
Does Cell-Type-Specific Silencing of Monoamine Oxidase B Interfere with the Development of Right Ventricle (RV) Hypertrophy or Right Ventricle Failure in Pulmonary Hypertension?

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Results
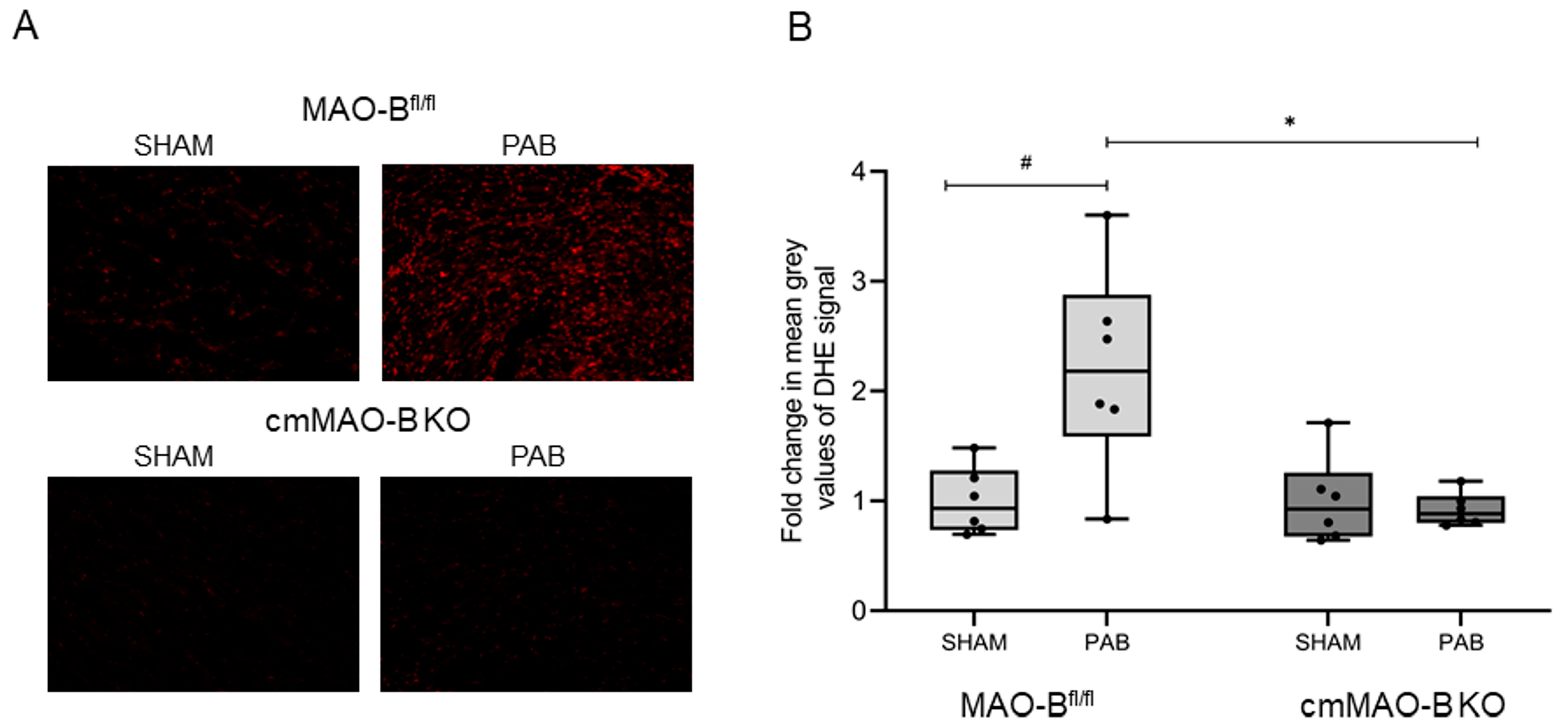
2.1. Effect of cmMAO-B Knockout on Reactive Oxygen Species Formation
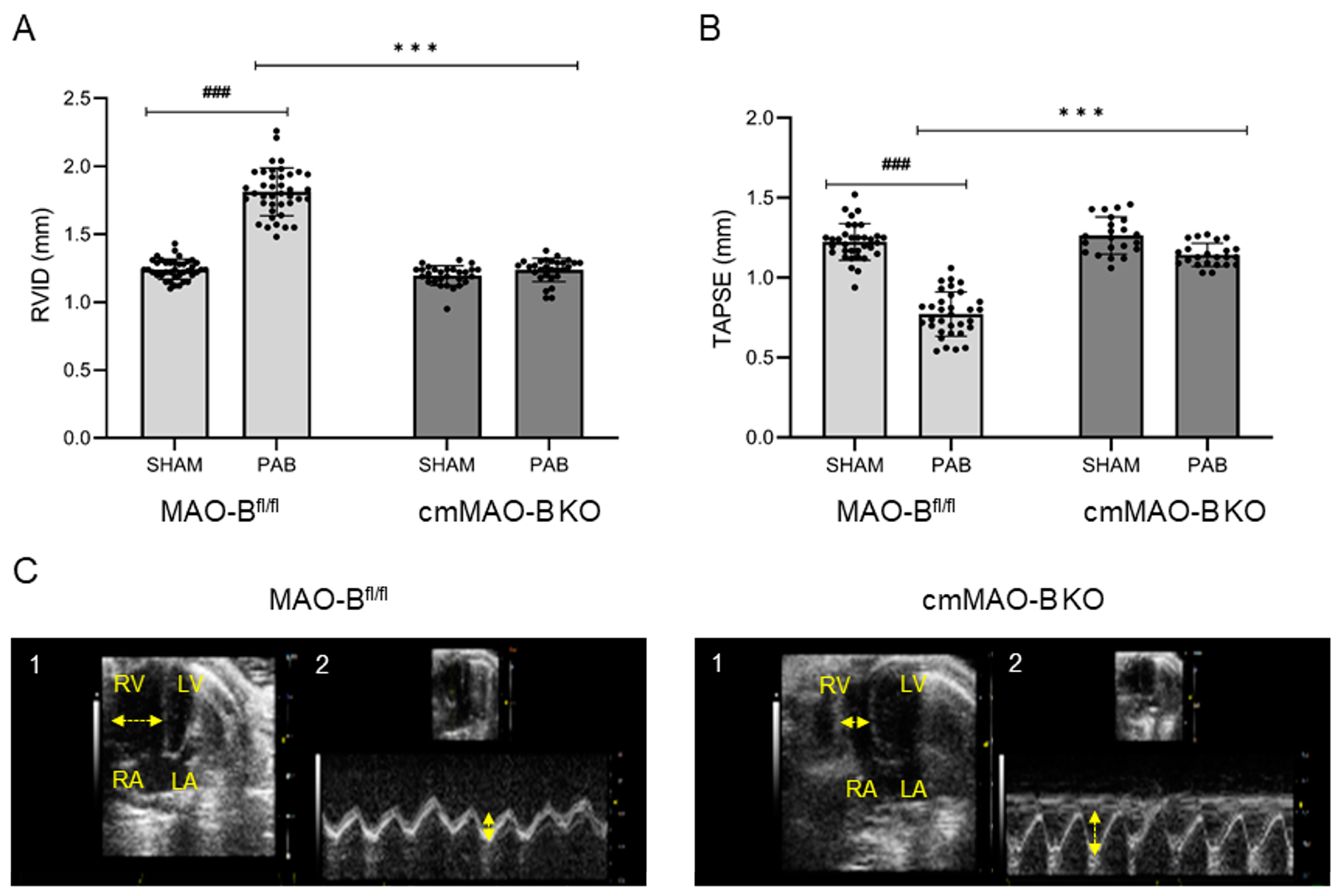
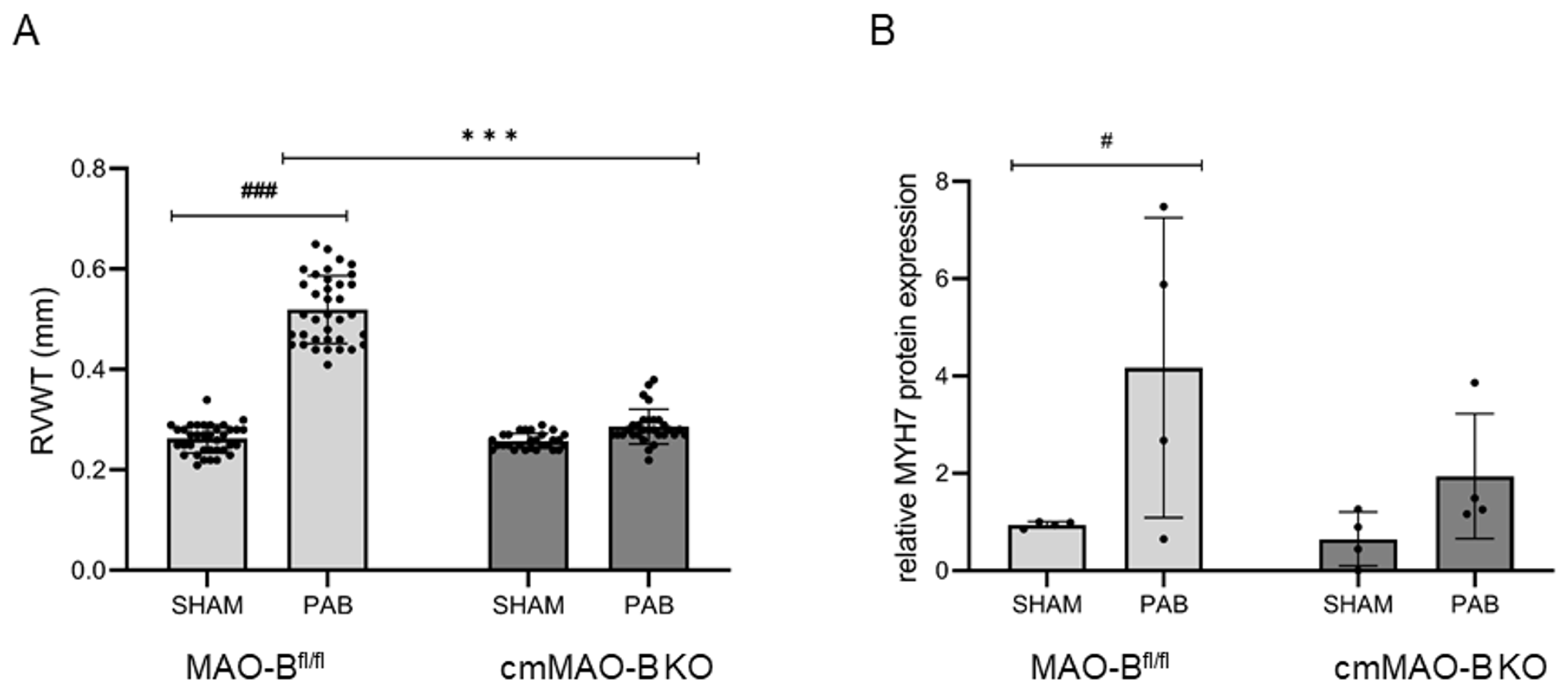
2.2. Effect of cmMAO-B Knockout on Right Ventricular Dimension and Function
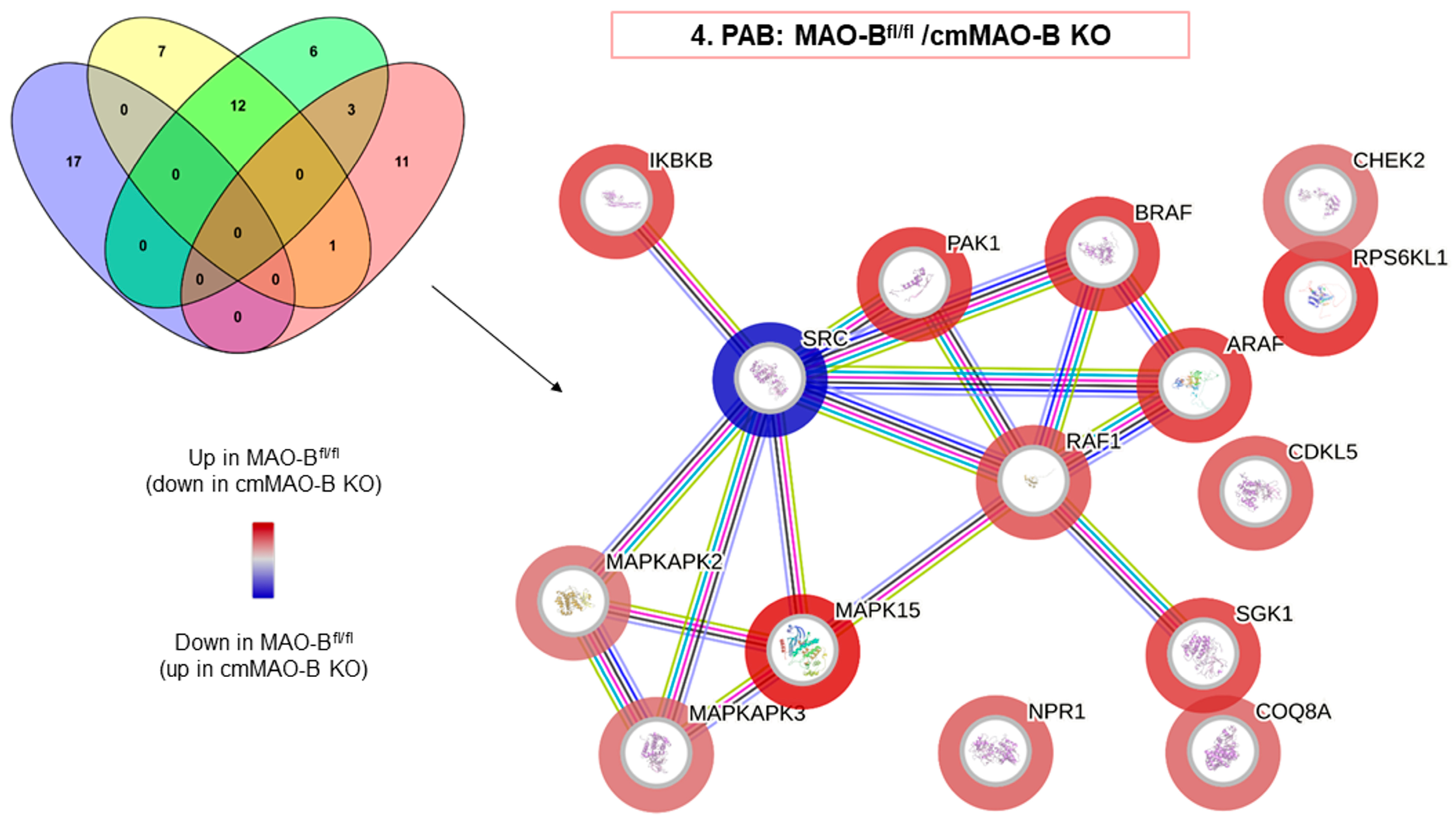
2.3. Effect of cmMAO-B Knockout on Protein Kinase Activity during PAB
2.4. Effect of cmMAO-B Knockout on Cardiomyocyte Function
3. Discussion
3.1. Main Findings
3.2. MAO and ROS Formation
3.3. MAO Substrates
3.4. MAO and Pulmonary Hypertension
3.5. MAO and RV Geometry
3.6. MAO and Cardiomyocyte Function
4. Materials and Methods
4.1. Animals and Ethical Concerns
4.2. Pulmonary Artery Banding (PAB) In Vivo
4.3. Echocardiography
4.4. Invasive Hemodynamic Measurement
4.5. Isolation and Culture of Adult Mouse Ventricular Cardiomyocytes
4.6. Determination of Cell Contraction
4.7. Metabolome Analysis: Sample Extraction
4.8. LC-MS/MS Analysis
4.9. Detection of Intracellular ROS
4.10. Peptide-Based Kinase Activity Assay
4.11. Jess Simple Western System (ProteinSimple, San Jose, CA, USA)
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maarman, G.J.; Schulz, R.; Sliwa, K.; Schermuly, R.T.; Lecour, S. Novel putative pharmacological therapies to protect the right ventricle in pulmonary hypertension: A review of current literature. Br. J. Pharmacol. 2017, 174, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, K.-D.; Kutsche, H.S.; Hirschhäuser, C.; Schreckenberg, R.; Schulz, R. Review on Chamber-Specific Differences in Right and Left Heart Reactive Oxygen Species Handling. Front. Physiol. 2018, 9, 1799. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Schlüter, K.-D. Importance of Mitochondria in Cardiac Pathologies: Focus on Uncoupling Proteins and Monoamine Oxidases. Int. J. Mol. Sci. 2023, 24, 6459. [Google Scholar] [CrossRef]
- Heusch, P.; Canton, M.; Aker, S.; Van De Sand, A.; Konietzka, I.; Rassaf, T.; Menazza, S.; Brodde, O.; Di Lisa, F.; Heusch, G.; et al. The contribution of reactive oxygen species and p38 mitogen-activated protein kinase to myofilament oxidation and progression of heart failure in rabbits. Br. J. Pharmacol. 2010, 160, 1408–1416. [Google Scholar] [CrossRef]
- Andreadou, I.; Schulz, R.; Papapetropoulos, A.; Turan, B.; Ytrehus, K.; Ferdinandy, P.; Daiber, A.; Di Lisa, F. The role of mitochondrial reactive oxygen species, NO and H 2 S in ischaemia/reperfusion injury and cardioprotection. J. Cell. Mol. Med. 2020, 24, 6510–6522. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F.; et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2018, 51, 1701024. [Google Scholar] [CrossRef]
- Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Hirschhäuser, C.; Rohrbach, S.; Li, L.; Niemann, B.; Schulz, R.; Schlüter, K.-D. Alterations in Glucose Metabolism During the Transition to Heart Failure: The Contribution of UCP-2. Cells 2020, 9, 552. [Google Scholar] [CrossRef]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The Role of Oxidative Stress and Inflammation in Cardiovascular Aging. BioMed Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef]
- Boengler, K.; Bornbaum, J.; Schlüter, K.-D.; Schulz, R. P66shc and its role in ischemic cardiovascular diseases. Basic Res. Cardiol. 2019, 114, 29. [Google Scholar] [CrossRef]
- Hirschhäuser, C.; Sydykov, A.; Wolf, A.; Esfandiary, A.; Bornbaum, J.; Kutsche, H.S.; Boengler, K.; Sommer, N.; Schreckenberg, R.; Schlüter, K.-D.; et al. Lack of Contribution of p66shc to Pressure Overload-Induced Right Heart Hypertrophy. Int. J. Mol. Sci. 2020, 21, 9339. [Google Scholar] [CrossRef]
- Obreztchikova, M.; Elouardighi, H.; Ho, M.; Wilson, B.A.; Gertsberg, Z.; Steinberg, S.F. Distinct Signaling Functions for Shc Isoforms in the Heart. J. Biol. Chem. 2006, 281, 20197–20204. [Google Scholar] [CrossRef]
- Addonizio, G.; Alexopoulos, G.S. Drug-induced dystonia in young and elderly patients. Am. J. Psychiatry 1988, 145, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.W.; Lan, N.C.; Johnson, D.L.; Abell, C.W.; Bembenek, M.E.; Kwan, S.W.; Seeburg, P.H.; Shih, J.C. cDNA cloning of human liver monoamine oxidase A and B: Molecular basis of differences in enzymatic properties. Proc. Natl. Acad. Sci. USA 1988, 85, 4934–4938. [Google Scholar] [CrossRef] [PubMed]
- Dorris, R.L. A simple method for screening Monoamine Oxidase (MAO) inhibitory drugs for type preference. J. Pharmacol. Methods 1982, 7, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Costiniti, V.; Spera, I.; Menabò, R.; Palmieri, E.M.; Menga, A.; Scarcia, P.; Porcelli, V.; Gissi, R.; Castegna, A.; Canton, M. Monoamine oxidase-dependent histamine catabolism accounts for post-ischemic cardiac redox imbalance and injury. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3050–3059. [Google Scholar] [CrossRef] [PubMed]
- Maintz, L.; Novak, N. Histamine and histamine intolerance. Am. J. Clin. Nutr. 2007, 85, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Neural Transm. 2018, 125, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Mialet-Perez, J.; Bianchi, P.; Kunduzova, O.; Parini, A. New insights on receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. J. Neural Transm. 2007, 114, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Deshwal, S.; Di Sante, M.; Di Lisa, F.; Kaludercic, N. Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr. Opin. Pharmacol. 2017, 33, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Deshwal, S.; Forkink, M.; Hu, C.-H.; Buonincontri, G.; Antonucci, S.; Di Sante, M.; Murphy, M.P.; Paolocci, N.; Mochly-Rosen, D.; Krieg, T.; et al. Monoamine oxidase-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes. Cell Death Differ. 2018, 25, 1671–1685. [Google Scholar] [CrossRef]
- Antonucci, S.; Di Sante, M.; Tonolo, F.; Pontarollo, L.; Scalcon, V.; Alanova, P.; Menabò, R.; Carpi, A.; Bindoli, A.; Rigobello, M.P.; et al. The Determining Role of Mitochondrial Reactive Oxygen Species Generation and Monoamine Oxidase Activity in Doxorubicin-Induced Cardiotoxicity. Antioxid. Redox Signal. 2021, 34, 531–550. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Arusei, R.J.; Di Lisa, F. Recent advances on the role of monoamine oxidases in cardiac pathophysiology. Basic Res. Cardiol. 2023, 118, 41. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Schulz, R. Mitochondrial Monoamine Oxidase: Another Player in Pulmonary Hypertension? Am. J. Respir. Cell Mol. Biol. 2021, 64, 277–278. [Google Scholar] [CrossRef] [PubMed]
- van Eif, V.W.W.; Bogaards, S.J.P.; van der Laarse, W.J. Intrinsic cardiac adrenergic (ICA) cell density and MAO-A activity in failing rat hearts. J. Muscle Res. Cell Motil. 2014, 35, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-Q.; Peters, E.L.; Schalij, I.; Axelsen, J.B.; Andersen, S.; Kurakula, K.; Gomez-Puerto, M.C.; Szulcek, R.; Pan, X.; da Silva Goncalves Bos, D.; et al. Increased MAO-A Activity Promotes Progression of Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2021, 64, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Malik, H.; Crawford, R.M.; Streeter, J.; Wang, J.; Huo, R.; Shih, J.C.; Chen, B.; Hall, D.; Abel, E.D.; et al. Cardiac MAO-A inhibition protects against catecholamine-induced ventricular arrhythmias via enhanced diastolic calcium control. Cardiovasc. Res. 2024, 120, cvae012. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Hirschhäuser, C.; Bornbaum, J.; Sydykov, A.; Dempfle, A.; Schneider, A.; Braun, T.; Schlüter, K.-D.; Schulz, R. Cardiomyocytes-specific deletion of monoamine oxidase B reduces irreversible myocardial ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Szabados, T.; Brosinsky, P.; Bencsik, P.; Ferdinandy, P.; Schulz, R. Sex Difference in Cardioprotection against Acute Myocardial Infarction in MAO-B Knockout Mice In Vivo. Int. J. Mol. Sci. 2023, 24, 6443. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Carpi, A.; Nagayama, T.; Sivakumaran, V.; Zhu, G.; Lai, E.W.; Bedja, D.; De Mario, A.; Chen, K.; Gabrielson, K.L.; et al. Monoamine Oxidase B Prompts Mitochondrial and Cardiac Dysfunction in Pressure Overloaded Hearts. Antioxid. Redox Signal. 2014, 20, 267–280. [Google Scholar] [CrossRef]
- Clerk, A.; Meijles, D.N.; Hardyman, M.A.; Fuller, S.J.; Chothani, S.P.; Cull, J.J.; Cooper, S.T.E.; Alharbi, H.O.; Vanezis, K.; Felkin, L.E.; et al. Cardiomyocyte BRAF and type 1 RAF inhibitors promote cardiomyocyte and cardiac hypertrophy in mice in vivo. Biochem. J. 2022, 479, 401–424. [Google Scholar] [CrossRef]
- Wen, Y.; Liu, R.; Lin, N.; Luo, H.; Tang, J.; Huang, Q.; Sun, H.; Tang, L. NADPH Oxidase Hyperactivity Contributes to Cardiac Dysfunction and Apoptosis in Rats with Severe Experimental Pancreatitis through ROS-Mediated MAPK Signaling Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 4578175. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, H.J.; Abe, K.; Vonk Noordegraaf, A.; Voelkel, N.F. The Right Ventricle Under Pressure. Chest 2009, 135, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Mitogen-Activated Protein Kinases in Heart Development and Diseases. Circulation 2007, 116, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Maulik, S.K.; Kumar, S. Oxidative stress and cardiac hypertrophy: A review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef]
- Alharbi, H.O.; Hardyman, M.A.; Cull, J.J.; Markou, T.; Cooper, S.T.E.; Glennon, P.E.; Fuller, S.J.; Sugden, P.H.; Clerk, A. Cardiomyocyte BRAF is a key signalling intermediate in cardiac hypertrophy in mice. Clin. Sci. 2022, 136, 1661–1681. [Google Scholar] [CrossRef] [PubMed]
- Holschneider, D.P.; Scremin, O.U.; Roos, K.P.; Chialvo, D.R.; Chen, K.; Shih, J.C. Increased baroreceptor response in mice deficient in monoamine oxidase A and B. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H964–H972. [Google Scholar] [CrossRef] [PubMed]
- Lairez, O.; Calise, D.; Bianchi, P.; Ordener, C.; Spreux-Varoquaux, O.; Guilbeau-Frugier, C.; Escourrou, G.; Seif, I.; Roncalli, J.; Pizzinat, N.; et al. Genetic deletion of MAO-A promotes serotonin-dependent ventricular hypertrophy by pressure overload. J. Mol. Cell. Cardiol. 2009, 46, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Grobe, J.M.; Weisgut, J.; Schwelberger, H.G.; Fogel, W.A.; Marušáková, M.; Wache, H.; Bähre, H.; Buchwalow, I.B.; Dhein, S.; et al. Histamine can be Formed and Degraded in the Human and Mouse Heart. Front. Pharmacol. 2021, 12, 582916. [Google Scholar] [CrossRef] [PubMed]
- Saheera, S.; Potnuri, A.G.; Guha, A.; Palaniyandi, S.S.; Thandavarayan, R.A. Histamine 2 receptors in cardiovascular biology: A friend for the heart. Drug Discov. Today 2022, 27, 234–245. [Google Scholar] [CrossRef]
- Potnuri, A.G.; Allakonda, L.; Appavoo, A.; Saheera, S.; Nair, R.R. Association of histamine with hypertension-induced cardiac remodeling and reduction of hypertrophy with the histamine-2-receptor antagonist famotidine compared with the beta-blocker metoprolol. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2018, 41, 1023–1035. [Google Scholar] [CrossRef]
- Leary, P.J.; Kronmal, R.A.; Bluemke, D.A.; Buttrick, P.M.; Jones, K.L.; Kao, D.P.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Minobe, W.; et al. Histamine H2 Receptor Polymorphisms, Myocardial Transcripts, and Heart Failure (from the Multi-Ethnic Study of Atherosclerosis and Beta-Blocker Effect on Remodeling and Gene Expression Trial). Am. J. Cardiol. 2018, 121, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Tanijiri, H. Cardiac hypertrophy in spontaneously hypertensive rats. Jpn. Heart J. 1975, 16, 174–188. [Google Scholar] [CrossRef]
- Pino, R.; Failli, P.; Mazzetti, L.; Buffoni, F. Monoamine oxidase and semicarbazide-sensitive amine oxidase activities in isolated cardiomyocytes of spontaneously hypertensive rats. Biochem. Mol. Med. 1997, 62, 188–196. [Google Scholar] [CrossRef]
- Fischer, Y.; Thomas, J.; Kamp, J.; Jüngling, E.; Rose, H.; Carpéné, C.; Kammermeier, H. 5-hydroxytryptamine stimulates glucose transport in cardiomyocytes via a monoamine oxidase-dependent reaction. Biochem. J. 1995, 311, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Universal child immunization by 1990: A realistic goal. World Ir. Nurs. 1986, 15, 5–6.
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef]
- Gibb, A.A.; Lorkiewicz, P.K.; Zheng, Y.-T.; Zhang, X.; Bhatnagar, A.; Jones, S.P.; Hill, B.G. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem. J. 2017, 474, 2785–2801. [Google Scholar] [CrossRef]
- Spruijt, O.A.; de Man, F.S.; Groepenhoff, H.; Oosterveer, F.; Westerhof, N.; Vonk-Noordegraaf, A.; Bogaard, H.-J. The effects of exercise on right ventricular contractility and right ventricular-arterial coupling in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2015, 191, 1050–1057. [Google Scholar] [CrossRef]
- Wang, Z.; Schreier, D.A.; Hacker, T.A.; Chesler, N.C. Progressive right ventricular functional and structural changes in a mouse model of pulmonary arterial hypertension. Physiol. Rep. 2013, 1, e00184. [Google Scholar] [CrossRef]
- Esfandiary, A.; Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Pak, O.; Kojonazarov, B.; Sydykov, A.; Hirschhäuser, C.; Wolf, A.; Haag, D.; et al. Protection against pressure overload-induced right heart failure by uncoupling protein 2 silencing. Cardiovasc. Res. 2019, 115, 1217–1227. [Google Scholar] [CrossRef]
- Canton, M.; Skyschally, A.; Menabò, R.; Boengler, K.; Gres, P.; Schulz, R.; Haude, M.; Erbel, R.; Di Lisa, F.; Heusch, G. Oxidative modification of tropomyosin and myocardial dysfunction following coronary microembolization. Eur. Heart J. 2006, 27, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart Failure with Preserved Ejection Fraction: Mechanisms, Clinical Features, and Therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Mamazhakypov, A.; Veith, C.; Schermuly, R.T.; Sydykov, A. Surgical protocol for pulmonary artery banding in mice to generate a model of pressure-overload-induced right ventricular failure. STAR Protoc. 2023, 4, 102660. [Google Scholar] [CrossRef] [PubMed]
- Bøtker, H.E.; Hausenloy, D.; Andreadou, I.; Antonucci, S.; Boengler, K.; Davidson, S.M.; Deshwal, S.; Devaux, Y.; Di Lisa, F.; Di Sante, M.; et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res. Cardiol. 2018, 113, 39. [Google Scholar] [CrossRef] [PubMed]
- Alack, K.; Weiss, A.; Krüger, K.; Höret, M.; Schermuly, R.; Frech, T.; Eggert, M.; Mooren, F.-C. Profiling of human lymphocytes reveals a specific network of protein kinases modulated by endurance training status. Sci. Rep. 2020, 10, 888. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Neubauer, M.C.; Yerabolu, D.; Kojonazarov, B.; Schlueter, B.C.; Neubert, L.; Jonigk, D.; Baal, N.; Ruppert, C.; Dorfmuller, P.; et al. Targeting cyclin-dependent kinases for the treatment of pulmonary arterial hypertension. Nat. Commun. 2019, 10, 2204. [Google Scholar] [CrossRef] [PubMed]
- Yerabolu, D.; Weiss, A.; Kojonazarov, B.; Boehm, M.; Schlueter, B.C.; Ruppert, C.; Günther, A.; Jonigk, D.; Grimminger, F.; Ghofrani, H.-A.; et al. Targeting Jak-Stat Signaling in Experimental Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2021, 64, 100–114. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RV | Ldiast (µm) | Con Vel (µm/s) | Rel Vel (µm/s) | dL/L (%) | |
|---|---|---|---|---|---|
| MAO-Bfl/fl | SHAM | 109.93 ± 9.79 | 202.63 ± 51.36 | 166.05 ± 45.05 | 8.53 ± 1.82 |
| PAB | 111.32 ± 7.23 | 182.99 ± 46.88 | 151.03 ± 45.33 | 7.65 ± 7.65 | |
| cmMAO-B KO | SHAM | 112.08 ± 11.66 | 215.58 ± 72.75 | 180.14 ± 59.07 | 9.10 ± 2.44 |
| PAB | 108.36 ± 11.00 | 183.64 ± 48.88 | 152.43 ± 45.10 | 8.30 ± 2.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brosinsky, P.; Heger, J.; Sydykov, A.; Weiss, A.; Klatt, S.; Czech, L.; Kraut, S.; Schermuly, R.T.; Schlüter, K.-D.; Schulz, R. Does Cell-Type-Specific Silencing of Monoamine Oxidase B Interfere with the Development of Right Ventricle (RV) Hypertrophy or Right Ventricle Failure in Pulmonary Hypertension? Int. J. Mol. Sci. 2024, 25, 6212. https://doi.org/10.3390/ijms25116212
Brosinsky P, Heger J, Sydykov A, Weiss A, Klatt S, Czech L, Kraut S, Schermuly RT, Schlüter K-D, Schulz R. Does Cell-Type-Specific Silencing of Monoamine Oxidase B Interfere with the Development of Right Ventricle (RV) Hypertrophy or Right Ventricle Failure in Pulmonary Hypertension? International Journal of Molecular Sciences. 2024; 25(11):6212. https://doi.org/10.3390/ijms25116212
Chicago/Turabian StyleBrosinsky, Paulin, Jacqueline Heger, Akylbek Sydykov, Astrid Weiss, Stephan Klatt, Laureen Czech, Simone Kraut, Ralph Theo Schermuly, Klaus-Dieter Schlüter, and Rainer Schulz. 2024. "Does Cell-Type-Specific Silencing of Monoamine Oxidase B Interfere with the Development of Right Ventricle (RV) Hypertrophy or Right Ventricle Failure in Pulmonary Hypertension?" International Journal of Molecular Sciences 25, no. 11: 6212. https://doi.org/10.3390/ijms25116212
APA StyleBrosinsky, P., Heger, J., Sydykov, A., Weiss, A., Klatt, S., Czech, L., Kraut, S., Schermuly, R. T., Schlüter, K.-D., & Schulz, R. (2024). Does Cell-Type-Specific Silencing of Monoamine Oxidase B Interfere with the Development of Right Ventricle (RV) Hypertrophy or Right Ventricle Failure in Pulmonary Hypertension? International Journal of Molecular Sciences, 25(11), 6212. https://doi.org/10.3390/ijms25116212