
Genetic Mutations and Mitochondrial Redox Signaling as Modulating Factors in Hypertrophic Cardiomyopathy: A Scoping Review

 ,
,  ,
,  , , ,
, , , 
Abstract
1. Introduction
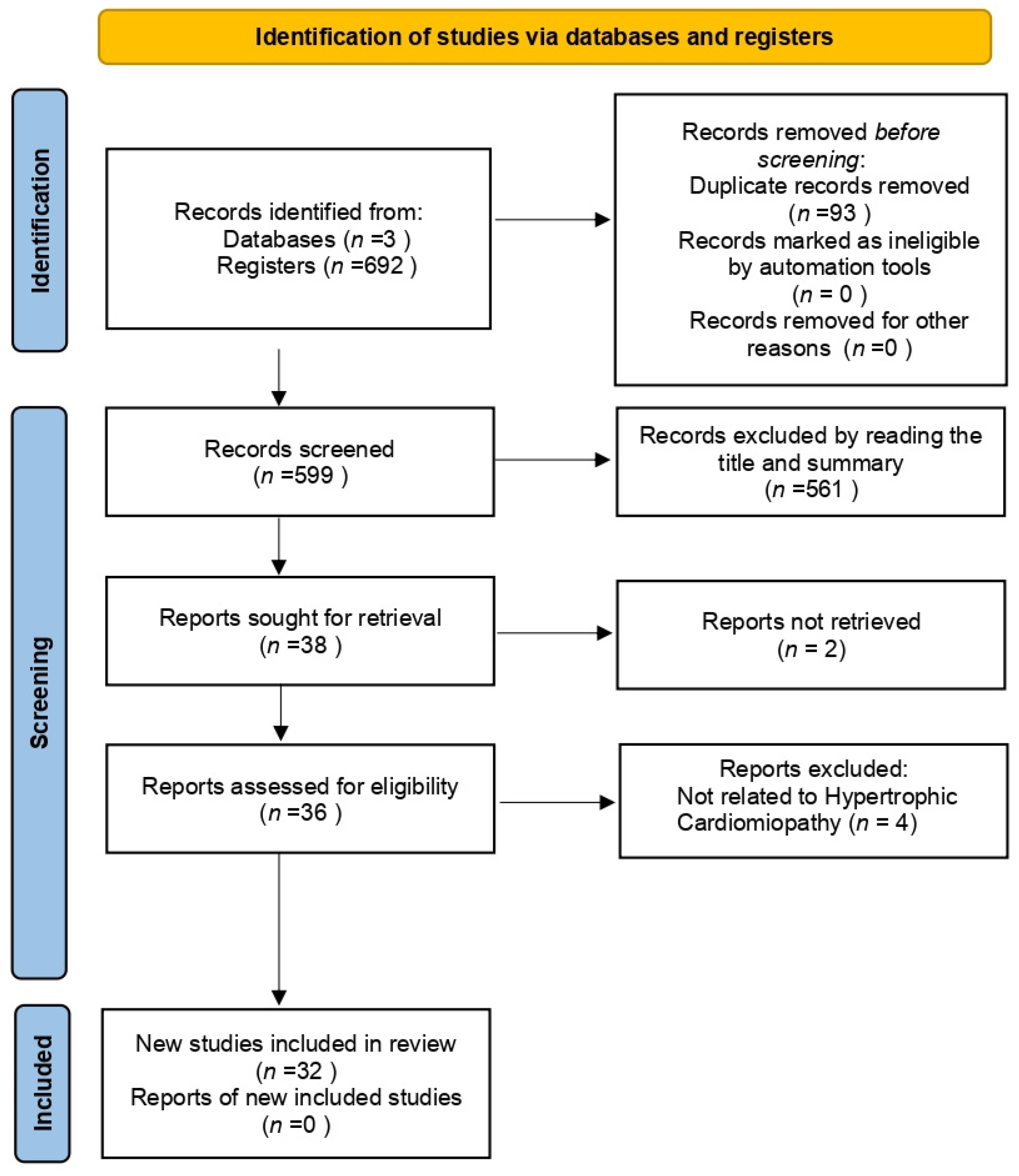
2. Materials and Methods
2.1. Protocol and Registration
2.2. Eligibility Criteria
2.3. Exclusion Criteria
2.4. Data Sources
2.5. Study Selection Process
2.6. Data Extraction Process
2.7. Data Synthesis
3. Results and Discussion
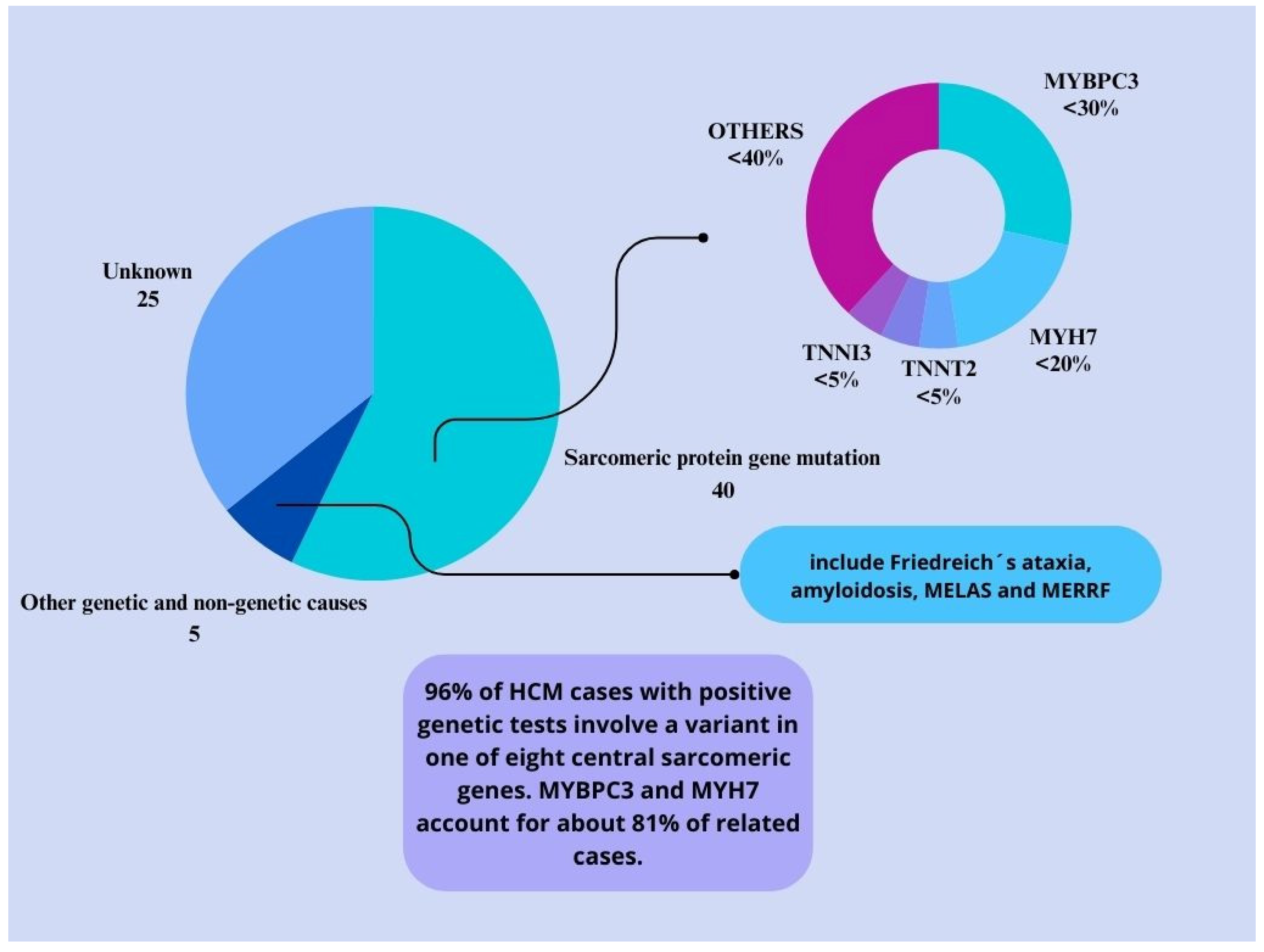
3.1. Genetic Mutations in HCM
3.1.1. Epigenetics and HCM
3.1.2. Myc Gene in HCM
3.1.3. Mitochondrial DNA Mutations and HCM
3.1.4. MicroRNAs and Gene Therapy in HCM
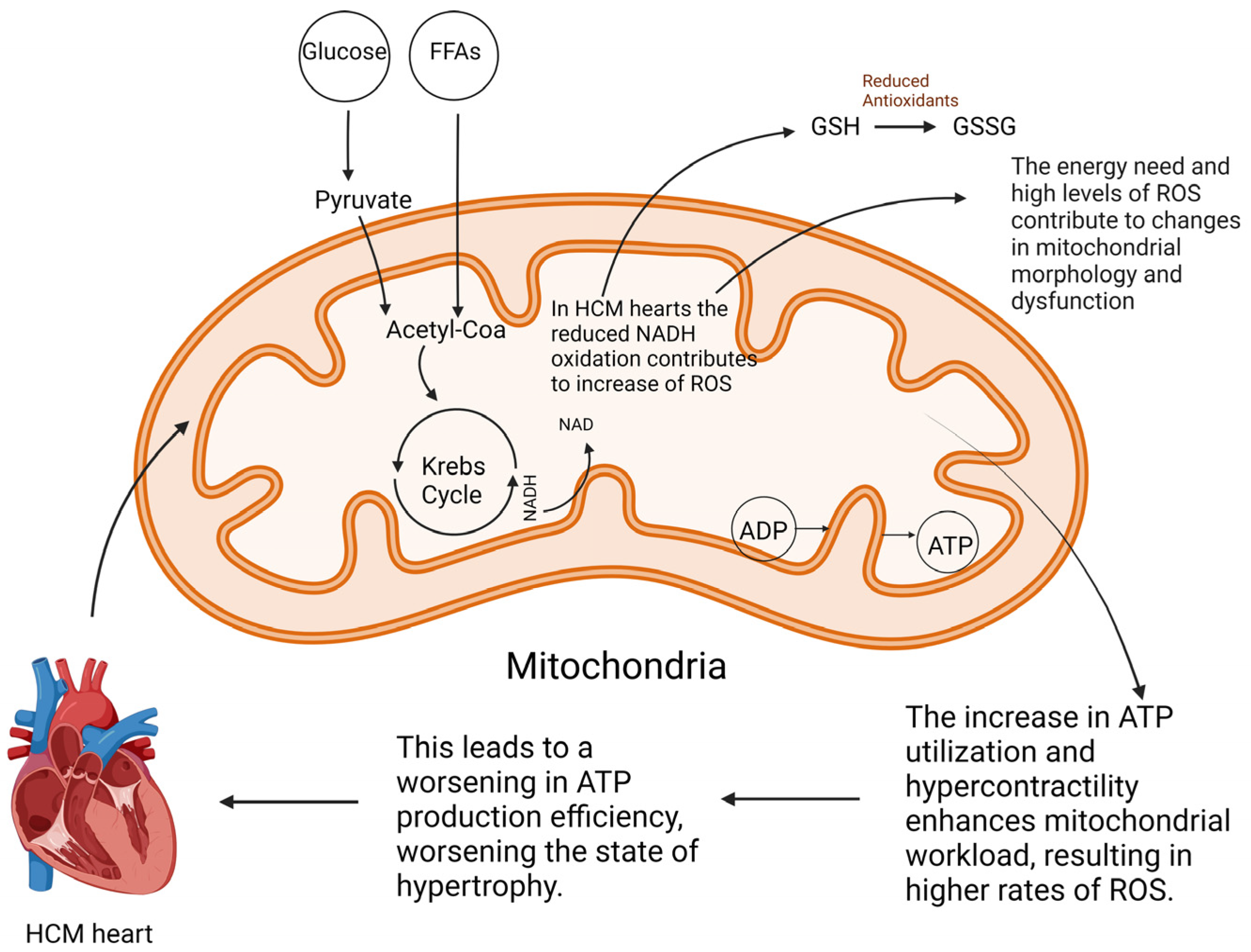
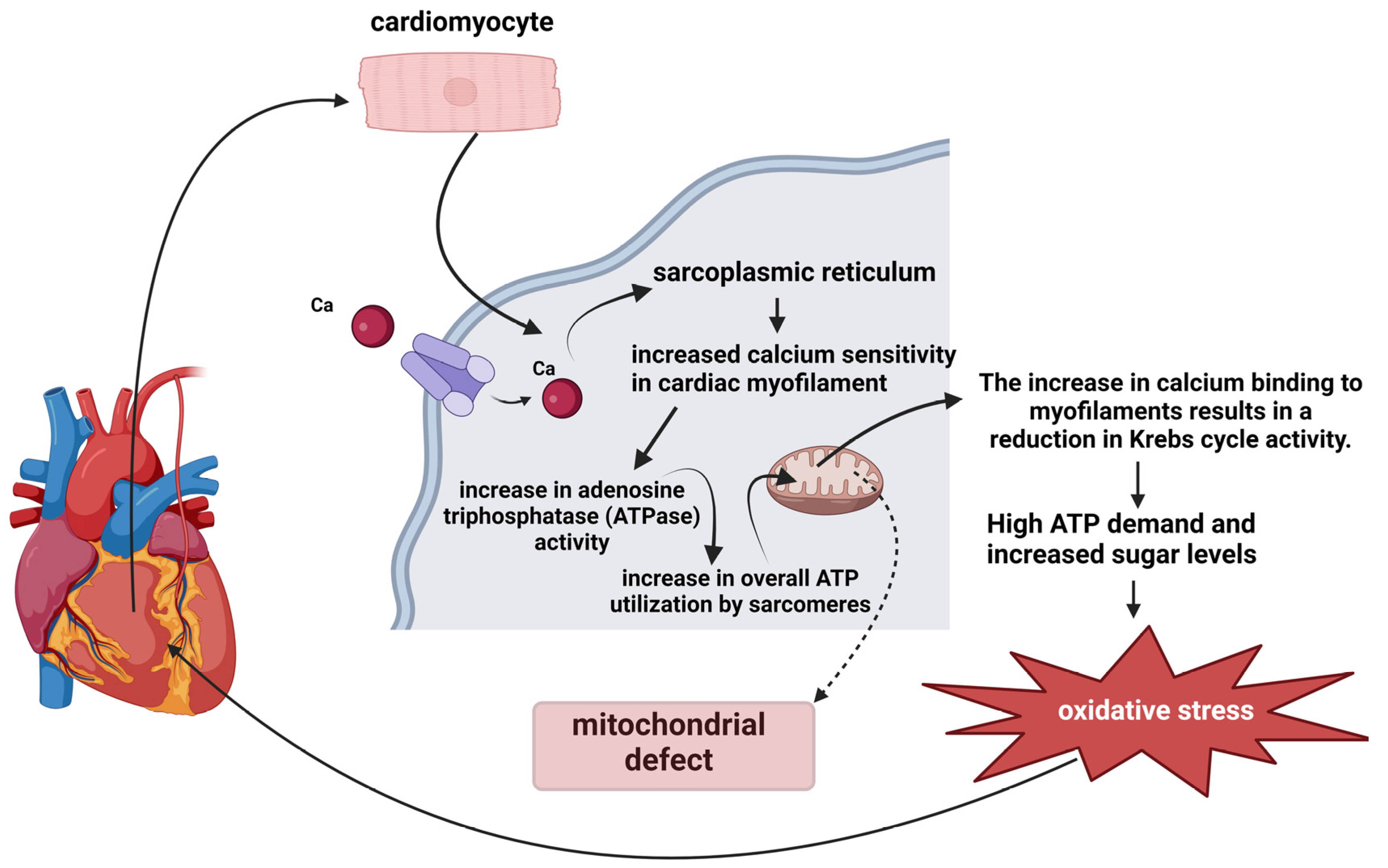
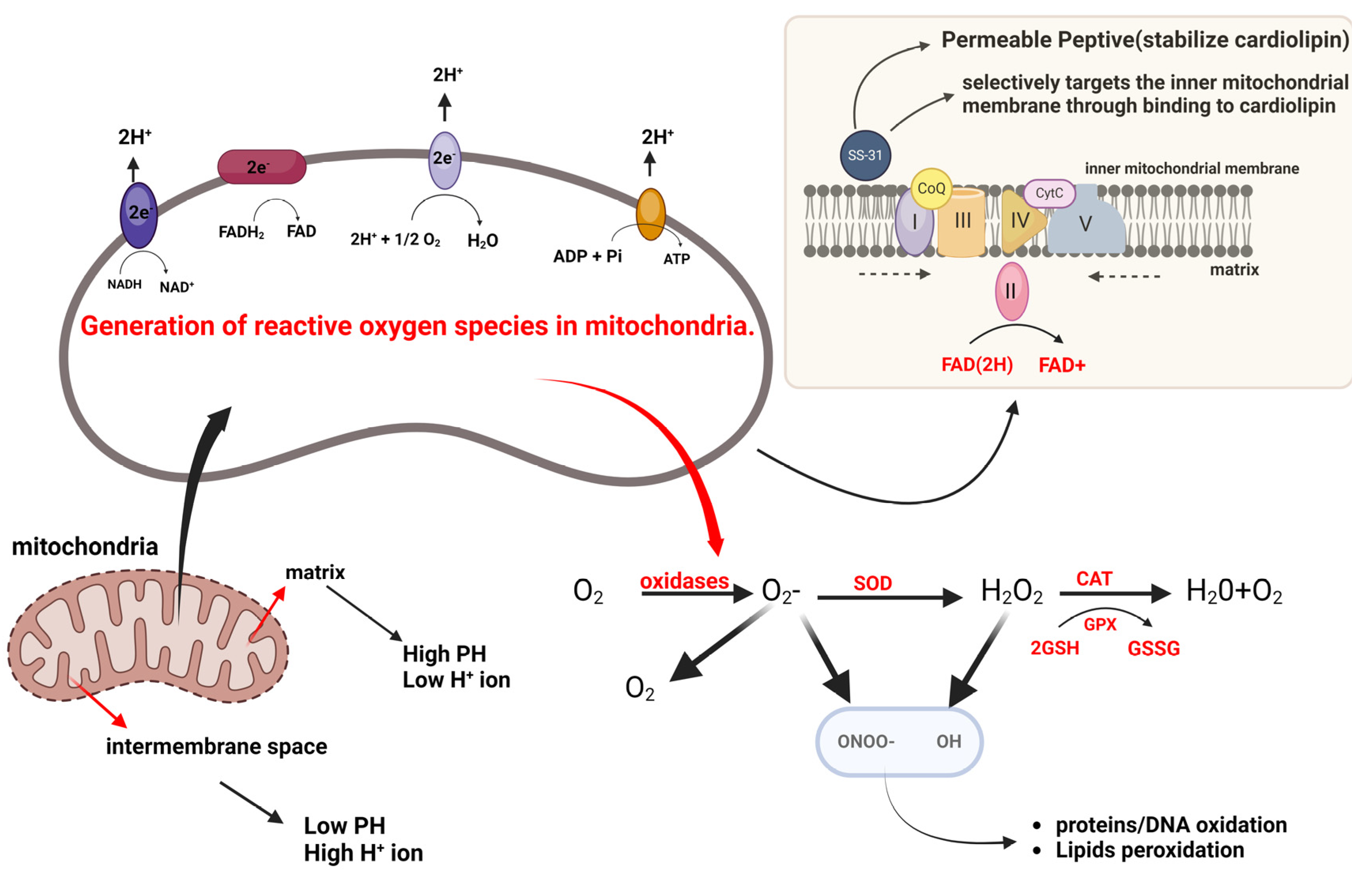
3.2. Mitochondrial Redox Signaling
3.2.1. Emerging Therapeutic Strategies
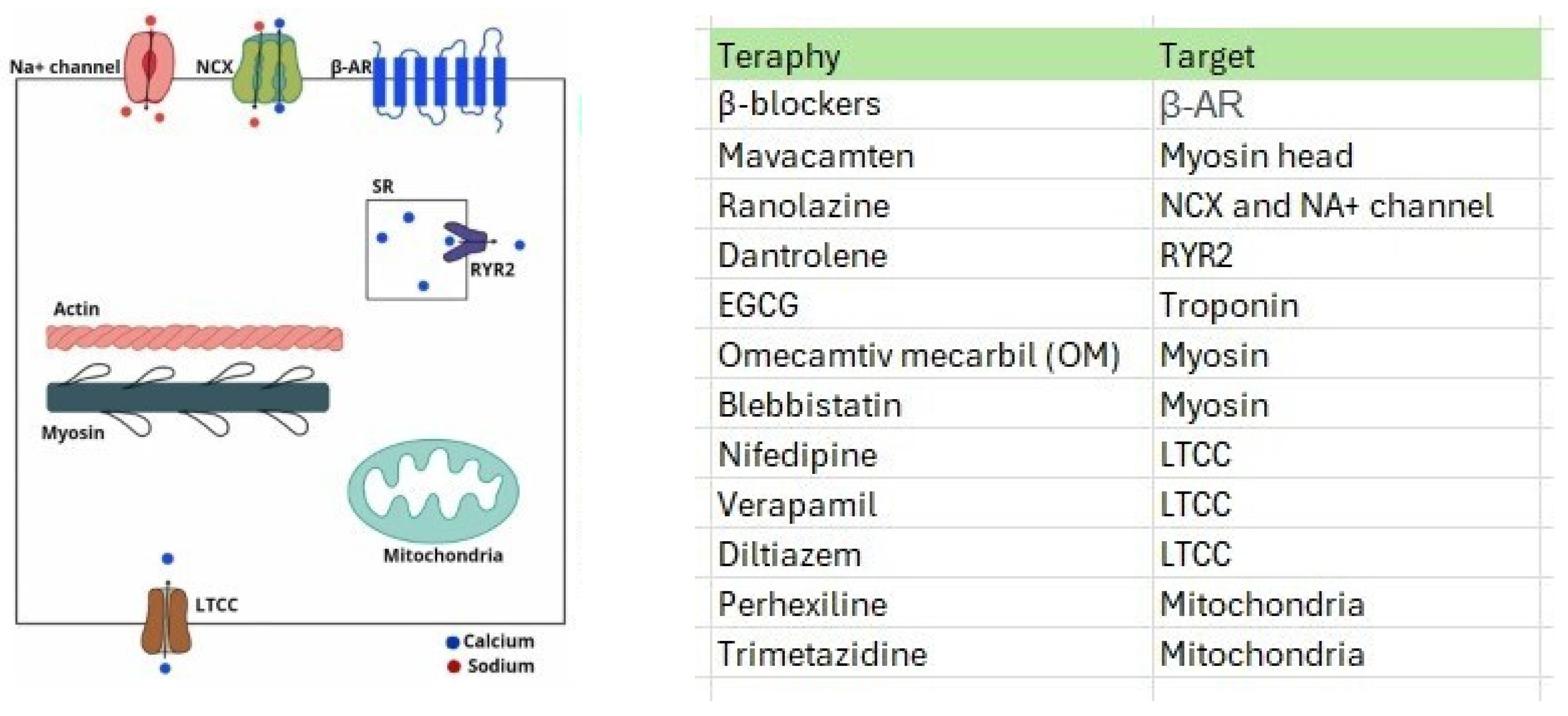
3.2.2. Emerging Drug Therapies for HCM
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dipchand, A.I.; Tein, I.; Robinson, B.; Benson, L.N. Maternally inherited hypertrophic cardiomyopathy: A manifestation of mitochondrial DNA mutations–clinical course in two families. Pediatr. Cardiol. 2001, 22, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, N.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Christiansen, L.B.; Dela, F. Impaired mitochondrial oxidative phosphorylation and fatty acid oxidation with enhanced mitochondrial oxidative stress in spontaneously occurring feline hypertrophic cardiomyopathy. J. Card. Fail. 2014, 20, S147. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, V.; Waddingham, M.T.; Tsuchimochi, H.; Maack, C.; Pearson, J.T. Mechano-energetic uncoupling in hypertrophic cardiomyopathy: Pathophysiological mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. Plus 2023, 4, 100036. [Google Scholar] [CrossRef]
- Elliott, P. Hypertrophic cardiomyopathy. Circulation 2018, 138, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T. Hypertrophic cardiomyopathy: Diverse pathophysiology revealed by genetic research, toward future therapy. Keio J. Med. 2020, 69, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Hool, L.C. Impaired calcium handling and mitochondrial metabolic dysfunction as early markers of hypertrophic cardiomyopathy. Arch. Biochem. Biophys. 2019, 665, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction, and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Wasmus, C.; Dudek, J. Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies. Life 2020, 10, 277. [Google Scholar] [CrossRef]
- Weissman, D.; Maack, C. Redox signaling in heart failure and therapeutic implications. Free Radic. Biol. Med. 2021, 171, 345–364. [Google Scholar] [PubMed]
- Lee, H.G.; Chen, Q.; Wolfram, J.A.; Richardson, S.L.; Liner, A.; Siedlak, S.L.; Zhu, X.; Ziats, N.P.; Fujioka, H.; Felsher, D.W.; et al. Cell cycle re-entry and mitochondrial defects in myc-mediated hypertrophic cardiomyopathy and heart failure. PLoS ONE 2009, 4, e7172. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, L.B.; Dela, F.; Koch, J.; Hansen, C.N.; Leifsson, P.S.; Yokota, T. Impaired cardiac mitochondrial oxidative phosphorylation and enhanced mitochondrial oxidative stress in feline hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1237–H1247. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Pan, H.; Tan, C.; Sun, Y.; Song, Y.; Zhang, X.; Yang, W.; Wang, X.; Li, D.; Dai, Y.; et al. Mitochondrial Dysfunctions Contribute to Hypertrophic Cardiomyopathy in Patient iPSC-Derived Cardiomyocytes with MT-RNR2 Mutation. Stem Cell Rep. 2018, 10, 808–821. [Google Scholar] [CrossRef]
- van der Velden, J.; Tocchetti, C.G.; Varricchi, G.; Bianco, A.; Sequeira, V.; Hilfiker-Kleiner, D.; Hamdani, N.; Leite-Moreira, A.F.; Mayr, M.; Falcão-Pires, I.; et al. Metabolic changes in hypertrophic cardiomyopathies: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1273–1280. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Z.; Chen, C.; Yao, S.; Yang, Q.; Li, F.; He, X.; Ai, C.; Wang, M.; Guan, M.X. Deletion of Gtpbp3 in zebrafish revealed the hypertrophic cardiomyopathy manifested by aberrant mitochondrial tRNA metabolism. Nucleic Acids Res. 2019, 47, 5341–5355. [Google Scholar] [CrossRef]
- Li, X.; Lu, W.J.; Li, Y.N.; Wu, F.; Bai, R.; Ma, S.; Dong, T.; Zhang, H.; Lee, A.S.; Wang, Y.; et al. MLP-deficient human pluripotent stem cell-derived cardiomyocytes develop hypertrophic cardiomyopathy and heart failure phenotypes due to abnormal calcium handling. Cell Death Dis. 2019, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Saoura, M.; Powell, C.A.; Kopajtich, R.; Alahmad, A.; AL-Balool, H.H.; Albash, B.; Alfadhel, M.; Alston, C.L.; Bertini, E.; Bonnen, P.E.; et al. Mutations in ELAC2 associated with hypertrophic cardiomyopathy impair mitochondrial tRNA 3′-end processing. Hum. Mutat. 2019, 40, 1731–1748. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Roest, A.S.V.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef]
- Zhang, Q.; He, X.; Yao, S.; Lin, T.; Zhang, L.; Chen, D.; Chen, C.; Yang, Q.; Li, F.; Zhu, Y.M.; et al. Ablation of Mto1 in zebrafish exhibited hypertrophic cardiomyopathy manifested by mitochondrion RNA maturation deficiency. Nucleic Acids Res. 2021, 49, 4689–4704. [Google Scholar] [CrossRef]
- Previs, M.J.; O’Leary, T.S.; Morley, M.P.; Palmer, B.M.; LeWinter, M.; Yob, J.M.; Pagani, F.D.; Petucci, C.; Kim, M.S.; Margulies, K.B.; et al. Defects in the proteome and metabolome in human hypertrophic cardiomyopathy. Circ. Heart Fail. 2022, 15, e009521. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.; Ewoldt, J.; Venturini, G.; Pereira, A.C.; Padilha, K.; Lawton, M.; Lin, W.; Goel, R.; Luptak, I.; Perissi, V.; et al. Multi-omics profiling of hypertrophic cardiomyopathy reveals altered mechanisms in mitochondrial dynamics and excitation–contraction coupling. Int. J. Mol. Sci. 2023, 24, 4724. [Google Scholar] [CrossRef] [PubMed]
- Nollet, E.E.; Duursma, I.; Rozenbaum, A.; Eggelbusch, M.; Wuest, R.C.; Schoonvelde, S.A.C.; Michels, M.; Jansen, M.; van der Wel, N.N.; Bedi, K.C.; et al. Mitochondrial dysfunction in human hypertrophic cardiomyopathy is linked to cardiomyocyte architecture disruption and corrected by improving NADH-driven mitochondrial respiration. Eur. Heart J. 2023, 44, 1170–1185. [Google Scholar] [CrossRef] [PubMed]
- Marin-Garcia, J.; Ananthakrishnan, R.; Goldenthal, M.J.; Filiano, J.J.; Perez-Atayde, A. Cardiac mitochondrial dysfunction and DNA depletion in children with hypertrophic cardiomyopathy. J. Inherit. Metab. Dis. 1997, 20, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2013, 40, 385–394. [Google Scholar]
- Kiselev, I.; Kozin, M.; Baulina, N.; Pisklova, M.; Danilova, L.; Zotov, A.; Chumakova, O.; Zateyshchikov, D.; Favorova, O. Novel Genes Involved in Hypertrophic Cardiomyopathy: Data of Transcriptome and Methylome Profiling. Int. J. Mol. Sci. 2022, 23, 15280. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, J.O.; Frenneaux, M.P.; Sherrid, M.V. Myocardial energy depletion and dynamic systolic dysfunction in hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 2016, 13, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Gollapudi, S.K.; Del Rio, C.L.; Spudich, J.A.; McDowell, R. Mavacamten, a precision medicine for hypertrophic cardiomyopathy: From a motor protein to patients. Sci. Adv. 2023, 9, eabo7622. [Google Scholar] [CrossRef]
- Wijnker, P.J.M.; Sequeira, V.; Kuster, D.W.D.; van der Velden, J. Hypertrophic cardiomyopathy: A vicious cycle triggered by sarcomere mutations and secondary disease hits. Antioxid. Redox Signal. 2019, 31, 318–358. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Tugrul, O.F.; Lai, A.; Amr, A.; Haas, J.; Proctor, T.; Ehlermann, P.; Jensen, K.; Katus, H.A.; et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin. Res. Cardiol. 2018, 107, 30–41. [Google Scholar] [CrossRef]
- Glavaški, M.; Velicki, L.; Vučinić, N. Hypertrophic cardiomyopathy: Genetic foundations, outcomes, interconnections, and their modifiers. Medicina 2023, 59, 1424. [Google Scholar] [CrossRef]
- Chou, C.; Chin, M.T. Genetic and Molecular Mechanisms of Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 2522. [Google Scholar] [CrossRef] [PubMed]
- Tuohy, C.V.; Kaul, S.; Song, H.K.; Nazer, B.; Heitner, S.B. Hypertrophic cardiomyopathy: The future of treatment. Eur. J. Heart Fail. 2020, 22, 228–240. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.; Montag, J.; Radocaj, A.; Brenner, B. Hypertrophic cardiomyopathy: Cell-to-cell imbalance in gene expression and contraction force as trigger for disease phenotype development. Circ. Res. 2016, 119, 992–995. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Schlesinger, J.; Schoenhals, S.; Tönjes, M.; Dunkel, I.; Meierhofer, D.; Cano, E.; Schulz, K.; Berger, M.F.; Haack, T.; et al. Phosphorylation of the chromatin remodeling factor DPF3a induces cardiac hypertrophy through releasing HEY repressors from DNA. Nucleic Acids Res. 2016, 44, 2538–2553. [Google Scholar] [CrossRef]
- Wolf, C.M. Hypertrophic cardiomyopathy: Genetics and clinical perspectives. Cardiovasc. Diagn. Ther. 2019, 9 (Suppl. S2), S388–S415. [Google Scholar] [CrossRef]
- Guan, F.; Yang, X.; Li, J.; Dong, W.; Zhang, X.; Liu, N.; Gao, S.; Wang, J.; Zhang, L.; Lu, D.; et al. New molecular mechanism underlying myc-mediated cytochrome P450 2E1 upregulation in apoptosis and energy metabolism in the myocardium. J. Am. Heart Assoc. 2019, 8, e009871. [Google Scholar] [CrossRef]
- Wang, L.; Lu, F.; Xu, J. Identification of Potential miRNA-mRNA Regulatory Network Contributing to Hypertrophic Cardiomyopathy (HCM). Front. Cardiovasc. Med. 2021, 8, 660372. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhuang, Q.; Guo, F.; Fu, L.; Dong, Y.; Xie, S.; Ding, X.; Hu, S.; Zhou, X.D.; Jiang, Y.; Zhou, H.; et al. 1-deoxynojirimycin promotes cardiac function and rescues mitochondrial cristae in mitochondrial hypertrophic cardiomyopathy. J. Clin. Investig. 2023, 133, e164660. [Google Scholar] [CrossRef]
- García-Díaz, L.; Coserria, F.; Antiñolo, G. Hypertrophic cardiomyopathy due to mitochondrial disease: Prenatal diagnosis, management, and outcome. Case Rep. Obstet. Gynecol. 2013, 2013, 472356. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, J.; Zhang, Q.; Yang, Q.; Yi, S.; Shen, Y.; Luo, J.; Qin, Z. Clinical and genetic analysis of combined oxidative phosphorylation defificiency-10 caused by MTO1 mutation. Clin. Chim. Acta 2022, 526, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Prondzynski, M.; Mearini, G.; Carrier, L. Gene therapy strategies in the treatment of hypertrophic cardiomyopathy. Pflug. Arch. 2019, 471, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Paratz, E.D.; Mundisugih, J.; Rowe, S.J.; Kizana, E.; Semsarian, C. Gene therapy in cardiology: Is a cure for hypertrophic cardiomyopathy on the horizon? Can. J. Cardiol. 2023, 40, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Marti-Gutierrez, N.; Park, S.W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef]
- Halkein, J.; Tabruyn, S.P.; Ricke-Hoch, M.; Haghikia, A.; Nguyen, N.-Q.; Scherr, M.; Castermans, K.; Malvaux, L.; Lambert, V.; Thiry, M.; et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J. Clin. Investig. 2013, 123, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Heggermont, W.A.; Papageorgiou, A.P.; Quaegebeur, A.; Deckx, S.; Carai, P.; Verhesen, W.; Eelen, G.; Schoors, S.; van Leeuwen, R.; Alekseev, S. Inhibition of microRNA-146a and overexpression of its target dihydrolipoyl succinyltransferase protect against pressure overload-Induced cardiac hypertrophy and dysfunction. Circulation 2017, 136, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wakimoto, H.; Seidman, J.G.; Seidman, C.E. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 2013, 342, 111–114. [Google Scholar] [CrossRef]
- Mearini, G.; Stimpel, D.; Geertz, B.; Weinberger, F.; Krämer, E.; Schlossarek, S.; Mourot-Filiatre, J.; Stoehr, A.; Dutsch, A.; Wijnker, P.J.M.; et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 2014, 5, 5515. [Google Scholar] [CrossRef]
- Tardiff, J.C.; Carrier, L.; Bers, D.M.; Poggesi, C.; Ferrantini, C.; Coppini, R.; Maier, L.S.; Ashrafian, H.; Huke, S.; van der Velden, J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 2015, 105, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Prondzynski, M.; Krämer, E.; Laufer, S.D.; Shibamiya, A.; Pless, O.; Flenner, F.; Müller, O.J.; Münch, J.; Redwood, C.; Hansen, A.; et al. Evaluation of MYBPC3 trans-Splicing and Gene Replacement as Therapeutic Options in Human iPSC-Derived Cardiomyocytes. Mol. Ther. Nucleic Acids 2017, 7, 475–486. [Google Scholar] [CrossRef] [PubMed]
- German, D.M.; Mitalipov, S.; Mishra, A.; Kaul, S. Therapeutic Genome Editing in Cardiovascular Diseases. JACC Basic Transl. Sci. 2019, 4, 122–131. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vakrou, S.; Abraham, M.R. Hypertrophic cardiomyopathy: A heart in need of an energy bar? Front. Physiol. 2014, 5, 309. [Google Scholar] [CrossRef]
- Nollet, E.E.; Algül, S.; Goebel, M.; Schlossarek, S.; van der Wel, N.N.; Jans, J.J.; van de Wiel, M.A.; Knol, J.C.; Pham, T.V.; Piersma, S.R.; et al. Western diet triggers cardiac dysfunction in a heterozygous Mybpc3-targeted knock-in hypertrophic cardiomyopa-thy mouse model. Eur. J. Heart Fail. 2023, 25, 31–32. [Google Scholar]
- Chen, Y.; Zhang, Z.; Hu, F.; Yang, W.; Yuan, J.; Cui, J.; Hao, S.; Hu, J.; Zhou, Y.; Qiao, S. 17β-estradiol prevents cardiac diastolic dysfunction by stimulating mitochondrial function: A preclinical study in a mouse model of a human hypertrophic cardiomyopathy mutation. J. Steroid Biochem. Mol. Biol. 2015, 147, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.G.; Hussey, J.W.; Dick, I.E. CACNA1C-Related Channelopathies. Handb. Exp. Pharmacol. 2023, 279, 159–181. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Ye, D.; Jin, F.; Tester, D.J.; Huseby, A.; Bos, J.M.; Johnson, A.J.; Kanter, R.; Ackerman, M.J. Identification and functional characterization of a novel CACNA1C-mediated cardiac disorder characterized by prolonged QT intervals with hypertrophic cardiomyopathy, congenital heart defects, and sudden cardiac death. Circ. Arrhythm. Electrophysiol. 2015, 8, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Sun, Y.; Zhuang, Q.; Song, Y.R.; Wu, B.F.; Jia, Z.X.; Pan, H.Y.; Zhou, H.; Hu, S.Y.; Zhang, B.T.; et al. Mitochondrial dysfunction caused by m.2336T>C mutation with hypertrophic cardiomyopathy in cybrid cell lines. Mitochondrion 2019, 46, 313–320. [Google Scholar] [CrossRef]
- Riaz, M.; Park, J.; Sewanan, L.R.; Ren, Y.; Schwan, J.; Das, S.K.; Pomianowski, P.T.; Huang, Y.; Ellis, M.W.; Luo, J.; et al. Muscle LIM Protein Force-Sensing Mediates Sarcomeric Biomechanical Signaling in Human Familial Hypertrophic Cardiomyopathy. Circulation 2022, 145, 1238–1253. [Google Scholar] [CrossRef]
- Walsh, R.; Offerhaus, J.A.; Tadros, R.; Bezzina, C.R. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat. Rev. Cardiol. 2022, 19, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.T.; Aryal, P.; Szweda, L.I.; Koch, W.J.; Leinwand, L.A. Alterations in mitochondrial function in a mouse model of hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H575–H583. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, V.; Bertero, E.; Maack, C. Energetic drain driving hypertrophic cardiomyopathy. FEBS Lett. 2019, 593, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Feridooni, H.A.; Dibb, K.M.; Howlett, S.E. How cardiomyocyte excitation, calcium release and contraction become altered with age. J. Mol. Cell. Cardiol. 2015, 83, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Coppini, R.; Ferrantini, C.; Mugelli, A.; Poggesi, C.; Cerbai, E. Altered Ca2+ and Na+ homeostasis in human hypertrophic cardiomyopathy: Implications for arrhythmogenesis. Front. Physiol. 2018, 9, 1391. [Google Scholar] [CrossRef] [PubMed]
- Farhana, A.; Lappin, S.L. Biochemistry, Lactate Dehydrogenase. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Christiansen, L.B.; Prats, C.; Hyttel, P.; Koch, J. Ultrastructural myocardial changes in seven cats with spontaneous hypertrophic cardiomyopathy. J. Vet. Cardiol. 2015, 17 (Suppl. S1), S220–S232. [Google Scholar] [CrossRef]
- Povos, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Disfunção mitocondrial e estresse oxidativo nas cardiopatias. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Lombardi, M.; Lazzeroni, D.; Pisano, A.; Girolami, F.; Alfieri, O.; La Canna, G.; D’amati, G.; Olivotto, I.; Rimoldi, O.E.; Foglieni, C.; et al. Mitochondrial energetics and Ca2+-activated ATPase in obstructive hypertrophic cardiomyopathy. J. Clin. Med. 2020, 9, 1799. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, H.; Liu, J. P66Shc Deletion Ameliorates Oxidative Stress and Cardiac Dysfunction in Pressure Overload-Induced Heart Failure. J. Card. Fail. 2020, 26, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.L.; Fu, Y.; Wu, C.W.; Zhang, Y.; Ren, H.; Zhou, S.-S. Signaling Pathways Related to Oxidative Stress in Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 907757. [Google Scholar] [CrossRef]
- Modanloo, M.; Shokrzadeh, M. Analyzing Mitochondrial Dysfunction, Oxidative Stress, and Apoptosis: Potential Role of L-carnitine. Iran. J. Kidney Dis. 2019, 13, 74–86. [Google Scholar] [PubMed]
- Liu, X.; Ye, B.; Miller, S.; Yuan, H.; Zhang, H.; Tian, L.; Nie, J.; Imae, R.; Arai, H.; Li, Y.; et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell. Biol. 2012, 32, 4493–4504. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Buonincontri, G.; Hu, C.-H.; Logan, A.; Sawiak, S.J.; Murphy, M.P.; Krieg, T. Complex I deficiency due to selective loss of Ndufs4 in the mouse heart results in severe hypertrophic cardiomyopathy. PLoS ONE 2014, 9, e94157. [Google Scholar] [CrossRef] [PubMed]
- Adeniran, I.; MacIver, D.H.; Zhang, H. Myocardial electrophysiological, contractile, and metabolic properties of hypertrophic cardiomyopathy: Insights from modelling. In Proceedings of the Computing in Cardiology 2014, Cambridge, MA, USA, 7–10 September 2014; IEEE: Piscataway, NJ, USA, 2014. [Google Scholar]
- Zhou, B.; Tian, R. Disfunção mitocondrial na fisiopatologia da insuficiência cardíaca. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Metabolismo das ERO mitocondriais: 10 anos depois. Bioquímica 2015, 80, 517–531. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxirredoxinas: Guardiões contra o estresse oxidativo e moduladores da sinalização de peróxidos. Tend. Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef]
- Wang, L.; Lu, P.; Yin, J.; Xu, K.; Xiang, D.; Zhang, Z.; Zhang, H.; Zheng, B.; Zhou, W.; Wang, C.; et al. Case report: Rare novel MIPEP compound heterozygous variants presenting with hypertrophic cardiomyopathy, severe lactic acidosis and hypotonia in a Chinese infant. Front. Cardiovasc. Med. 2022, 9, 1095882. [Google Scholar] [CrossRef] [PubMed]
- Nollet, E.; Burdzina, A.; Wüst, R.C.I.; Michels, M.; Asselbergs, F.W.; van der Wel, N.; Bedi, K.; Margulies, K.; Nirschl, J.; Kuster, D.; et al. “Disentangling” mitochondrial dysfunction in hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2022, 173, S113–S115. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, W.; Dai, Y.; Chen, K. Identification and verification of IGFBP3 and YTHDC1 as biomarkers associated with immune infiltration and mitophagy in hypertrophic cardiomyopathy. Front. Genet. 2022, 13, 986995. [Google Scholar] [CrossRef]
- Cibi, D.M.; Bi-Lin, K.W.; Shekeran, S.G.; Sandireddy, R.; Tee, N.; Singh, A.; Wu, Y.; Srinivasan, D.K.; Kovalik, J.-P.; Ghosh, S.; et al. Prdm16 deficiency leads to age-dependent cardiac hypertrophy, adverse remodeling, mitochondrial dysfunction, and heart failure. Cell Rep. 2020, 33, 108288. [Google Scholar] [CrossRef]
- Michałek, M.; Tabiś, A.; Pasławska, U.; Noszczyk-Nowak, A. Antioxidant defence and oxidative stress markers in cats with asymptomatic and symptomatic hypertrophic cardiomyopathy: A pilot study. BMC Vet. Res. 2020, 16, 26. [Google Scholar] [CrossRef]
- Lin, C.S.; Sun, Y.L.; Liu, C.Y. Structural and biochemical evidence of mitochondrial depletion in pigs with hypertrophic cardiomyopathy. Res. Vet. Sci. 2003, 74, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Szyguła-Jurkiewicz, B.; Szczurek-Wasilewicz, W.; Osadnik, T.; Macioł-Skurk, K.; Małyszek-Tumidajewicz, J.; Skrzypek, M.; Romuk, E.; Gąsior, M.; Banach, M.; Jóźwiak, J.J. Oxidative Stress Markers in Hypertrophic Cardiomyopathy. Medicina 2021, 58, 31. [Google Scholar] [CrossRef]
- Sabbah, H.N. Barth syndrome cardiomyopathy: Targeting the mitochondria with elamipretide. Heart Fail. Rev. 2021, 26, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Birk, A.V. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef]
- Tse, G.; Yan, B.P.; Chan, Y.W.; Tian, X.Y.; Huang, Y. Reactive oxygen species, endoplasmic reticulum stress and mitochondrial dysfunction: The link with cardiac arrhythmogenesis. Front. Physiol. 2016, 7, 313. [Google Scholar] [CrossRef]
- Mailloux, R.J. Application of mitochondria-targeted pharmaceuticals for the treatment of heart disease. Curr. Pharm. Des. 2016, 22, 4763–4779. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.-C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef]
- Silva, F.S.; Simoes, R.F.; Couto, R.; Oliveira, P. Targeting mitochondria in cardiovascular diseases. Curr. Pharm. Des. 2016, 22, 5698–5717. [Google Scholar] [CrossRef] [PubMed]
- Kadoya, T.; Sakakibara, A.; Kitayama, K.; Yamada, Y.; Higuchi, S.; Kawakita, R.; Kawasaki, Y.; Fujino, M.; Murakami, Y.; Shimura, M.; et al. Successful treatment of infantile-onset ACAD9-related cardiomyopathy with a combination of sodium pyruvate, beta-blocker, and coenzyme Q10. J. Pediatr. Endocrinol. Metab. 2019, 32, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Değerliyurt, A.; Gülleroğlu, N.B.; Kibar Gül, A.E. Primary CoQ10 deficiency with a severe phenotype due to the c.901 C > T (p.R301W) mutation in the COQ8A gene. Int. J. Neurosci. 2022, 134, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Saifudeen, I.; Subhadra, L.; Konnottil, R.; Nair, R.R. Metabolic modulation by medium-chain triglycerides reduces oxidative stress and ameliorates CD36-mediated cardiac remodeling in spontaneously hypertensive rat in the initial and established stages of hypertrophy. J. Card. Fail. 2017, 23, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Abozguia, K.; Elliott, P.; McKenna, W.; Phan, T.T.; Nallur-Shivu, G.; Ahmed, I.; Maher, A.R.; Kaur, K.; Taylor, J.; Henning, A.; et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 2010, 122, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Gehmlich, K.; Dodd, M.S.; Allwood, J.W.; Kelly, M.; Bellahcene, M.; Lad, H.V.; Stockenhuber, A.; Hooper, C.; Ashrafian, H.; Redwood, C.S.; et al. Changes in the cardiac metabolome caused by perhexiline treatment in a mouse model of hypertrophic cardiomyopathy. Mol. Biosyst. 2015, 11, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Coats, C.J.; Pavlou, M.; Watkinson, O.T.; Protonotarios, A.; Moss, L.; Hyland, R.; Rantell, K.; Pantazis, A.A.; Tome, M.; McKenna, W.J.; et al. Effect of trimetazidine dihydrochloride therapy on exercise capacity in patients with nonobstructive hypertrophic cardiomyopathy: A randomized clinical trial. JAMA Cardiol. 2019, 4, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Kelly, D.P.; Margulies, K.B. Implications of altered ketone metabolism and therapeutic ketosis in heart failure. Circulation 2020, 141, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Yurista, S.R.; Chong, C.R.; Badimon, J.J.; Kelly, D.P.; de Boer, R.A.; Westenbrink, B.D. Therapeutic potential of ketone bodies for patients with cardiovascular disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2021, 77, 1660–1669. [Google Scholar] [CrossRef]
- Yurista, S.R.; Matsuura, T.R.; Silljé, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone ester treatment improves cardiac function and reduces pathologic remodeling in preclinical models of heart failure. Circ. Heart Fail. 2021, 14, e007684. [Google Scholar] [CrossRef]
- Belge, C.; Hammond, J.; Dubois-Deruy, E.; Manoury, B.; Hamelet, J.; Beauloye, C.; Markl, A.; Pouleur, A.-C.; Bertrand, L.; Esfahani, H.; et al. Enhanced expression of β3-adrenoceptors in cardiac myocytes attenuates neurohormone-induced hypertrophic remodeling through nitric oxide synthase. Circulation 2014, 129, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, J.; Wang, Y.; Sun, K.; Wang, H.; Zou, Y.; Tian, T.; Liu, Y.; Zou, J.; Hui, R.; et al. Plasma uric acid as a prognostic marker in patients with hypertrophic cardiomyopathy. Can. J. Cardiol. 2015, 31, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Namai-Takahashi, A.; Sakuyama, A.; Nakamura, T.; Miura, T.; Takahashi, J.; Kurosawa, R.; Kohzuki, M.; Ito, O. Xanthine Oxidase Inhibitor, Febuxostat Ameliorates the High Salt Intake-Induced Cardiac Hypertrophy and Fibrosis in Dahl Salt-Sensitive Rats. Am. J. Hypertens. 2019, 32, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Bakkehaug, J.P.; Kildal, A.B.; Engstad, E.T.; Boardman, N.; Næsheim, T.; Rønning, L.; Aasum, E.; Larsen, T.S.; Myrmel, T.; How, O.-J.; et al. Myosin activator omecamtiv mecarbil increases myocardial oxygen consumption and impairs cardiac efficiency mediated by resting myosin ATPase activity. Circ. Heart Fail. 2015, 8, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef]
- Kawas, R.F.; Anderson, R.L.; Ingle, S.R.B.; Song, Y.; Sran, A.S.; Rodriguez, H.M. A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J. Biol. Chem. 2017, 292, 16571–16577. [Google Scholar] [CrossRef]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: A Clinical Trial. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Lakdawala, N.K.; Cirino, A.L.; Lipshultz, S.E.; Sparks, E.; Abbasi, S.A.; Kwong, R.Y.; Antman, E.M.; Semsarian, C.; González, A.; et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: A pilot randomized trial to modify disease expression. JACC Heart Fail. 2015, 3, 180–188. [Google Scholar] [CrossRef]
- Axelsson Raja, A.; Shi, L.; Day, S.M.; Russell, M.; Zahka, K.; Lever, H.; Colan, S.D.; Margossian, R.; Hall, E.K.; Becker, J.; et al. Baseline characteristics of the VANISH cohort. Circ. Heart Fail. 2019, 12, e006231. [Google Scholar] [CrossRef]
- Vissing, C.R.; Axelsson Raja, A.; Day, S.M.; Russell, M.W.; Zahka, K.; Lever, H.M.; Pereira, A.C.; Colan, S.D.; Margossian, R.; Murphy, A.M.; et al. Cardiac remodeling in subclinical hypertrophic cardiomyopathy: The VANISH randomized clinical trial. JAMA Cardiol. 2023, 8, 1083–1088. [Google Scholar] [CrossRef]
- Zhuang, L.; Jia, K.; Chen, C.; Li, Z.; Zhao, J.; Hu, J.; Zhang, H.; Fan, Q.; Huang, C.; Xie, H.; et al. DYRK1B-STAT3 Drives Cardiac Hypertrophy and Heart Failure by Impairing Mitochondrial Bioenergetics. Circulation 2022, 145, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Filipovska, A.; Hool, L.; Viola, H. Preventative therapeutic approaches for hypertrophic cardiomyopathy. J. Physiol. 2021, 599, 3495–3512. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Shah, A.A.; Johnstone, V.P.A.; Szappanos, H.C.; Hodson, M.P.; Hool, L.C. Characterization and validation of a preventative therapy for hypertrophic cardiomyopathy in a murine model of the disease. Proc. Natl. Acad. Sci. USA 2020, 117, 23113–23124. [Google Scholar] [CrossRef]
- Wilder, T.; Ryba, D.M.; Wieczorek, D.F.; Wolska, B.M.; Solaro, R.J. N-acetylcysteine reverses diastolic dysfunction and hypertrophy in familial hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1720–H1730. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Tan, Y.; Li, L.; Chang, J.; Syrris, P.; Hessabi, M.; Rahbar, M.H.; Willerson, J.T.; Cheong, B.Y.; Liu, C.-Y.; et al. Hypertrophy Regression with N-Acetylcysteine in Hypertrophic Cardiomyopathy (HALT-HCM): A Randomized, Placebo-Controlled, Double-Blind Pilot Study. Circ. Res. 2018, 122, 1109–1118. [Google Scholar] [CrossRef]
- Horowitz, J.D.; Chirkov, Y.Y. Perhexiline and hypertrophic cardiomyopathy: A new horizon for metabolic modulation. Circulation 2010, 122, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Jeremic, J.; Govoruskina, N.; Bradic, J.; Milosavljevic, I.; Srejovic, I.; Zivkovic, V.; Jeremic, N.; Turnic, T.N.; Tanaskovic, I.; Bolevich, S.; et al. Sacubitril/valsartan reverses cardiac structure and function in experimental model of hypertension-induced hypertrophic cardiomyopathy. Mol. Cell. Biochem. 2023, 478, 2645–2656. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Possibilities | References | Primary Effects |
|---|---|---|
| Alpha-lipoic acid | [9] | Neutralizes free radicals, reduces oxidative stress, improves endothelial function, increases nitric oxide production, and influences cellular signaling pathways associated with cardiovascular health. |
| N-acetylcysteine | [9,116,117] | Has mucolytic and fluidizing activity. Reduces oxidative damage to heart tissues and increases the production of nitric oxide, which helps dilate veins and improve blood flow. |
| MitoQ | [9] | Neutralizes free radicals within the mitochondria, reduces oxidative stress, and preserves mitochondrial function. |
| MitoVit E | [9] | Has antioxidant capacity aimed at protecting mitochondria against damage caused by free radicals and reducing oxidative stress. |
| Coenzyme Q | [9,94,95] | CoQ is involved in the mitochondrial electron transport chain, where it aids in generating adenosine triphosphate (ATP), the cellular energy currency. Additionally, CoQ possesses antioxidant properties, shielding the mitochondria and the heart against damage caused by free radicals during the energy production process. |
| Elamipretide/SS-31 | [87,88,89,90] | It concentrates on mitochondrial membranes and helps regulate energy production by enhancing the efficiency of the electron transport chain. Additionally, Elamipretide can reduce oxidative stress, stabilize mitochondrial membranes, and preserve mitochondrial integrity. |
| 17b-estradiol | [56] | It influences cardiac function by regulating genes, interacting with specific receptors, modulating vascular response, and indirectly affecting metabolic and cellular signaling processes. |
| 1-Deoxynojirimycin | [41] | Inhibits metabolic pathways involved in protein glycosylation. |
| Tricaprylin | [96] | It acts as a rapid source of fuel that can be used for ATP production, thereby aiding in the heart’s contractile function. This can be beneficial, especially in situations where the heart has an increased energy demand, such as certain cardiac conditions or intense physical exercise. |
| Perhexiline | [97,98,118] | Inhibits cardiac fatty acid uptake, promoting glucose utilization and reducing oxygen demand. |
| Trimetazidine | [99] | Blocks the mitochondrial protein responsible for transporting fatty acids into mitochondria. This action favors glucose metabolism, preserving cardiac function during low-oxygen-supply conditions such as angina. It improves cardiac efficiency and reduces angina pain. |
| Beta-3 adrenergic receptors | [103] | It is primarily activated by neurotransmitters like norepinephrine and adrenaline. It is involved in regulating fat metabolism and thermogenesis (heat production) and can influence cardiac function, lipolysis (fat breakdown), and muscle contraction in specific tissues, such as the smooth muscle of the bladder. |
| Omecamtiv Mecarbil | [106] | A myocardial activator that increases the sensitivity of the cardiac muscle (myocardium) to calcium concentration, resulting in greater efficiency in cardiac muscle contraction. Binds to myosin in the myocardium, altering its conformation and allowing a more effective interaction with calcium. |
| Mavacamten | [34,106,107,108] | An allosteric inhibitor of heart-specific myosin adenosine triphosphatase. |
| Diltiazem | [110] | A non-selective calcium channel antagonist that affects the heart and blood vessels. |
| Valsartan | [111,112] | Displaces angiotensin II from the AT1 receptor and produces its blood-pressure-lowering effects by antagonizing AT1-induced vasoconstriction, aldosterone release, catecholamine release, arginine vasopressin release, water intake, and hypertrophic responses. |
| Sacubitril | [119] | Simultaneously inhibits neprilysin (neutral endopeptidase; NEP) through sacubitrilat, the active metabolite of the prodrug sacubitril. |
| AID-TAT | [114,115] | This is related to activating the enzyme AMPK (AMP-activated protein kinase). This leads to a series of cellular events, including increased glucose uptake and increased ATP production. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menezes Junior, A.d.S.; França-e-Silva, A.L.G.d.; Oliveira, H.L.d.; Lima, K.B.A.d.; Porto, I.d.O.P.; Pedroso, T.M.A.; Silva, D.d.M.e.; Freitas, A.F., Jr. Genetic Mutations and Mitochondrial Redox Signaling as Modulating Factors in Hypertrophic Cardiomyopathy: A Scoping Review. Int. J. Mol. Sci. 2024, 25, 5855. https://doi.org/10.3390/ijms25115855
Menezes Junior AdS, França-e-Silva ALGd, Oliveira HLd, Lima KBAd, Porto IdOP, Pedroso TMA, Silva DdMe, Freitas AF Jr. Genetic Mutations and Mitochondrial Redox Signaling as Modulating Factors in Hypertrophic Cardiomyopathy: A Scoping Review. International Journal of Molecular Sciences. 2024; 25(11):5855. https://doi.org/10.3390/ijms25115855
Chicago/Turabian StyleMenezes Junior, Antonio da Silva, Ana Luísa Guedes de França-e-Silva, Henrique Lima de Oliveira, Khissya Beatryz Alves de Lima, Iane de Oliveira Pires Porto, Thays Millena Alves Pedroso, Daniela de Melo e Silva, and Aguinaldo F. Freitas, Jr. 2024. "Genetic Mutations and Mitochondrial Redox Signaling as Modulating Factors in Hypertrophic Cardiomyopathy: A Scoping Review" International Journal of Molecular Sciences 25, no. 11: 5855. https://doi.org/10.3390/ijms25115855
APA StyleMenezes Junior, A. d. S., França-e-Silva, A. L. G. d., Oliveira, H. L. d., Lima, K. B. A. d., Porto, I. d. O. P., Pedroso, T. M. A., Silva, D. d. M. e., & Freitas, A. F., Jr. (2024). Genetic Mutations and Mitochondrial Redox Signaling as Modulating Factors in Hypertrophic Cardiomyopathy: A Scoping Review. International Journal of Molecular Sciences, 25(11), 5855. https://doi.org/10.3390/ijms25115855