mediated Myelination Deficits. Front. Cell. Neurosci. 2021, 15, 777542. [Google Scholar] [CrossRef]
- Ved, R.; Sharouf, F.; Harari, B.; Muzaffar, M.; Manivannan, S.; Ormonde, C.; Gray, W.P.; Zaben, M. Disulfide HMGB1 Acts via TLR2/4 Receptors to Reduce the Numbers of Oligodendrocyte Progenitor Cells after Traumatic Injury in Vitro. Sci. Rep. 2021, 11, 6181. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liang, J.; Cui, M.; Zhang, L.; Ren, S.; Zheng, W.; Dong, X.; Zhang, B. Saturated Fatty Acids Activate the Inflammatory Signalling Pathway in Schwann Cells: Implication in Sciatic Nerve Injury. Scand. J. Immunol. 2020, 92, e12896. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Cheng, Y.; Wang, Y.; Wu, J.; Rong, Z.; Sun, L.; Zhou, Y.; Zhang, K. Involvement of Abnormal P-α-Syn Accumulation and TLR2-Mediated Inflammation of Schwann Cells in Enteric Autonomic Nerve Dysfunction of Parkinson’s Disease: An Animal Model Study. Mol. Neurobiol. 2023, 60, 4738–4752. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Jiang, W.W.; Wang, Y.; Yuan, Y.S.; Rong, Z.; Wu, J.; Fan, Y.; Lu, M.; Zhang, K.Z. Phosphorylated α-Synuclein Aggregated in Schwann Cells Exacerbates Peripheral Neuroinflammation and Nerve Dysfunction in Parkinson’s Disease through TLR2/NF-ΚB Pathway. Cell Death Discov. 2021, 7, 289. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Di Liegro, C.M.; Di Liegro, I. Cell-to-Cell Communication in Learning and Memory: From Neuro- and Glio-Transmission to Information Exchange Mediated by Extracellular Vesicles. Int. J. Mol. Sci. 2019, 21, 266. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.X.; Liu, S.Y.; Zheng, X.; Chen, X.; Lu, L.X.; Chen, B.; Xiong, X.Y.; Shu, H.F.; Yang, Q.W.; Yang, H. TLR1 Expression in Mouse Brain Was Increased in a KA-Induced Seizure Model. Inflamm. Res. 2015, 64, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Dong, G.J.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal Role for Neuronal Toll-like Receptors in Ischemic Brain Injury and Functional Deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef] [PubMed]
- Casula, M.; Iyer, A.M.; Spliet, W.G.M.; Anink, J.J.; Steentjes, K.; Sta, M.; Troost, D.; Aronica, E. Toll-like Receptor Signaling in Amyotrophic Lateral Sclerosis Spinal Cord Tissue. Neuroscience 2011, 179, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Préhaud, C.; Mégret, F.; Lafage, M.; Lafon, M. Virus Infection Switches TLR-3-Positive Human Neurons To Become Strong Producers of Beta Interferon. J. Virol. 2005, 79, 12893. [Google Scholar] [CrossRef]
- Jackson, A.C.; Rossiter, J.P.; Lafon, M. Expression of Toll-like Receptor 3 in the Human Cerebellar Cortex in Rabies, Herpes Simplex Encephalitis, and Other Neurological Diseases. J. Neurovirol. 2006, 12, 229–234. [Google Scholar] [CrossRef]
- Seong, K.J.; Choi, S.; Lee, H.G.; Rhee, J.H.; Lee, J.H.; Koh, J.T.; Kim, S.H.; Choi, W.S.; Jung, J.Y.; Kim, W.J. Toll-Like Receptor 5 Promotes the Neurogenesis From Embryonic Stem Cells and Adult Hippocampal Neural Stem Cells in Mice. Stem Cells 2022, 40, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Bansode, Y.D.; Chattopadhyay, D.; Saha, B. Transcriptomic Analysis of Interferon Response in Toll-Like Receptor 2 Ligand-Treated and Herpes Simplex Virus 1-Infected Neurons and Astrocytes. Viral Immunol. 2021, 34, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Zhao, W.; Sun, L.; Zhang, R.; Zhu, S.; Xie, K.; Feng, X.; Wu, X.; Sun, Z.; et al. Food Reward Depends on TLR4 Activation in Dopaminergic Neurons. Pharmacol. Res. 2021, 169, 105659. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, L.; Tate, R.; Chamberlain, L.H.; Robertson, G.; Zagnoni, M.; Sposito, T.; Wray, S.; Wright, J.A.; Bryant, C.E.; Gay, N.J.; et al. Toll-like Receptor 3 Activation Impairs Excitability and Synaptic Activity via TRIF Signalling in Immature Rat and Human Neurons. Neuropharmacology 2018, 135, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Ying, J.; Qiu, X.; Yu, L.; Yue, Y.; Liu, Q.; Shi, J.; Li, X.; Qu, Y.; Mu, D.; et al. A New Cell Death Program Regulated by Toll-like Receptor 9 through P38 Mitogen-Activated Protein Kinase Signaling Pathway in a Neonatal Rat Model with Sepsis Associated Encephalopathy. Chin. Med. J. 2022, 135, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Han, G.; Wu, S.; Du, S.; Zhang, Y.; Liu, W.; Jiang, B.; Zhang, L.; Xia, S.; Jia, S.; et al. Toll-like Receptor 7 Contributes to Neuropathic Pain by Activating NF-ΚB in Primary Sensory Neurons. Brain Behav. Immun. 2020, 87, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ye, L.; Wan, Q.; Zhou, L.; Wang, X.; Li, J.; Hu, S.; Zhou, D.; Ho, W. Activation of Toll-like Receptors Inhibits Herpes Simplex Virus-1 Infection of Human Neuronal Cells. J. Neurosci. Res. 2009, 87, 2916–2925. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood-Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Chen, Z.; Li, G. Immune Response and Blood-Brain Barrier Dysfunction during Viral Neuroinvasion. Innate Immun. 2021, 27, 109–117. [Google Scholar] [CrossRef]
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The Blood-Brain Barrier: Structure, Regulation, and Drug Delivery. Signal Transduct. Target. Ther. 2023, 8, 217. [Google Scholar] [CrossRef]
- Halder, S.K.; Sapkota, A.; Milner, R. The Importance of Laminin at the Blood-Brain Barrier. Neural Regen. Res. 2023, 18, 2557–2563. [Google Scholar] [CrossRef] [PubMed]
- Nagyoszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.K.; Haskó, J.; Krizbai, I.A. Expression and Regulation of Toll-like Receptors in Cerebral Endothelial Cells. Neurochem. Int. 2010, 57, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Johnson, R.H.; Kho, D.T.; O’Carroll, S.J.; Angel, C.E.; Graham, E.S. The Functional and Inflammatory Response of Brain Endothelial Cells to Toll-Like Receptor Agonists. Sci. Rep. 2018, 8, 10102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, X.; Chen, X.; Wei, Y. Neuronal Injuries in Cerebral Infarction and Ischemic Stroke: From Mechanisms to Treatment (Review). Int. J. Mol. Med. 2022, 49, 15. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, G.; Harhausen, D.; Schepers, C.; Hoffmann, O.; Röhr, C.; Prinz, V.; König, J.; Lehrach, H.; Nietfeld, W.; Trendelenburg, G. TLR2 Has a Detrimental Role in Mouse Transient Focal Cerebral Ischemia. Biochem. Biophys. Res. Commun. 2007, 359, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Wang, X.; Ye, L.; Zhou, Y.; Persidsky, Y.; Ho, W. Immune Activation of Human Brain Microvascular Endothelial Cells Inhibits HIV Replication in Macrophages. Blood 2013, 121, 2934–2942. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Muñoz, I.; Compte, M.; Álvarez-Cienfuegos, A.; Álvarez-Vallina, L.; Sanz, L. Lipopolysaccharide Activates Toll-like Receptor 4 (TLR4)-Mediated NF-ΚB Signaling Pathway and Proinflammatory Response in Human Pericytes. J. Biol. Chem. 2014, 289, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Andreeva, E.R.; Eremin, I.I.; Markin, A.M.; Nadelyaeva, I.I.; Orekhov, A.N.; Melnichenko, A.A. The Role of Pericytes in Regulation of Innate and Adaptive Immunity. Biomedicines 2023, 11, 600. [Google Scholar] [CrossRef]
- Nyúl-Tóth, Á.; Kozma, M.; Nagyőszi, P.; Nagy, K.; Fazakas, C.; Haskó, J.; Molnár, K.; Farkas, A.E.; Végh, A.G.; Váró, G.; et al. Expression of Pattern Recognition Receptors and Activation of the Non-Canonical Inflammasome Pathway in Brain Pericytes. Brain Behav. Immun. 2017, 64, 220–231. [Google Scholar] [CrossRef]
- Zhang, L.; Chopp, M.; Liu, X.; Teng, H.; Tang, T.; Kassis, H.; Zhang, Z.G. Combination Therapy with VELCADE and Tissue Plasminogen Activator Is Neuroprotective in Aged Rats after Stroke and Targets MicroRNA-146a and the Toll-like Receptor Signaling Pathway. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Li, J.R.; Ou, Y.C.; Lin, S.Y.; Wang, Y.Y.; Chen, W.Y.; Hu, Y.H.; Lai, C.Y.; Chang, C.J.; Chen, C.J. Interplay of Inflammatory Gene Expression in Pericytes following Japanese Encephalitis Virus Infection. Brain Behav. Immun. 2017, 66, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Hoi, M.P.M. Emerging Roles of Astrocytes in Blood-Brain Barrier Disruption upon Amyloid-Beta Insults in Alzheimer’s Disease. Neural Regen. Res. 2023, 18, 1890. [Google Scholar] [CrossRef] [PubMed]
- Macvicar, B.A.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Hong, J.; Cho, I.H.; Jang, Y.H.; Lee, H.; Kim, D.; Yu, S.W.; Lee, S.; Lee, S.J. TLR2-Induced Astrocyte MMP9 Activation Compromises the Blood Brain Barrier and Exacerbates Intracerebral Hemorrhage in Animal Models. Mol. Brain 2015, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Xu, J.; Zheng, Y.; Wei, Y.; Zhu, X.; Lo, E.H.; Moskowitz, M.A.; Sims, J.R. High-Mobility Group Box 1 Promotes Metalloproteinase-9 Upregulation through Toll-like Receptor 4 after Cerebral Ischemia. Stroke 2010, 41, 2077–2082. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, M.; Zhang, T.; Liu, W.; Xu, H.; Mu, F.; Ren, D.; Jia, N.; Li, Z.; Ding, Y.; et al. Z-Guggulsterone Attenuates Astrocytes-Mediated Neuroinflammation after Ischemia by Inhibiting Toll-like Receptor 4 Pathway. J. Neurochem. 2018, 147, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Caso, J.R.; Pradillo, J.M.; Hurtado, O.; Lorenzo, P.; Moro, M.A.; Lizasoain, I. Toll-like Receptor 4 Is Involved in Brain Damage and Inflammation after Experimental Stroke. Circulation 2007, 115, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Wu, F.; Zhan, R.Y.; Zhou, H.J. Inflammatory Role of Microglia in Brain Injury Caused by Subarachnoid Hemorrhage. Front. Cell. Neurosci. 2022, 16, 956185. [Google Scholar] [CrossRef]
- Okada, T.; Kawakita, F.; Nishikawa, H.; Nakano, F.; Liu, L.; Suzuki, H. Selective Toll-Like Receptor 4 Antagonists Prevent Acute Blood-Brain Barrier Disruption after Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2019, 56, 976–985. [Google Scholar] [CrossRef]
- Lehnardt, S.; Lehmann, S.; Kaul, D.; Tschimmel, K.; Hoffmann, O.; Cho, S.; Krueger, C.; Nitsch, R.; Meisel, A.; Weber, J.R. Toll-like Receptor 2 Mediates CNS Injury in Focal Cerebral Ischemia. J. Neuroimmunol. 2007, 190, 28–33. [Google Scholar] [CrossRef]
- Kumar, A.; Pareek, V.; Faiq, M.A.; Ghosh, S.K.; Kumari, C. Adult Neurogenesis in Humans: A Review of Basic Concepts, History, Current Research, and Clinical Implications. Innov. Clin. Neurosci. 2019, 16, 30. [Google Scholar]
- Kempermann, G.; Gage, F.H.; Aigner, L.; Song, H.; Curtis, M.A.; Thuret, S.; Kuhn, H.G.; Jessberger, S.; Frankland, P.W.; Cameron, H.A.; et al. Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell 2018, 23, 25–30. [Google Scholar] [CrossRef]
- Gage, F.H. Neurogenesis in the Adult Brain. J. Neurosci. 2002, 22, 612. [Google Scholar] [CrossRef]
- Bergmann, O.; Spalding, K.L.; Frisén, J. Adult Neurogenesis in Humans. Cold Spring Harb. Perspect. Biol. 2015, 7, a018994. [Google Scholar] [CrossRef]
- Stiles, J.; Jernigan, T.L. The Basics of Brain Development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef]
- Zengeler, K.E.; Lukens, J.R. Innate Immunity at the Crossroads of Healthy Brain Maturation and Neurodevelopmental Disorders. Nat. Rev. Immunol. 2021, 21, 454–468. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Cardona, A.E. The Myeloid Cells of the Central Nervous System Parenchyma. Nature 2010, 468, 253–262. [Google Scholar] [CrossRef]
- Barak, B.; Feldman, N.; Okun, E. Toll-like Receptors as Developmental Tools That Regulate Neurogenesis during Development: An Update. Front. Neurosci. 2014, 8, 91968. [Google Scholar] [CrossRef]
- Sakamoto, M.; Kageyama, R.; Imayoshi, I. The Functional Significance of Newly Born Neurons Integrated into Olfactory Bulb Circuits. Front. Neurosci. 2014, 8, 87675. [Google Scholar] [CrossRef]
- Shen, Y.; Qin, H.; Chen, J.; Mou, L.; He, Y.; Yan, Y.; Zhou, H.; Lv, Y.; Chen, Z.; Wang, J.; et al. Postnatal Activation of TLR4 in Astrocytes Promotes Excitatory Synaptogenesis in Hippocampal Neurons. J. Cell Biol. 2016, 215, 719. [Google Scholar] [CrossRef]
- Hung, Y.F.; Chen, C.Y.; Li, W.C.; Wang, T.F.; Hsueh, Y.P. Tlr7 Deletion Alters Expression Profiles of Genes Related to Neural Function and Regulates Mouse Behaviors and Contextual Memory. Brain Behav. Immun. 2018, 72, 101–113. [Google Scholar] [CrossRef]
- Hui, C.W.; St-Pierre, A.; El Hajj, H.; Remy, Y.; Hébert, S.S.; Luheshi, G.N.; Srivastava, L.K.; Tremblay, M.È. Prenatal Immune Challenge in Mice Leads to Partly Sex-Dependent Behavioral, Microglial, and Molecular Abnormalities Associated with Schizophrenia. Front. Mol. Neurosci. 2018, 11, 13. [Google Scholar] [CrossRef]
- Rolls, A.; Shechter, R.; London, A.; Ziv, Y.; Ronen, A.; Levy, R.; Schwartz, M. Toll-like Receptors Modulate Adult Hippocampal Neurogenesis. Nat. Cell Biol. 2007, 9, 1081–1088. [Google Scholar] [CrossRef]
- Zhang, X.; Hei, Y.; Bai, W.; Huang, T.; Kang, E.; Chen, H.; Kong, C.; Yang, Y.; Ye, Y.; He, X. Toll-Like Receptor 2 Attenuates Traumatic Brain Injury-Induced Neural Stem Cell Proliferation in Dentate Gyrus of Rats. Neural Plast. 2020, 2020, 9814978. [Google Scholar] [CrossRef]
- Li, G.; Forero, M.G.; Wentzell, J.; Durmus, I.; Wolf, R.; Anthoney, N.; Parker, M.; Jiang, R.; Hasenauer, J.; Strausfeld, N.; et al. A Toll-Receptor Map Underlies Structural Brain Plasticity. Elife 2020, 9, e52743. [Google Scholar] [CrossRef]
- Sanchez-Petidier, M.; Guerri, C.; Moreno-Manzano, V. Toll-like Receptors 2 and 4 Differentially Regulate the Self-Renewal and Differentiation of Spinal Cord Neural Precursor Cells. Stem Cell Res. Ther. 2022, 13, 117. [Google Scholar] [CrossRef]
- Lathia, J.D.; Okun, E.; Tang, S.C.; Griffioen, K.; Cheng, A.; Mughal, M.R.; Laryea, G.; Selvaraj, P.K.; Ffrench-Constant, C.; Magnus, T.; et al. Toll-like Receptor 3 Is a Negative Regulator of Embryonic Neural Progenitor Cell Proliferation. J. Neurosci. 2008, 28, 13978–13984. [Google Scholar] [CrossRef]
- Okun, E.; Griffioen, K.; Barak, B.; Roberts, N.J.; Castro, K.; Pita, M.A.; Cheng, A.; Mughal, M.R.; Wan, R.; Ashery, U.; et al. Toll-like Receptor 3 Inhibits Memory Retention and Constrains Adult Hippocampal Neurogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15625–15630. [Google Scholar] [CrossRef]
- Connolly, M.G.; Yost, O.L.; Potter, O.V.; Giedraitis, M.E.; Kohman, R.A. Toll-like Receptor 4 Differentially Regulates Adult Hippocampal Neurogenesis in an Age- and Sex-Dependent Manner. Hippocampus 2020, 30, 958–969. [Google Scholar] [CrossRef]
- Moraga, A.; Pradillo, J.M.; Cuartero, M.I.; Hernández-Jiménez, M.; Oses, M.; Moro, M.A.; Lizasoain, I. Toll-like Receptor 4 Modulates Cell Migration and Cortical Neurogenesis after Focal Cerebral Ischemia. FASEB J. 2014, 28, 4710–4718. [Google Scholar] [CrossRef]
- Palma-Tortosa, S.; Hurtado, O.; Pradillo, J.M.; Ferreras-Martín, R.; García-Yébenes, I.; García-Culebras, A.; Moraga, A.; Moro, M.Á.; Lizasoain, I. Toll-like Receptor 4 Regulates Subventricular Zone Proliferation and Neuroblast Migration after Experimental Stroke. Brain Behav. Immun. 2019, 80, 573–582. [Google Scholar] [CrossRef]
- Shechter, R.; Ronen, A.; Rolls, A.; London, A.; Bakalash, S.; Young, M.J.; Schwartz, M. Toll-like Receptor 4 Restricts Retinal Progenitor Cell Proliferation. J. Cell Biol. 2008, 183, 393. [Google Scholar] [CrossRef]
- Hung, Y.F.; Chen, C.Y.; Shih, Y.C.; Liu, H.Y.; Huang, C.M.; Hsueh, Y.P. Endosomal TLR3, TLR7, and TLR8 Control Neuronal Morphology through Different Transcriptional Programs. J. Cell Biol. 2018, 217, 2727–2742. [Google Scholar] [CrossRef]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like Receptor Signaling in Neural Plasticity and Disease. Trends Neurosci. 2011, 34, 269. [Google Scholar] [CrossRef]
- Schroeder, P.; Rivalan, M.; Zaqout, S.; Krüger, C.; Schüler, J.; Long, M.; Meisel, A.; Winter, Y.; Kaindl, A.M.; Lehnardt, S. Abnormal Brain Structure and Behavior in MyD88-Deficient Mice. Brain Behav. Immun. 2021, 91, 181–193. [Google Scholar] [CrossRef]
- Liu, H.Y.; Hong, Y.F.; Huang, C.M.; Chen, C.Y.; Huang, T.N.; Hsueh, Y.P. TLR7 Negatively Regulates Dendrite Outgrowth through the Myd88–c-Fos–IL-6 Pathway. J. Neurosci. 2013, 33, 11479. [Google Scholar] [CrossRef]
- Chen, C.; Liu, H.; Hsueh, Y. TLR3 Downregulates Expression of Schizophrenia Gene Disc1 via MYD88 to Control Neuronal Morphology. EMBO Rep. 2017, 18, 169–183. [Google Scholar] [CrossRef]
- Ma, Y.; Li, J.; Chiu, I.; Wang, Y.; Sloane, J.A.; Lü, J.; Kosaras, B.; Sidman, R.L.; Volpe, J.J.; Vartanian, T. Toll-like Receptor 8 Functions as a Negative Regulator of Neurite Outgrowth and Inducer of Neuronal Apoptosis. J. Cell Biol. 2006, 175, 209. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, J.Y.; Kim, S.J.; Choi, S.Y.; Yune, T.Y.; Ryu, J.H. Toll-like Receptor-2 Deficiency Induces Schizophrenia-like Behaviors in Mice. Sci. Rep. 2015, 5, 8502. [Google Scholar] [CrossRef]
- Shechter, R.; London, A.; Kuperman, Y.; Ronen, A.; Rolls, A.; Chen, A.; Schwartz, M. Hypothalamic Neuronal Toll-like Receptor 2 Protects against Age-Induced Obesity. Sci. Rep. 2013, 3, 1254. [Google Scholar] [CrossRef]
- Potter, O.V.; Giedraitis, M.E.; Johnson, C.D.; Cox, M.N.; Kohman, R.A. Young and Aged TLR4 Deficient Mice Show Sex-Dependent Enhancements in Spatial Memory and Alterations in Interleukin-1 Related Genes. Brain Behav. Immun. 2019, 76, 37–47. [Google Scholar] [CrossRef]
- Fei, X.; Wang, J.X.; Wu, Y.; Dong, N.; Sheng, Z.Y. Sevoflurane-Induced Cognitive Decline in Aged Mice: Involvement of Toll-like Receptors 4. Brain Res. Bull. 2020, 165, 23–29. [Google Scholar] [CrossRef]
- Squillace, S.; Niehoff, M.L.; Doyle, T.M.; Green, M.; Esposito, E.; Cuzzocrea, S.; Arnatt, C.K.; Spiegel, S.; Farr, S.A.; Salvemini, D. Sphingosine-1-Phosphate Receptor 1 Activation in the Central Nervous System Drives Cisplatin-Induced Cognitive Impairment. J. Clin. Investig. 2022, 132, e157738. [Google Scholar] [CrossRef]
- Schilling, S.; Chausse, B.; Dikmen, H.O.; Almouhanna, F.; Hollnagel, J.O.; Lewen, A.; Kann, O. TLR2- and TLR3-Activated Microglia Induce Different Levels of Neuronal Network Dysfunction in a Context-Dependent Manner. Brain Behav. Immun. 2021, 96, 80–91. [Google Scholar] [CrossRef]
- Nie, X.; Kitaoka, S.; Tanaka, K.; Segi-Nishida, E.; Imoto, Y.; Ogawa, A.; Nakano, F.; Tomohiro, A.; Nakayama, K.; Taniguchi, M.; et al. The Innate Immune Receptors TLR2/4 Mediate Repeated Social Defeat Stress-Induced Social Avoidance through Prefrontal Microglial Activation. Neuron 2018, 99, 464–479.e7. [Google Scholar] [CrossRef]
- Weber, M.D.; Frank, M.G.; Sobesky, J.L.; Watkins, L.R.; Maier, S.F. Blocking Toll-like Receptor 2 and 4 Signaling during a Stressor Prevents Stress-Induced Priming of Neuroinflammatory Responses to a Subsequent Immune Challenge. Brain Behav. Immun. 2013, 32, 112–121. [Google Scholar] [CrossRef]
- Figueroa-Hall, L.K.; Paulus, M.P.; Savitz, J. Toll-Like Receptor Signaling in Depression. Psychoneuroendocrinology 2020, 121, 104843. [Google Scholar] [CrossRef]
- Missig, G.; Robbins, J.O.; Mokler, E.L.; McCullough, K.M.; Bilbo, S.D.; McDougle, C.J.; Carlezon, W.A. Sex-Dependent Neurobiological Features of Prenatal Immune Activation via TLR7. Mol. Psychiatry 2020, 25, 2330–2341. [Google Scholar] [CrossRef]
- Khariv, V.; Pang, K.; Servatius, R.J.; David, B.T.; Goodus, M.T.; Beck, K.D.; Heary, R.F.; Elkabes, S. Toll-like Receptor 9 Deficiency Impacts Sensory and Motor Behaviors. Brain Behav. Immun. 2013, 32, 164–172. [Google Scholar] [CrossRef]
- Matsuda, T.; Murao, N.; Katano, Y.; Juliandi, B.; Kohyama, J.; Akira, S.; Kawai, T.; Nakashima, K. TLR9 Signalling in Microglia Attenuates Seizure-Induced Aberrant Neurogenesis in the Adult Hippocampus. Nat. Commun. 2015, 6, 6514. [Google Scholar] [CrossRef]
- Gárate, I.; García-Bueno, B.; Madrigal, J.L.M.; Bravo, L.; Berrocoso, E.; Caso, J.R.; Micó, J.A.; Leza, J.C. Origin and Consequences of Brain Toll-like Receptor 4 Pathway Stimulation in an Experimental Model of Depression. J. Neuroinflamm. 2011, 8, 151. [Google Scholar] [CrossRef]
- Martín-Hernández, D.; Caso, J.R.; Bris, Á.G.; Maus, S.R.; Madrigal, J.L.M.; García-Bueno, B.; MacDowell, K.S.; Alou, L.; Gómez-Lus, M.L.; Leza, J.C. Bacterial Translocation Affects Intracellular Neuroinflammatory Pathways in a Depression-like Model in Rats. Neuropharmacology 2016, 103, 122–133. [Google Scholar] [CrossRef]
- Zhang, K.; Lin, W.; Zhang, J.; Zhao, Y.; Wang, X.; Zhao, M. Effect of Toll-like Receptor 4 on Depressive-like Behaviors Induced by Chronic Social Defeat Stress. Brain Behav. 2020, 10, e01525. [Google Scholar] [CrossRef]
- Li, H.; Chen, W.; Gou, M.; Li, W.; Tong, J.; Zhou, Y.; Xie, T.; Yu, T.; Feng, W.; Li, Y.; et al. The Relationship between TLR4/NF-ΚB/IL-1β Signaling, Cognitive Impairment, and White-Matter Integrity in Patients with Stable Chronic Schizophrenia. Front. Psychiatry 2022, 13, 966657. [Google Scholar] [CrossRef]
- Grantham, E.K.; Warden, A.S.; McCarthy, G.S.; DaCosta, A.; Mason, S.; Blednov, Y.; Mayfield, R.D.; Harris, R.A. Role of Toll-like Receptor 7 (TLR7) in Voluntary Alcohol Consumption. Brain Behav. Immun. 2020, 89, 423–432. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Benavidez, J.M.; Geil, C.; Perra, S.; Morikawa, H.; Harris, R.A. Activation of Inflammatory Signaling by Lipopolysaccharide Produces a Prolonged Increase of Voluntary Alcohol Intake in Mice. Brain Behav. Immun. 2011, 25 (Suppl. S1), S92–S105. [Google Scholar] [CrossRef]
- Truitt, J.M.; Blednov, Y.A.; Benavidez, J.M.; Black, M.; Ponomareva, O.; Law, J.; Merriman, M.; Horani, S.; Jameson, K.; Lasek, A.W.; et al. Inhibition of IKKβ Reduces Ethanol Consumption in C57BL/6J Mice. eNeuro 2016, 3, ENEURO.0256-16.2016. [Google Scholar] [CrossRef]
- Warden, A.S.; Azzam, M.; DaCosta, A.; Mason, S.; Blednov, Y.A.; Messing, R.O.; Mayfield, R.D.; Harris, R.A. Toll-like Receptor 3 Activation Increases Voluntary Alcohol Intake in C57BL/6J Male Mice. Brain Behav. Immun. 2019, 77, 55–65. [Google Scholar] [CrossRef]
- Gabr, M.M.; Saeed, I.; Miles, J.A.; Ross, B.P.; Shaw, P.N.; Hollmann, M.W.; Parat, M.O. Interaction of Opioids with TLR4-Mechanisms and Ramifications. Cancers 2021, 13, 5274. [Google Scholar] [CrossRef]
- Rubio-Araiz, A.; Porcu, F.; Pérez-Hernández, M.; García-Gutiérrez, M.S.; Aracil-Fernández, M.A.; Gutierrez-López, M.D.; Guerri, C.; Manzanares, J.; O’Shea, E.; Colado, M.I. Disruption of Blood-Brain Barrier Integrity in Postmortem Alcoholic Brain: Preclinical Evidence of TLR4 Involvement from a Binge-like Drinking Model. Addict. Biol. 2017, 22, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation Pathways: A General Review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Chugh, H.; Sakharkar, M.K.; Dhawan, U.; Chidambaram, S.B.; Chandra, R. Neuroinflammation Mechanisms and Phytotherapeutic Intervention: A Systematic Review. ACS Chem. Neurosci. 2020, 11, 3707–3731. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef]
- Moyse, E.; Krantic, S.; Djellouli, N.; Roger, S.; Angoulvant, D.; Debacq, C.; Leroy, V.; Fougere, B.; Aidoud, A. Neuroinflammation: A Possible Link between Chronic Vascular Disorders and Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 827263. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Koury, J.; Kaul, M. Innate Immune Sensing of Viruses and Its Consequences for the Central Nervous System. Viruses 2021, 13, 170. [Google Scholar] [CrossRef] [PubMed]
- Gern, O.L.; Mulenge, F.; Pavlou, A.; Ghita, L.; Steffen, I.; Stangel, M.; Kalinke, U. Toll-like Receptors in Viral Encephalitis. Viruses 2021, 13, 2065. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Gao, Y.; Zhang, J.; Zhang, H.; Xie, C.; Nan, F.; Feng, S.; Ha, Z.; Li, C.; Zhu, X.; et al. Toll-like Receptor 2 Signaling Pathway Activation Contributes to a Highly Efficient Inflammatory Response in Japanese Encephalitis Virus-Infected Mouse Microglial Cells by Proteomics. Front. Microbiol. 2022, 13, 989183. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Akbar, I.; Kumari, B.; Vrati, S.; Basu, A.; Banerjee, A. Japanese Encephalitis Virus-Induced Let-7a/b Interacted with the NOTCH-TLR7 Pathway in Microglia and Facilitated Neuronal Death via Caspase Activation. J. Neurochem. 2019, 149, 518–534. [Google Scholar] [CrossRef]
- Shen, Y.; Yang, R.; Zhao, J.; Chen, M.; Chen, S.; Ji, B.; Chen, H.; Liu, D.; Li, L.; Du, G. The Histone Deacetylase Inhibitor Belinostat Ameliorates Experimental Autoimmune Encephalomyelitis in Mice by Inhibiting TLR2/MyD88 and HDAC3/NF-ΚB P65-Mediated Neuroinflammation. Pharmacol. Res. 2022, 176, 105969. [Google Scholar] [CrossRef]
- Kwilasz, A.J.; Green Fulgham, S.M.; Duran-Malle, J.C.; Schrama, A.E.W.; Mitten, E.H.; Todd, L.S.; Patel, H.P.; Larson, T.A.; Clements, M.A.; Harris, K.M.; et al. Toll-like Receptor 2 and 4 Antagonism for the Treatment of Experimental Autoimmune Encephalomyelitis (EAE)-Related Pain. Brain Behav. Immun. 2021, 93, 80–95. [Google Scholar] [CrossRef]
- Chu, Y.; Jing, Y.; Zhao, X.; Wang, M.; Zhang, M.; Ma, R.; Ma, W.; Lv, Y.; Zhu, L. Modulation of the HMGB1/TLR4/NF-ΚB Signaling Pathway in the CNS by Matrine in Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2021, 352, 577480. [Google Scholar] [CrossRef]
- Vitturi, B.K.; Rosemberg, S.; Arita, F.N.; da Rocha, A.J.; Forte, W.C.N.; Tilbery, C.P. Multiphasic Disseminated Encephalomyelitis Associated with Herpes Virus Infection in a Patient with TLR3 Deficiency. Mult. Scler. Relat. Disord. 2019, 36, 101379. [Google Scholar] [CrossRef] [PubMed]
- Partanen, T.; Chen, J.; Lehtonen, J.; Kuismin, O.; Rusanen, H.; Vapalahti, O.; Vaheri, A.; Anttila, V.J.; Bode, M.; Hautala, N.; et al. Heterozygous TLR3 Mutation in Patients with Hantavirus Encephalitis. J. Clin. Immunol. 2020, 40, 1156–1162. [Google Scholar] [CrossRef]
- Su, W.; Ju, J.; Gu, M.; Wang, X.; Liu, S.; Yu, J.; Mu, D. SARS-CoV-2 Envelope Protein Triggers Depression-like Behaviors and Dysosmia via TLR2-Mediated Neuroinflammation in Mice. J. Neuroinflamm. 2023, 20, 110. [Google Scholar] [CrossRef]
- Fontes-Dantas, F.L.; Fernandes, G.G.; Gutman, E.G.; De Lima, E.V.; Antonio, L.S.; Hammerle, M.B.; Mota-Araujo, H.P.; Colodeti, L.C.; Araújo, S.M.B.; Froz, G.M.; et al. SARS-CoV-2 Spike Protein Induces TLR4-Mediated Long-Term Cognitive Dysfunction Recapitulating Post-COVID-19 Syndrome in Mice. Cell Rep. 2023, 42, 112189. [Google Scholar] [CrossRef]
- Wang, P.; Liu, J.B.; Wang, X.; Meng, F.Z.; Xiao, Q.H.; Liu, L.; Zhu, J.; Hu, W.H.; Ho, W.Z. Activation of Toll-like Receptor 3 Inhibits HIV Infection of Human IPSC-Derived Microglia. J. Med. Virol. 2023, 95, e29217. [Google Scholar] [CrossRef]
- Jin, X.; Yin, S.; Zhang, Y.; Chen, X. Association between TLR2 + 2477G/A Polymorphism and Bacterial Meningitis: A Meta-Analysis. Epidemiol. Infect. 2018, 146, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Tenhu, E.; Teräsjärvi, J.; Cruzeiro, M.L.; Savonius, O.; Rugemalira, E.; Roine, I.; He, Q.; Pelkonen, T. Gene Polymorphisms of TLR4 and TLR9 and Haemophilus Influenzae Meningitis in Angolan Children. Genes 2020, 11, 1099. [Google Scholar] [CrossRef]
- Xue, H.; Peng, H.; Li, J.; Li, M.; Lu, S. TLR9 Rs352140 Polymorphism Contributes to a Decreased Risk of Bacterial Meningitis: Evidence from a Meta-Analysis. Epidemiol. Infect. 2020, 148, e294. [Google Scholar] [CrossRef]
- Too, L.K.; Yau, B.; Baxter, A.G.; McGregor, I.S.; Hunt, N.H. Double Deficiency of Toll-like Receptors 2 and 4 Alters Long-Term Neurological Sequelae in Mice Cured of Pneumococcal Meningitis. Sci. Rep. 2019, 9, 16189. [Google Scholar] [CrossRef] [PubMed]
- Ribes, S.; Zacke, L.; Nessler, S.; Saiepour, N.; Avendaño-Guzmán, E.; Ballüer, M.; Hanisch, U.K.; Nau, R. Oligodeoxynucleotides Containing Unmethylated Cytosine-Guanine Motifs Are Effective Immunostimulants against Pneumococcal Meningitis in the Immunocompetent and Neutropenic Host. J. Neuroinflamm. 2021, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, X.; Sun, Y.; Gong, P.; Yu, H. Promotion Properties of TLR7 in Pediatric Meningitis via the NF-ΚB Pathway. J. Bioenerg. Biomembr. 2021, 53, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, Y.; Zhao, Y.; Zhang, Y.; Liu, Y.; Ma, F.; Wang, X.; Fu, J. Andrographolide Ameliorates Neuroinflammation in APP/PS1 Transgenic Mice. Int. Immunopharmacol. 2021, 96, 107808. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and Therapeutic Modulation of a Pro-Inflammatory Subset of Disease-Associated-Microglia in Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Tang, T.M.; Lue, L.F. Increased Expression of Toll-like Receptor 3, an Anti-Viral Signaling Molecule, and Related Genes in Alzheimer’s Disease Brains. Exp. Neurol. 2018, 309, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Jana, M.; Paidi, R.K.; Majumder, M.; Raha, S.; Dasarathy, S.; Pahan, K. Tau Fibrils Induce Glial Inflammation and Neuropathology via TLR2 in Alzheimer’s Disease–Related Mouse Models. J. Clin. Investig. 2023, 133, e161987. [Google Scholar] [CrossRef] [PubMed]
- Lax, N.; Fainstein, N.; Nishri, Y.; Ben-Zvi, A.; Ben-Hur, T. Systemic Microbial TLR2 Agonists Induce Neurodegeneration in Alzheimer’s Disease Mice. J. Neuroinflamm. 2020, 17, 55. [Google Scholar] [CrossRef]
- Viola, T.W.; Creutzberg, K.C.; Zaparte, A.; Kestering-Ferreira, É.; Tractenberg, S.G.; Centeno-Silva, A.; Orso, R.; Lumertz, F.S.; Brietzke, E.; Wearick-Silva, L.E.; et al. Acute Neuroinflammation Elicited by TLR-3 Systemic Activation Combined with Early Life Stress Induces Working Memory Impairments in Male Adolescent Mice. Behav. Brain Res. 2019, 376, 112221. [Google Scholar] [CrossRef]
- Rubio-Araiz, A.; Finucane, O.M.; Keogh, S.; Lynch, M.A. Anti-TLR2 Antibody Triggers Oxidative Phosphorylation in Microglia and Increases Phagocytosis of β-Amyloid. J. Neuroinflamm. 2018, 15, 247. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.B.; Jana, M.; Roy, A.; Corbett, G.T.; Kundu, M.; Chandra, S.; Mondal, S.; Dasarathi, S.; Mufson, E.J.; Mishra, R.K.; et al. Selective Disruption of TLR2-MyD88 Interaction Inhibits Inflammation and Attenuates Alzheimer’s Pathology. J. Clin. Investig. 2018, 128, 4297–4312. [Google Scholar] [CrossRef]
- Zhou, C.; Sun, X.; Hu, Y.; Song, J.; Dong, S.; Kong, D.; Wang, Y.; Hua, X.; Han, J.; Zhou, Y.; et al. Genomic Deletion of TLR2 Induces Aggravated White Matter Damage and Deteriorated Neurobehavioral Functions in Mouse Models of Alzheimer’s Disease. Aging 2019, 11, 7257–7273. [Google Scholar] [CrossRef]
- Pourbadie, H.G.; Sayyah, M.; Khoshkholgh-Sima, B.; Choopani, S.; Nategh, M.; Motamedi, F.; Shokrgozar, M.A. Early Minor Stimulation of Microglial TLR2 and TLR4 Receptors Attenuates Alzheimer’s Disease-Related Cognitive Deficit in Rats: Behavioral, Molecular, and Electrophysiological Evidence. Neurobiol. Aging 2018, 70, 203–216. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, T.; Ni, W.; Zhou, C.; Zhou, H.; Lin, L.; Hu, Y.; Sun, X.; Han, J.; Zhou, Y.; et al. Early Activation of Toll-like Receptor-3 Reduces the Pathological Progression of Alzheimer’s Disease in APP/PS1 Mouse. Alzheimer’s Res. Ther. 2023, 15, 33. [Google Scholar] [CrossRef]
- Scholtzova, H.; Do, E.; Dhakal, S.; Sun, Y.; Liu, S.; Mehta, P.D.; Wisniewski, T. Innate Immunity Stimulation via Toll-Like Receptor 9 Ameliorates Vascular Amyloid Pathology in Tg-SwDI Mice with Associated Cognitive Benefits. J. Neurosci. 2017, 37, 936–959. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. 2023, 18, 95–121. [Google Scholar] [CrossRef]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation-An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef] [PubMed]
- Conte, C.; Ingrassia, A.; Breve, J.; Bol, J.J.; Timmermans-Huisman, E.; van Dam, A.M.; Beccari, T.; van de Berg, W.D.J. Toll-like Receptor 4 Is Upregulated in Parkinson’s Disease Patients and Co-Localizes with PSer129αSyn: A Possible Link with the Pathology. Cells 2023, 12, 1368. [Google Scholar] [CrossRef]
- Chung, L.Y.R.; Lin, Y.T.; Liu, C.; Tai, Y.C.; Lin, H.Y.; Lin, C.H.; Chen, C.C. Neuroinflammation Upregulated Neuronal Toll-Like Receptors 2 and 4 to Drive Synucleinopathy in Neurodegeneration. Front. Pharmacol. 2022, 13, 845930. [Google Scholar] [CrossRef]
- He, Y.; Zhao, J.; Dong, H.; Zhang, X.; Duan, Y.; Ma, Y.; Yu, M.; Fei, J.; Huang, F. TLR2 Deficiency Is Beneficial at the Late Phase in MPTP-Induced Parkinson’ Disease Mice. Life Sci. 2023, 333, 122171. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 Absence Reduces Neuroinflammation and Inflammasome Activation in Parkinson’s Diseases in Vivo Model. Brain Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Venezia, S.; Kaufmann, W.A.; Wenning, G.K.; Stefanova, N. Toll-like Receptor 4 Deficiency Facilitates α-Synuclein Propagation and Neurodegeneration in a Mouse Model of Prodromal Parkinson’s Disease. Park. Relat. Disord. 2021, 91, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Filippone, A.; Biondo, C.; Mancuso, G.; Casili, G.; Lanza, M.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. TLR7/8 in the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 9384. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Liu, Y.; Zhu, K.; Xie, A. The Association between TLR3 Rs3775290 Polymorphism and Sporadic Parkinson’s Disease in Chinese Han Population. Neurosci. Lett. 2020, 728, 135005. [Google Scholar] [CrossRef] [PubMed]
- Miri, N.S.; Saadat, P.; Azadmehr, A.; Oladnabi, M.; Daraei, A. Toll-Like Receptor (TLR)-9 Rs352140 Polymorphism Is an Immunopathology Protective Factor in Parkinson’s Disease in the Northern Iranian Population. Iran. J. Immunol. 2020, 17, 313–323. [Google Scholar] [CrossRef]
- Li, Z.; Song, A.; Yu, H. Interaction between Toll-like Receptor 4 (TLR4) Gene and Alcohol Drinking on Parkinson’s Disease Risk in Chinese Han Population. J. Clin. Neurosci. 2019, 62, 128–132. [Google Scholar] [CrossRef]
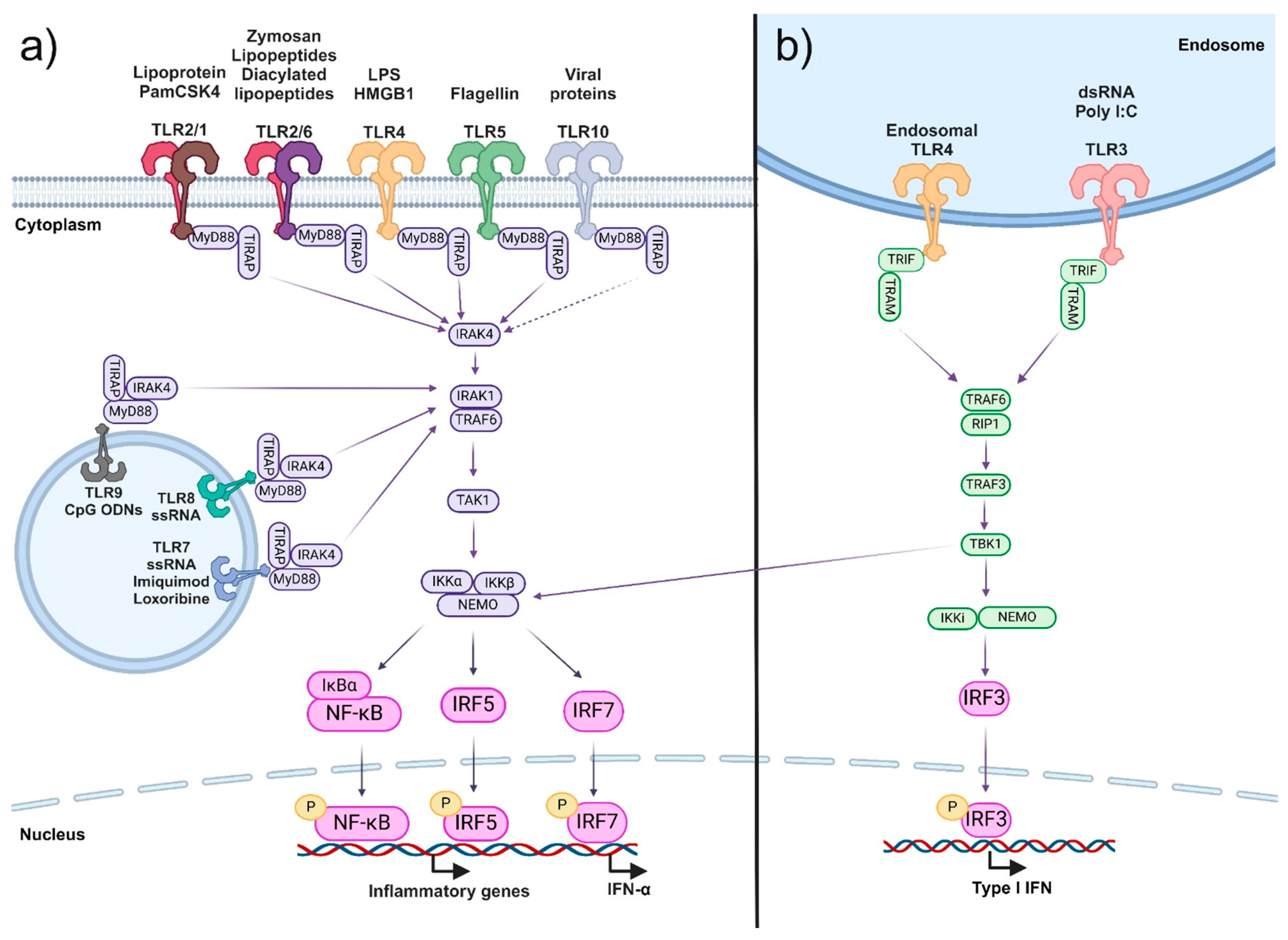
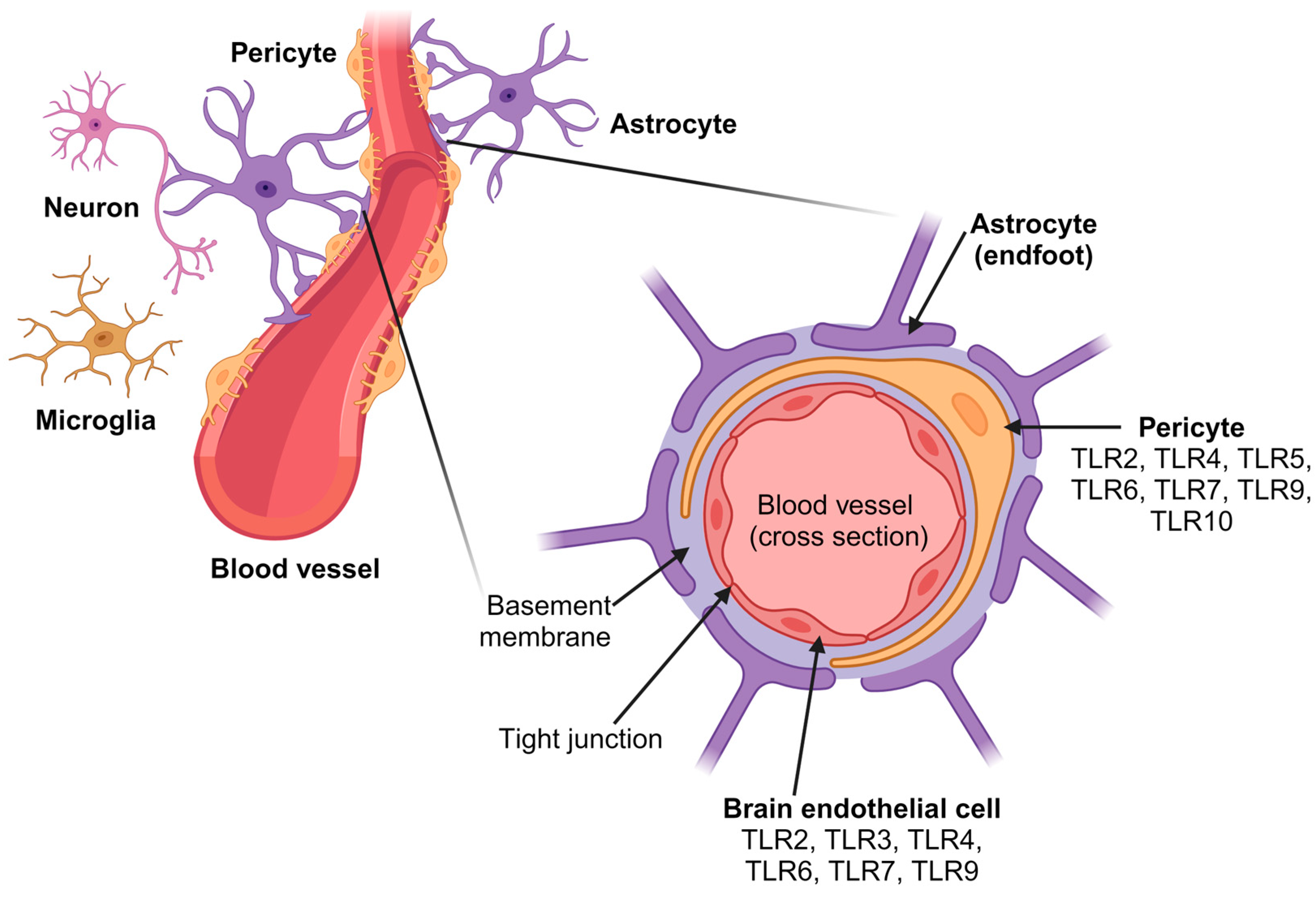
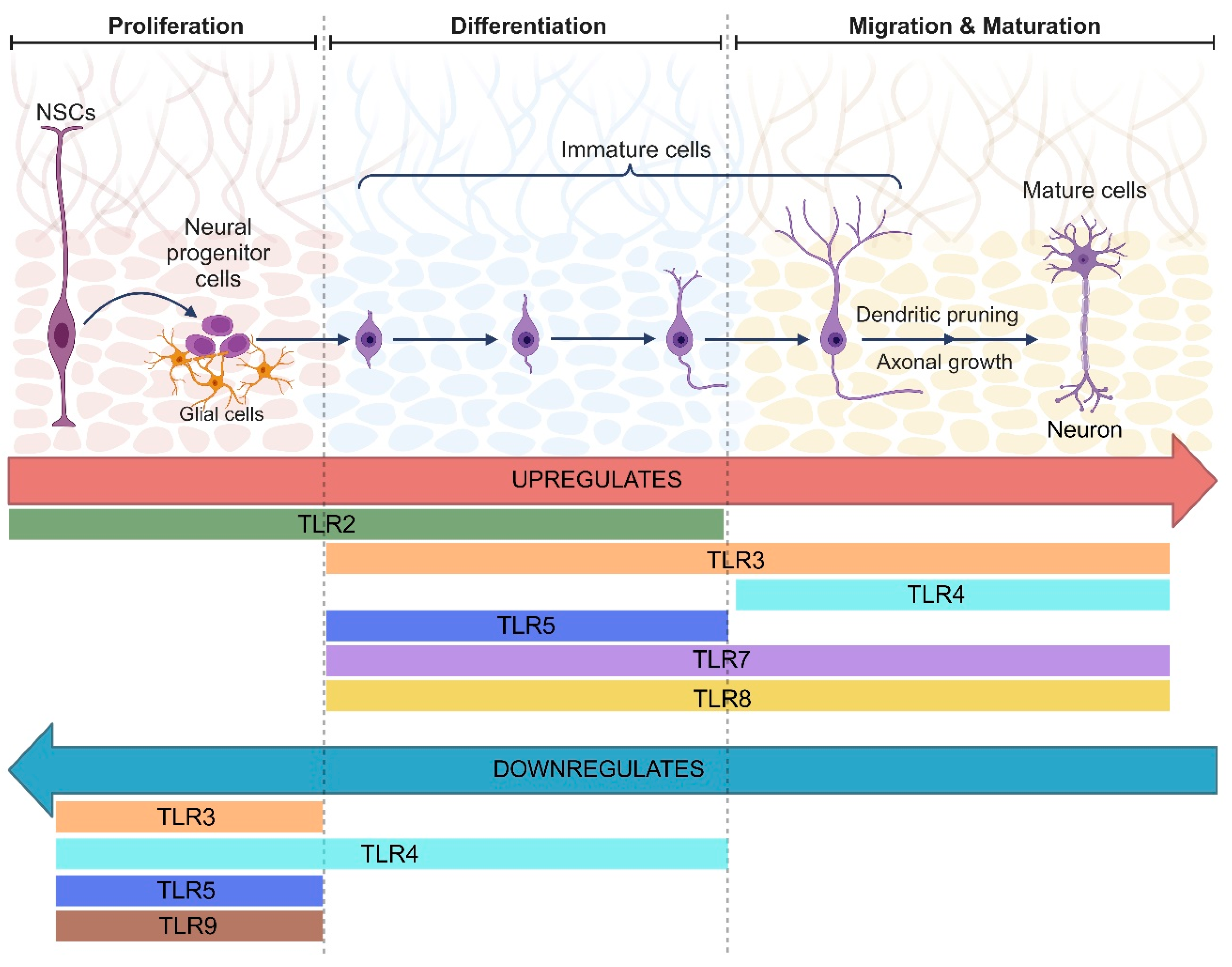

{kind=link}
{kind=link}
{kind=link}
| Receptor | Outcome | Related Disease | Model | Refs. |
|---|---|---|---|---|
| TLR1 | Increased receptor expression. | Epilepsy | Mouse model | [36] |
| TLR2 | Negatively regulated maturation of oligodendrocyte precursor cells. | Multiple sclerosis | KO mouse | [26] |
| Receptor deficiency decreased neuronal death, inflammation, and brain injury. | Cerebral ischemia | KO mouse | [37,56,71] | |
| Increased receptor expression. | Amyotrophic lateral sclerosis | Postmortem human spinal cord sample | [38] | |
| Receptor activation increased transendothelial permeability. | Not | Rat and human cerebral endothelial cells | [52] | |
| Receptor deficiency reduced MMP-9 activation in astrocytes. | Intracerebral hemorrhage | KO mouse | [65] | |
| Receptor deficiency induced schizophrenia-like symptoms. | Schizophrenia | KO mouse | [100] | |
| Regulated metabolic processes. | Obesity | Mouse neuron cell line | [101] | |
| Administration of antagonist decreased proinflammatory cytokines. | Multiple sclerosis | EAE mouse model | [131] | |
| Regulated neuroinflammation, depression-like behaviors, and dysosmia. | COVID-19 | Mouse | [135] | |
| Receptor activation with tau fibrils induced stimulated microglial inflammation. | Tauopathy | Mouse | [148] | |
| Induced death of neurons and inflammation. | AD | AD mouse model | [149] | |
| Increased accumulation of α-synuclein proteins and the shortening of neurites. | PD | Dopaminergic neuronal cell line | [160] | |
| TLR3 | Increased receptor expression. | Multiple sclerosis | Human postmortem brain tissue | [8] |
| Receptor activation decreased myelin expression and increased cytokine and chemokine production. | Maternal/fetal infection | Mouse oligodendrocyte precursor cells | [29] | |
| Receptor activation induced IL-1β secretion. | Viral infection | Human neurons | [39] | |
| Increased receptor expression. | Viral encephalitis | Human postmortem brain tissue | [40] | |
| Induced interferon expression. | HIV infection | Microvascular endothelial cells | [57] | |
| Negatively regulated NSPC proliferation. | Not | KO mouse | [88] | |
| Receptor activation increased alcohol intake. | Alcohol use disorder | Mouse | [119] | |
| Receptor activation in early stage decreased neuron loss and improved neurobehavioral functions. | AD | AD mouse model | [155] | |
| TLR4 | Increased receptor expression. | Multiple sclerosis | Human postmortem brain tissue | [8] |
| Receptor activation inhibited autophagy and phagocytic function. | Not | Mouse microglial cells | [14] | |
| Induced neuronal death. | Cerebral ischemia | KO mouse | [37] | |
| Receptor deficiency decreased infarct size, improvements in neurological and behavioral outcomes, and inflammation. | Cerebral ischemia | TLR4-deficient mutant mouse | [66,68] | |
| Inhibited spatial memory and induced inflammation. | Not | KO mouse | [102,103] | |
| Receptor activation through cisplatin resulted in cognitive dysfunction. | Cancer | KO mouse | [104] | |
| Receptor deficiency decreased ethanol-induced impairment of BBB. | Alcohol use disorder | KO mouse | [121] | |
| Receptor blockade prevented detrimental effects on synapse and memory. | COVID-19 | Mouse | [136] | |
| Receptor deficiency decreased inflammation, dopaminergic neuron loss, and autophagy and induced α-synuclein aggregate formation. | PD | KO mouse | [162,163] | |
| TLR5 | Induced inflammatory factors and neuronal apoptosis. | Not | Murine microglial and neuronal cultures | [15] |
| Increased neuronal differentiation. | Not | KO mouse | [41] | |
| TLR6 | Activation of TLR2/TLR6 heterodimer increased interferon production. | HSV-1 infection | Mouse neuronal cell line | [42] |
| TLR7 | Receptor activation induced microglial inflammatory cytokine production and antitumor effect. | Glioblastoma | Mouse glioblastoma | [16] |
| Induced chemotaxis and prostaglandin production. | JEV infection | Pericyte cell line | [62] | |
| Receptor deficiency reduced anxiety, aggression, and contextual memory. | Not | KO mouse | [82] | |
| Acute activation of receptor reduced ethanol intake. Chronic activation induced tolerance to the adverse effects and an increase in ethanol consumption. | Alcohol use disorder | Mouse | [116] | |
| eceptor overexpression promoted meningitis. | Meningitis | Mouse | [143] | |
| TLR8 | TLR7 and TLR8 deficiency reduced astrogliosis, microgliosis, α-synuclein aggregate formation, and T-cell activation. | PD | TLR7/TLR8 KO mouse | [164] |
| TLR9 | Induced antitumor effect. | Glioblastoma | Mouse microglial cell line | [13] |
| Receptor inhibition decreased neurodegeneration. | Sepsis-associated encephalopathy | Sepsis rat model | [45] | |
| Induced TNF-α production in microglia and inhibition of seizure-induced aberrant neurogenesis. | Epilepsy | KO mouse | [111] | |
| Administration of ODN decreased bacterial burden in the cerebellum and bacteremia. | Meningitis | Mouse | [142] | |
| Receptor activation induced cognitive improvements and amyloid angiopathy reduction. | AD | AD mouse model | [156] | |
| TLR10 | Metabolite of vitamin D increased the expression of receptor. | Not | Human microglial cell line | [17] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abarca-Merlin, D.M.; Martínez-Durán, J.A.; Medina-Pérez, J.D.; Rodríguez-Santos, G.; Alvarez-Arellano, L. From Immunity to Neurogenesis: Toll-like Receptors as Versatile Regulators in the Nervous System. Int. J. Mol. Sci. 2024, 25, 5711. https://doi.org/10.3390/ijms25115711
Abarca-Merlin DM, Martínez-Durán JA, Medina-Pérez JD, Rodríguez-Santos G, Alvarez-Arellano L. From Immunity to Neurogenesis: Toll-like Receptors as Versatile Regulators in the Nervous System. International Journal of Molecular Sciences. 2024; 25(11):5711. https://doi.org/10.3390/ijms25115711
Chicago/Turabian StyleAbarca-Merlin, Daniela Melissa, J. Abigail Martínez-Durán, J. David Medina-Pérez, Guadalupe Rodríguez-Santos, and Lourdes Alvarez-Arellano. 2024. "From Immunity to Neurogenesis: Toll-like Receptors as Versatile Regulators in the Nervous System" International Journal of Molecular Sciences 25, no. 11: 5711. https://doi.org/10.3390/ijms25115711
APA StyleAbarca-Merlin, D. M., Martínez-Durán, J. A., Medina-Pérez, J. D., Rodríguez-Santos, G., & Alvarez-Arellano, L. (2024). From Immunity to Neurogenesis: Toll-like Receptors as Versatile Regulators in the Nervous System. International Journal of Molecular Sciences, 25(11), 5711. https://doi.org/10.3390/ijms25115711