
Granulocyte Colony Stimulating Factor-Mobilized Peripheral Blood Mononuclear Cells: An Alternative Cellular Source for Chimeric Antigen Receptor Therapy

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Adoptive Cell Therapy (ACT): The CAR Immunotherapy
3. New Cellular Sources for CAR Immunotherapy: G-CSF Mobilized PBSC Products
3.1. CAR-T Immunotherapy
3.2. CAR-NK Immunotherapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xu, Z.; Huang, X. Cellular Immunotherapy for Hematological Malignancy: Recent Progress and Future Perspectives. Cancer Biol. Med. 2021, 18, 966–980. [Google Scholar] [CrossRef] [PubMed]
- Minda, A.G.; Awel, F.S.; Seifudin, K.A.; Gezahegne, M.K. Immunotherapy against Cancer: A Comprehensive Review. J. Cancer Res. Exp. Oncol. 2016, 8, 15–25. [Google Scholar] [CrossRef]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive Cellular Therapies: The Current Landscape. Virchows Archiv 2019, 474, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.; Lin, C.Y.; Siegel, D.L.; Maus, M.V. CAR-T Cell Therapies from the Transfusion Medicine Perspective. Transfus. Med. Rev. 2016, 30, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human IPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Barone, M.; Spadea, M.; Saglio, F.; Pessolano, R.; Fagioli, F. HSCT with Mismatched Unrelated Donors: Bone Marrow versus Peripheral Blood Stem Cells Sources in Pediatric Patients. Pediatr. Transplant. 2022, 26, e14233. [Google Scholar] [CrossRef] [PubMed]
- Grieco, D.; Lacetera, N.; Macis, M.; Di Martino, D. Motivating Cord Blood Donation with Information and Behavioral Nudges. Sci. Rep. 2018, 8, 252. [Google Scholar] [CrossRef]
- Spanholtz, J.; Preijers, F.; Tordoir, M.; Trilsbeek, C.; Paardekooper, J.; de Witte, T.; Schaap, N.; Dolstra, H. Clinical-Grade Generation of Active NK Cells from Cord Blood Hematopoietic Progenitor Cells for Immunotherapy Using a Closed-System Culture Process. PLoS ONE 2011, 6, e20740. [Google Scholar] [CrossRef] [PubMed]
- Cany, J.; van der Waart, A.B.; Spanholtz, J.; Tordoir, M.; Jansen, J.H.; van der Voort, R.; Schaap, N.M.; Dolstra, H. Combined IL-15 and IL-12 Drives the Generation of CD34+-Derived Natural Killer Cells with Superior Maturation and Alloreactivity Potential Following Adoptive Transfer. Oncoimmunology 2015, 4, e1017701. [Google Scholar] [CrossRef]
- Künkele, A.; Brown, C.; Beebe, A.; Mgebroff, S.; Johnson, A.J.; Taraseviciute, A.; Rolczynski, L.S.; Chang, C.A.; Finney, O.C.; Park, J.R.; et al. Manufacture of Chimeric Antigen Receptor T Cells from Mobilized Cyropreserved Peripheral Blood Stem Cell Units Depends on Monocyte Depletion. Biol. Blood Marrow Transplant. 2019, 25, 223–232. [Google Scholar] [CrossRef]
- Cummins, K.D.; Gupta, A.; Beyar-Katz, O.; Li, Y.; Chatterjee, P.; Kippner, L.; Shestova, O.; Eiva, M.A.; Salas-Mckee, J.; Yeago, C.; et al. G-CSF Mobilized Apheresis as an Alternative Source of CAR T-Cells. Blood 2022, 140, 7415–7416. [Google Scholar] [CrossRef]
- Rettinger, E.; Kuçi, S.; Naumann, I.; Becker, P.; Kreyenberg, H.; Anzaghe, M.; Willasch, A.; Koehl, U.; Bug, G.; Ruthardt, M.; et al. The Cytotoxic Potential of Interleukin-15-Stimulated Cytokine-Induced Killer Cells against Leukemia Cells. Cytotherapy 2012, 14, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Borch, T.H.; van den Berg, J.H.; Met, Ö.; Kessels, R.; Geukes Foppen, M.H.; Stoltenborg Granhøj, J.; Nuijen, B.; Nijenhuis, C.; Jedema, I.; et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2022, 387, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Perko, R.; Kang, G.; Sunkara, A.; Leung, W.; Thomas, P.G.; Dallas, M.H. Gamma Delta T Cell Reconstitution Is Associated with Fewer Infections and Improved Event-Free Survival after Hematopoietic Stem Cell Transplantation for Pediatric Leukemia. Biol. Blood Marrow Transplant. 2015, 21, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Douka, S.; Brandenburg, L.E.; Casadidio, C.; Walther, J.; Garcia, B.B.M.; Spanholtz, J.; Raimo, M.; Hennink, W.E.; Mastrobattista, E.; Caiazzo, M. Lipid Nanoparticle-Mediated Messenger RNA Delivery for Ex Vivo Engineering of Natural Killer Cells. J. Control. Release 2023, 361, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Lamers-Kok, N.; Panella, D.; Georgoudaki, A.M.; Liu, H.; Özkazanc, D.; Kučerová, L.; Duru, A.D.; Spanholtz, J.; Raimo, M. Natural Killer Cells in Clinical Development as Non-Engineered, Engineered, and Combination Therapies. J. Hematol. Oncol. 2022, 15, 164. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Agrawal, A.; Borthakur, G.; Battula, V.L.; Maiti, A. Gamma Delta T Cells in Acute Myeloid Leukemia: Biology and Emerging Therapeutic Strategies. J. Immunother. Cancer 2024, 12, e007981. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the γ or ζ Subunits of the Immunoglobulin and T-Cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z. Tumor-Specific T-Bodies: Towards Clinical Application. Cancer Immunol. Immunother. 1997, 45, 131–136. [Google Scholar] [CrossRef]
- Alnefaie, A.; Albogami, S.; Asiri, Y.; Ahmad, T.; Alotaibi, S.S.; Al-Sanea, M.M.; Althobaiti, H. Chimeric Antigen Receptor T-Cells: An Overview of Concepts, Applications, Limitations, and Proposed Solutions. Front. Bioeng. Biotechnol. 2022, 10, 797440. [Google Scholar] [CrossRef]
- Hollyman, D.; Stefanski, J.; Przybylowski, M.; Bartido, S.; Borquez-Ojeda, O.; Taylor, C.; Yeh, R.; Capacio, V.; Olszewska, M.; Hosey, J.; et al. Manufacturing Validation of Biologically Functional T Cells Targeted to CD19 Antigen for Autologous Adoptive Cell Therapy. J. Immunother. 2009, 32, 169–180. [Google Scholar] [CrossRef]
- Davila, M.L.; Brentjens, R.; Wang, X.; Rivière, I.; Sadelain, M. How Do CARs Work? Oncoimmunology 2012, 1, 1577–1583. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCRζ/CD28 Receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of Large, Established Tumor Xenografts with Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The Fourth Generation of CARs. Expert. Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKS, the Fourth-generation CAR T Cells: Current Developments and Clinical Translation. Adv. Cell Gene Ther. 2020, 3, 1005116. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1–2 Trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Qayed, M.; McGuirk, J.P.; Myers, G.D.; Parameswaran, V.; Waller, E.K.; Holman, P.; Rodrigues, M.; Clough, L.F.; Willert, J. Leukapheresis Guidance and Best Practices for Optimal Chimeric Antigen Receptor T-Cell Manufacturing. Cytotherapy 2022, 24, 869–878. [Google Scholar] [CrossRef]
- Wang, X.; Rivière, I. Clinical Manufacturing of CAR T Cells: Foundation of a Promising Therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene Maraleucel for Patients with Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-Term Outcomes Following CAR T Cell Therapy: What We Know so Far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Grover, P.; Veilleux, O.; Tian, L.; Sun, R.; Previtera, M.; Curran, E.; Muffly, L. Chimeric Antigen Receptor T-Cell Therapy in Adults with B-Cell Acute Lymphoblastic Leukemia. Blood Adv. 2022, 6, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Nath, K.; Devlin, S.M.; Sauter, C.S.; Palomba, M.L.; Shah, G.; Dahi, P.; Lin, R.J.; Scordo, M.; Perales, M.A.; et al. CD19 CAR T-Cell Therapy and Prophylactic Anakinra in Relapsed or Refractory Lymphoma: Phase 2 Trial Interim Results. Nat. Med. 2023, 29, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Cliff, E.R.S.; Kelkar, A.H.; Russler-Germain, D.A.; Tessema, F.A.; Raymakers, A.J.N.; Feldman, W.B.; Kesselheim, A.S. High Cost of Chimeric Antigen Receptor T-Cells: Challenges and Solutions. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e397912. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-Specific Chimeric Antigen Receptor MRNA-Engineered T Cells Induce Anti-Tumor Activity in Solid Malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase i Trial of Regional Mesothelin-Targeted Car t-Cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–Pd-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, S.; Zhong, S.; An, H.; Yin, H.; McGowan, E. Phase I Clinical Trial of PD-1 Knockout Anti-MUC1 CAR-T Cells in the Treatment of Patients with Non-Small Cell Lung Cancer. Ann. Oncol. 2019, 30, xi12. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.H.; Leung, W. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef]
- Moreno, D.F.; Cid, J. Graft-versus-Host Disease. Med. Clínica (Engl. Ed.) 2019, 152, 22–28. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK Cell Platform for Cancer Immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 2017, 47, 820–833. [Google Scholar] [CrossRef]
- Carreras, E.; Dufour, C.; Mohty, M.; Kröger, N. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Peter Gale, R. E Donnall Thomas (1920–2012). Leukemia 2013, 27, 259. [Google Scholar] [CrossRef] [PubMed]
- Scheding, S.; Brugger, W.; Mertelsmann, R.; Kanz, L. Peripheral Blood Stem Cells: In Vivo Biology and Therapeutic Potential. Stem Cells 1994, 12, 203–211. [Google Scholar] [CrossRef]
- Theyab, A.; Alsharif, K.F.; Alzahrani, K.J.; Oyouni, A.A.A.; Hawsawi, Y.M.R.; Algahtani, M.; Alghamdi, S.; Alshammary, A.F. New Insight into Strategies Used to Develop Long-Acting G-CSF Biologics for Neutropenia Therapy. Front. Oncol. 2023, 12, 1026377. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Griffin, J.D. Granulocyte Colony-Stimulating Factor and Its Receptor. Blood 1991, 78, 131–141. [Google Scholar] [CrossRef]
- Bussolino, F.; Ziche, M.; Ming Wang, J.; Alessi, D.; Morbidelli, L.; Cremona, O.; Bosia, A.; Marchisio, P.C.; Mantovani, A. In Vitro and in Vivo Activation of Endothelial Cells by Colony-Stimulating Factors. J. Clin. Investig. 1991, 87, 986–995. [Google Scholar] [CrossRef]
- Dwivedi, P.; Greis, K.D. Granulocyte Colony-Stimulating Factor Receptor Signaling in Severe Congenital Neutropenia, Chronic Neutrophilic Leukemia, and Related Malignancies. Exp. Hematol. 2017, 46, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gabrilove, J.L.; Jakubowski, A.; Scher, H.; Sternberg, C.; Wong, G.; Grous, J.; Yagoda, A.; Fain, K.; Moore, M.A.S.; Clarkson, B.; et al. Effect of Granulocyte Colony-Stimulating Factor on Neutropenia and Associated Morbidity Due to Chemotherapy for Transitional-Cell Carcinoma of the Urothelium. N. Engl. J. Med. 1988, 318, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Deluca, E.; Sheridan, W.P.; Watson, D.; Szer, J.; Begley, C.G. Prior Chemotherapy Does Not Prevent Effective Mobilisation by G-Csf of Peripheral Blood Progenitor Cells. Br. J. Cancer 1992, 66, 893–899. [Google Scholar] [CrossRef] [PubMed]
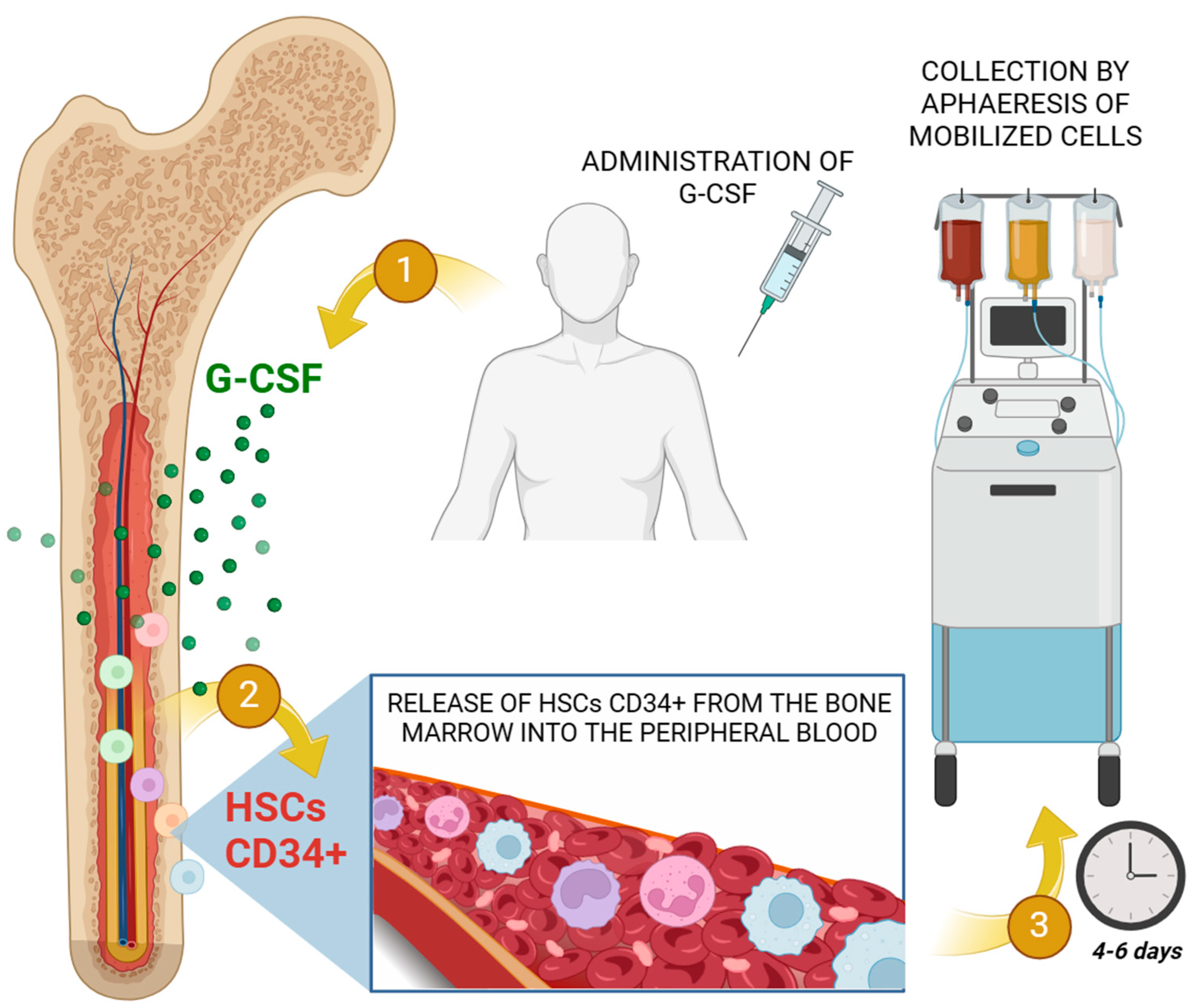
- Chang, H.H.; Liou, Y.S.; Sun, D.S. Hematopoietic Stem Cell Mobilization. Tzu Chi Med. J. 2022, 34, 270–275. [Google Scholar]
- Reyes, E.; García-Castro, I.; Esquivel, F.; Hornedo, J.; Cortes-Funes, H.; Solovera, J.; Alvarez-Mon, M. Granulocyte Colony-Stimulating Factor (G-CSF) Transiently Suppresses Mitogen-Stimulated T-Cell Proliferative Response. Br. J. Cancer 1999, 80, 229–235. [Google Scholar] [CrossRef]
- Russell, N.H.; Byrne, J.L. Allogeneic Transplantation Using Peripheral Blood Stem Cells. Best. Pract. Res. Clin. Haematol. 2001, 14, 685–700. [Google Scholar] [CrossRef]
- Favre, G.; Beksaç, M.; Bacigalupo, A.; Ruutu, T.; Nagler, A.; Gluckman, E.; Russell, N.; Apperley, J.; Szer, J.; Bradstock, K.; et al. Differences between Graft Product and Donor Side Effects Following Bone Marrow or Stem Cell Donation. Bone Marrow Transplant. 2003, 32, 873–880. [Google Scholar] [CrossRef]
- Bendall, L.J.; Bradstock, K.F. G-CSF: From Granulopoietic Stimulant to Bone Marrow Stem Cell Mobilizing Agent. Cytokine Growth Factor. Rev. 2014, 25, 355–367. [Google Scholar] [CrossRef]
- Fruehauf, S.; Tricot, G. Comparison of Unmobilized and Mobilized Graft Characteristics and the Implications of Cell Subsets on Autologous and Allogeneic Transplantation Outcomes. Biol. Blood Marrow Transpl. 2010, 16, 1629–1648. [Google Scholar] [CrossRef]
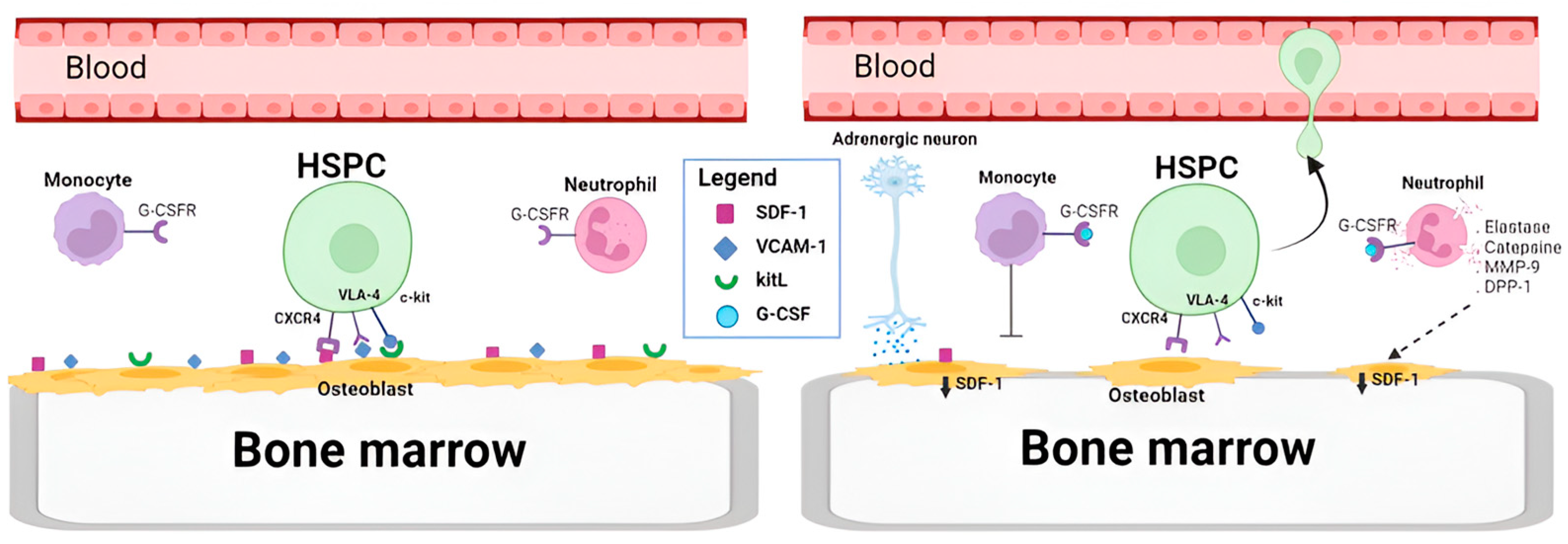
- Lévesque, J.P.; Hendy, J.; Takamatsu, Y.; Simmons, P.J.; Bendall, L.J. Disruption of the CXCR4/CXCL12 Chemotactic Interaction during Hematopoietic Stem Cell Mobilization Induced by Gcsf or Cyclophosphamide. J. Clin. Investig. 2003, 111, 187–196. [Google Scholar] [CrossRef]
- Heissig, B.; Hattori, K.; Dias, S.; Friedrich, M.; Ferris, B.; Hackett, N.R.; Crystal, R.G.; Besmer, P.; Lyden, D.; Moore, M.A.S.; et al. Recruitment of Stem and Progenitor Cells from the Bone Marrow Niche Requires MMP-9 Mediated Release of Kit-Ligand. Cell 2002, 109, 625–637. [Google Scholar] [CrossRef]
- Lévesque, J.P.; Takamatsu, Y.; Nilsson, S.K.; Haylock, D.N.; Simmons, P.J. Vascular Cell Adhesion Molecule-1 (CD106) Is Cleaved by Neutrophil Proteases in the Bone Marrow Following Hematopoietic Progenitor Cell Mobilization by Granulocyte Colony-Stimulating Factor. Blood 2001, 98, 1289–1297. [Google Scholar] [CrossRef]
- Semerad, C.L.; Christopher, M.J.; Liu, F.; Short, B.; Simmons, P.J.; Winkler, I.; Levesque, J.P.; Chappel, J.; Ross, F.P.; Link, D.C. G-CSF Potently Inhibits Osteoblast Activity and CXCL12 MRNA Expression in the Bone Marrow. Blood 2005, 106, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Yoshida, S.; Kawasumi, M.; Hashimoto, K.; Kimura, T.; Sato, Y.; Kobayashi, T.; Miyauchi, Y.; Hoshi, H.; Iwasaki, R.; et al. Osteoclasts Are Dispensable for Hematopoietic Stem Cell Maintenance and Mobilization. J. Exp. Med. 2011, 208, 2175–2181. [Google Scholar] [CrossRef]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; Van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone Marrow Macrophages Maintain Hematopoietic Stem Cell (HSC) Niches and Their Depletion Mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef] [PubMed]
- Asada, N.; Katayama, Y.; Sato, M.; Minagawa, K.; Wakahashi, K.; Kawano, H.; Kawano, Y.; Sada, A.; Ikeda, K.; Matsui, T.; et al. Matrix-Embedded Osteocytes Regulate Mobilization of Hematopoietic Stem/Progenitor Cells. Cell Stem Cell 2013, 12, 737–747. [Google Scholar] [CrossRef]
- Chow, A.; Lucas, D.; Hidalgo, A.; Méndez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; Van Rooijen, N.; et al. Bone Marrow CD169+ Macrophages Promote the Retention of Hematopoietic Stem and Progenitor Cells in the Mesenchymal Stem Cell Niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Suzuki, T.; Wakahashi, K.; Asada, N.; Kawano, Y.; Kawano, H.; Sada, A.; Minagawa, K.; Nakamura, Y.; Mizuno, S.; et al. FGF-23 from Erythroblasts Promotes Hematopoietic Progenitor Mobilization. Blood 2021, 137, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yan, L.; Shang, J.; Kang, L.; Yan, Z.; Jin, S.; Zhu, M.; Chang, H.; Gong, F.; Zhou, J.; et al. Anti-CD19 and Anti-BCMA CAR T Cell Therapy Followed by Lenalidomide Maintenance after Autologous Stem-Cell Transplantation for High-Risk Newly Diagnosed Multiple Myeloma. Am. J. Hematol. 2022, 97, 537–547. [Google Scholar] [CrossRef]
- Cao, X.Y.; Zhang, J.P.; Zhao, Y.L.; Xiong, M.; Zhou, J.R.; Lu, Y.; Sun, R.J.; Wei, Z.J.; Liu, D.Y.; Zhang, X.; et al. Analysis Benefits of a Second Allo-HSCT after CAR-T Cell Therapy in Patients with Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia Who Relapsed after Transplant. Front. Immunol. 2023, 14, 1191382. [Google Scholar] [CrossRef]
- Zurko, J.; Ramdial, J.; Shadman, M.; Ahmed, S.; Szabo, A.; Iovino, L.; Tomas, A.A.; Sauter, C.; Perales, M.A.; Shah, N.N.; et al. Allogeneic Transplant Following CAR T-Cell Therapy for Large B-Cell Lymphoma. Haematologica 2023, 108, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, C.; Cao, Y.; Wang, N.; Huang, L.; Shang, Z.; Wang, J.; Huang, L.; Xu, J.; Xiao, M.; et al. Sequential CAR T-Cell Therapy After Autologous Stem Cell Transplantation for the Treatment of Relapsed/Refractory Intravascular Large B-Cell Lymphoma with Central Nervous System Involvement: A Case Report. Front. Oncol. 2022, 12, 817969. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, B.; Lee, P.; Sasine, J.P. Chimeric Antigen Receptor T-Cell Therapy and Hematopoiesis. Cells 2023, 12, 531. [Google Scholar] [CrossRef]
- Rejeski, K.; Burchert, A.; Iacoboni, G.; Sesques, P.; Fransecky, L.; Bücklein, V.; Trenker, C.; Hernani, R.; Naumann, R.; Schäfer, J.; et al. Safety and Feasibility of Stem Cell Boost as a Salvage Therapy for Severe Hematotoxicity after CD19 CAR T-Cell Therapy. Blood Adv. 2022, 6, 4719–4725. [Google Scholar] [CrossRef]
- Ageitos, A.G.; Varney, M.L.; Bierman, P.J.; Vose, J.M.; Warkentin, P.I.; Talmadge, J.E. Comparison of Monocyte-Dependent T Cell Inhibitory Activity in GM-CSF vs. G-CSF Mobilized PSC Products. Bone Marrow Transpl. 1999, 23, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Young, M.R.I.; Young, M.E.; Wright, M.A. Stimulation of Immune-Suppressive Bone Marrow Cells by Colony-Stimulating Factors. Exp. Hematol. 1990, 18, 806–811. [Google Scholar] [PubMed]
- Ino, K.; Singh, R.K.; Talmadge, J.E. Monocytes from Mobilized Stem Cells Inhibit C Cell Function. J. Leukoc. Biol. 1997, 61, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Samuel, E.R.; Newton, K.; MacKinnon, S.; Lowdell, M.W. Successful Isolation and Expansion of CMV-Reactive T Cells from G-CSF Mobilized Donors That Retain a Strong Cytotoxic Effector Function. Br. J. Haematol. 2013, 160, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Gerdes, W.; Berthold, R.; Sack, U.; Koehl, U.; Hauschildt, S.; Grahnert, A. Comparison of Three CD3-Specific Separation Methods Leading to Labeled and Label-Free T Cells. Cells 2021, 10, 2824. [Google Scholar] [CrossRef]
- Tristán-Manzano, M.; Maldonado-Pérez, N.; Justicia-Lirio, P.; Muñoz, P.; Cortijo-Gutiérrez, M.; Pavlovic, K.; Jiménez-Moreno, R.; Nogueras, S.; Carmona, M.D.; Sánchez-Hernández, S.; et al. Physiological Lentiviral Vectors for the Generation of Improved CAR-T Cells. Mol. Ther. Oncolytics 2022, 25, 335–349. [Google Scholar] [CrossRef]
- Ji, S.Q.; Chen, H.R.; Wang, H.X.; Yan, H.M.; Pan, S.P.; Xun, C.Q. Comparison of Outcome of Allogeneic Bone Marrow Transplantation with and without Granulocyte Colony-Stimulating Factor (Lenograstim) Donor-Marrow Priming in Patients with Chronic Myelogenous Leukemia. Biol. Blood Marrow Transpl. 2002, 8, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Bonanno, G.; Pierelli, L.; Mariotti, A.; Capoluongo, E.; Contemi, A.M.; Ameglio, F.; Curti, A.; de Ritis, D.G.; Voso, M.T.; et al. Granulocyte Colony-Stimulating Factor Promotes the Generation of Regulatory DC through Induction of IL-10 and IFN-α. Eur. J. Immunol. 2004, 34, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.S.; MacDonald, K.P.A.; Rowe, V.; Johnson, D.H.; Banovic, T.; Clouston, A.D.; Hill, G.R. Donor Treatment with Pegylated G-CSF Augments the Generation of IL-10-Producing Regulatory T Cells and Promotes Transplantation Tolerance. Blood 2004, 103, 3573–3581. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Zavala, F.; Danese, S.; Kared, H.; Leone, G. Granulocyte Colony-Stimulating Factor: A Novel Mediator of T Cell Tolerance. J. Immunol. 2005, 175, 7085–7091. [Google Scholar] [CrossRef] [PubMed]
- Toh, H.C.; Sun, L.; Soe, Y.; Wu, Y.; Phoon, Y.P.; Chia, W.K.; Wu, J.; Wong, K.Y.; Tan, P. G-CSF Induces a Potentially Tolerant Gene and Immunophenotype Profile in T Cells in Vivo. Clin. Immunol. 2009, 132, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Bunse, C.E.; Borchers, S.; Varanasi, P.R.; Tischer, S.; Figueiredo, C.; Immenschuh, S.; Kalinke, U.; Köhl, U.; Goudeva, L.; Maecker-Kolhoff, B.; et al. Impaired Functionality of Antiviral T Cells in G-CSF Mobilized Stem Cell Donors: Implications for the Selection of CTL Donor. PLoS ONE 2013, 8, e77925. [Google Scholar] [CrossRef] [PubMed]
- Beloki, L.; Ramírez, N.; Olavarría, E.; Samuel, E.R.; Lowdell, M.W. Manufacturing of Highly Functional and Specific T Cells for Adoptive Immunotherapy against Virus from Granulocyte Colony-Stimulating Factor-Mobilized Donors. Cytotherapy 2014, 16, 1390–1408. [Google Scholar] [CrossRef] [PubMed]
- Clancy, L.E.; Blyth, E.; Simms, R.M.; Micklethwaite, K.P.; Ma, C.K.K.; Burgess, J.S.; Antonenas, V.; Shaw, P.J.; Gottlieb, D.J. Cytomegalovirus-Specific Cytotoxic t Lymphocytes Can Be Efficiently Expanded from Granulocyte Colony-Stimulating Factor-Mobilized Hemopoietic Progenitor Cell Products Ex Vivo and Safely Transferred to Stem Cell Transplantation Recipients to Facilitate Immune Reconstitution. Biol. Blood Marrow Transpl. 2013, 19, 725–734. [Google Scholar] [CrossRef]
- Samuel, E.R.; Beloki, L.; Newton, K.; Mackinnon, S.; Lowdell, M.W. Isolation of Highly Suppressive CD25 +FoxP3+ T Regulatory Cells from G-CSF-Mobilized Donors with Retention of Cytotoxic Anti-Viral CTLs: Application for Multi-Functional Immunotherapy Post Stem Cell Transplantation. PLoS ONE 2014, 9, e85911. [Google Scholar] [CrossRef]
- Ye, Y.; Yang, L.; Yuan, X.; Huang, H.; Luo, Y. Optimization of Donor Lymphocyte Infusion for AML Relapse After Allo-HCT in the Era of New Drugs and Cell Engineering. Front. Oncol. 2022, 11, 790299. [Google Scholar] [CrossRef]
- Caldemeyer, L.E.; Akard, L.P.; Edwards, J.R.; Tandra, A.; Wagenknecht, D.R.; Dugan, M.J. Donor Lymphocyte Infusions Used to Treat Mixed-Chimeric and High-Risk Patient Populations in the Relapsed and Nonrelapsed Settings after Allogeneic Transplantation for Hematologic Malignancies Are Associated with High Five-Year Survival If Persistent Full Donor Chimerism Is Obtained or Maintained. Biol. Blood Marrow Transpl. 2017, 23, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanam, H.; Buhmann, R.; Henschler, R. Influence of Culture Conditions on Ex Vivo Expansion of T Lymphocytes and Their Function for Therapy: Current Insights and Open Questions. Front. Bioeng. Biotechnol. 2022, 10, 886637. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. Sorting through Subsets: Which T-Cell Populations Mediate Highly Effective Adoptive Immunotherapy? J. Immunother. 2012, 35, 651–660. [Google Scholar] [CrossRef]
- Canesin, G.; Hoyt, H.; Williams, R.; Silva, M.; Chng, M.; Cummins, C.; Ung, M.; Qiu, H.; Shin, J.; Hu, J.; et al. G-CSF/Plerixafor Dual-Mobilized Donor Derived CD33CAR T-Cells as Potent and Effective AML Therapy in Pre-Clinical Models. Blood 2021, 138, 1716. [Google Scholar] [CrossRef]
- Battram, A.M.; Oliver-Caldés, A.; Suárez-Lledó, M.; Lozano, M.; Bosch i Crespo, M.; Martínez-Cibrián, N.; Cid, J.; Moreno, D.F.; Rodríguez-Lobato, L.G.; Urbano-Ispizua, A.; et al. T Cells Isolated from G-CSF-Treated Multiple Myeloma Patients Are Suitable for the Generation of BCMA-Directed CAR-T Cells. Mol. Ther. Methods Clin. Dev. 2022, 26, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Otto, M.; Barfield, R.C.; Iyengar, R.; Gatewood, J.; Müller, I.; Holladay, M.S.; Houston, J.; Leung, W.; Handgretinger, R. Human Γδ T Cells from G-CSF-Mobilized Donors Retain Strong Tumoricidal Activity and Produce Immunomodulatory Cytokines after Clinical-Scale Isolation. J. Immunother. 2005, 28, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Abken, H. Treatment with Living Drugs: Pharmaceutical Aspects of CAR T Cells. Pharmacology 2022, 107, 446–463. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H. The NK-92 Cell Line—30 Years Later: Its Impact on Natural Killer Cell Research and Treatment of Cancer. Cytotherapy 2023, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, M.; Battram, A.M.; Perez-Amill, L.; Martín-Antonio, B. Natural Killer Cells in Immunotherapy: Are We Nearly There? Cancers 2020, 12, 3139. [Google Scholar] [CrossRef]
- Cheng, Z.F.; Li, H.K.; Yang, H.P.; Lee, C.Y.; Tang, S.W.; Lin, Y.L.; Hsiao, S.C. A Novel Endogenous CD16-Expressing Natural Killer Cell for Cancer Immunotherapy. Biochem. Biophys. Rep. 2021, 26, 100935. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Aloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a Human Cell Line (NK-92) with Phenotypical and Functional Characteristics of Activated Natural Killer Cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Sarvaria, A.; Jawdat, D.; Madrigal, J.A.; Saudemont, A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front. Immunol. 2017, 8, 329. [Google Scholar] [CrossRef]
- Patel, A.; Truscott, L.C.; De Oliveira, S.N. Development of Optimized Protocol for Generation of NK Cells Expressing Chimeric Antigen Receptors from Hematopoietic Stem Cells for Cancer Immunotherapy. Blood 2015, 126, 2044. [Google Scholar] [CrossRef]
- Clausen, J.; Petzer, A.L.; Vergeiner, B.; Enk, M.; Stauder, R.; Gastl, G.; Gunsilius, E. Optimal Timing for the Collection and in Vitro Expansion of Cytotoxic CD56+ Lymphocytes from Patients Undergoing Autologous Peripheral Blood Stem Cell Transplantation. J. Hematother Stem Cell Res. 2001, 10, 513–521. [Google Scholar] [CrossRef]
- Clausen, J.; Enk, M.; Vergeiner, B.; Eisendle, K.; Petzer, A.L.; Gastl, G.; Gunsilius, E. Suppression of Natural Killer Cells in the Presence of CD34+ Blood Progenitor Cells and Peripheral Blood Lymphocytes. Biol. Blood Marrow Transpl. 2004, 10, 691–697. [Google Scholar] [CrossRef]
- Xiong, Y.; Mouginot, M.; Reppel, L.; Qian, C.; Stoltz, J.-F.; Bensoussan, D.; Decot, V. Modification of NK Cell Subset Repartition and Functions in Granulocyte Colony-Stimulating Factor-Mobilized Leukapheresis after Expansion with IL-15. Immunol. Res. 2017, 65, 1130–1138. [Google Scholar] [CrossRef]
- Pelosi, A.; Besi, F.; Tumino, N.; Merli, P.; Quatrini, L.; Li Pira, G.; Algeri, M.; Moretta, L.; Vacca, P. NK Cells and PMN-MDSCs in the Graft from G-CSF Mobilized Haploidentical Donors Display Distinct Gene Expression Profiles from Those of the Non-Mobilized Counterpart. Front. Immunol. 2021, 12, 657329. [Google Scholar] [CrossRef]
- Zhao, X.; Peng, T.; Cao, X.; Hou, Y.; Li, R.; Han, T.; Fan, Z.; Zhao, M.; Chang, Y.; Chen, H.; et al. In Vivo G-CSF Treatment Activates the GR-SOCS1 Axis to Suppress IFN-γ Secretion by Natural Killer Cells. Cell Rep. 2022, 40, 111342. [Google Scholar] [CrossRef] [PubMed]
- Gazitt, Y. Immunologic Profiles of Effector Cells and Peripheral Blood Stem Cells Mobilized with Different Hematopoietic Growth Factors. Stem Cells 2000, 18, 390–398. [Google Scholar] [CrossRef]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-Induced Memory-like Natural Killer Cells Exhibit Enhanced Responses against Myeloid Leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.P.; Miljkovic, M.D.; Fleisher, T.A.; Pittaluga, S.; Hsu-Albert, J.; Bryant, B.R.; Petrus, M.N.; Perera, L.P.; Müller, J.R.; Shih, J.H.; et al. Short-Course IL-15 given as a Continuous Infusion Led to a Massive Expansion of Effective NK Cells: Implications for Combination Therapy with Antitumor Antibodies. J. Immunother. Cancer 2021, 9, e002193. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, P.; Kamenjarin, K.; Ossa, J.F.V.; Uherek, B.; Bönig, H.; Wels, W.S. Directed Differentiation of Mobilized Hematopoietic Stem and Progenitor Cells into Functional NK Cells with Enhanced Antitumor Activity. Cells 2020, 9, 811. [Google Scholar] [CrossRef] [PubMed]
- Ojo, E.O.; Sharma, A.A.; Liu, R.; Moreton, S.; Checkley-Luttge, M.A.; Gupta, K.; Lee, G.; Lee, D.A.; Otegbeye, F.; Sekaly, R.P.; et al. Membrane Bound IL-21 Based NK Cell Feeder Cells Drive Robust Expansion and Metabolic Activation of NK Cells. Sci. Rep. 2019, 9, 14916. [Google Scholar] [CrossRef]
- Zhu, Y.; Smith, D.J.; Zhou, Y.; Li, Y.R.; Yu, J.; Lee, D.; Wang, Y.C.; Di Biase, S.; Wang, X.; Hardoy, C.; et al. Development of Hematopoietic Stem Cell-Engineered Invariant Natural Killer T Cell Therapy for Cancer. Cell Stem Cell 2019, 25, 542–557.e9. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballesteros-Ribelles, A.; Millán-López, A.; Carmona-Luque, M.; Herrera, C. Granulocyte Colony Stimulating Factor-Mobilized Peripheral Blood Mononuclear Cells: An Alternative Cellular Source for Chimeric Antigen Receptor Therapy. Int. J. Mol. Sci. 2024, 25, 5769. https://doi.org/10.3390/ijms25115769
Ballesteros-Ribelles A, Millán-López A, Carmona-Luque M, Herrera C. Granulocyte Colony Stimulating Factor-Mobilized Peripheral Blood Mononuclear Cells: An Alternative Cellular Source for Chimeric Antigen Receptor Therapy. International Journal of Molecular Sciences. 2024; 25(11):5769. https://doi.org/10.3390/ijms25115769
Chicago/Turabian StyleBallesteros-Ribelles, Antonio, Alejandro Millán-López, MDolores Carmona-Luque, and Concha Herrera. 2024. "Granulocyte Colony Stimulating Factor-Mobilized Peripheral Blood Mononuclear Cells: An Alternative Cellular Source for Chimeric Antigen Receptor Therapy" International Journal of Molecular Sciences 25, no. 11: 5769. https://doi.org/10.3390/ijms25115769
APA StyleBallesteros-Ribelles, A., Millán-López, A., Carmona-Luque, M., & Herrera, C. (2024). Granulocyte Colony Stimulating Factor-Mobilized Peripheral Blood Mononuclear Cells: An Alternative Cellular Source for Chimeric Antigen Receptor Therapy. International Journal of Molecular Sciences, 25(11), 5769. https://doi.org/10.3390/ijms25115769