
Pathological Changes and Metabolic Adaptation in the Myocardium of Rats in Response to Chronic Variable Mild Stress
 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
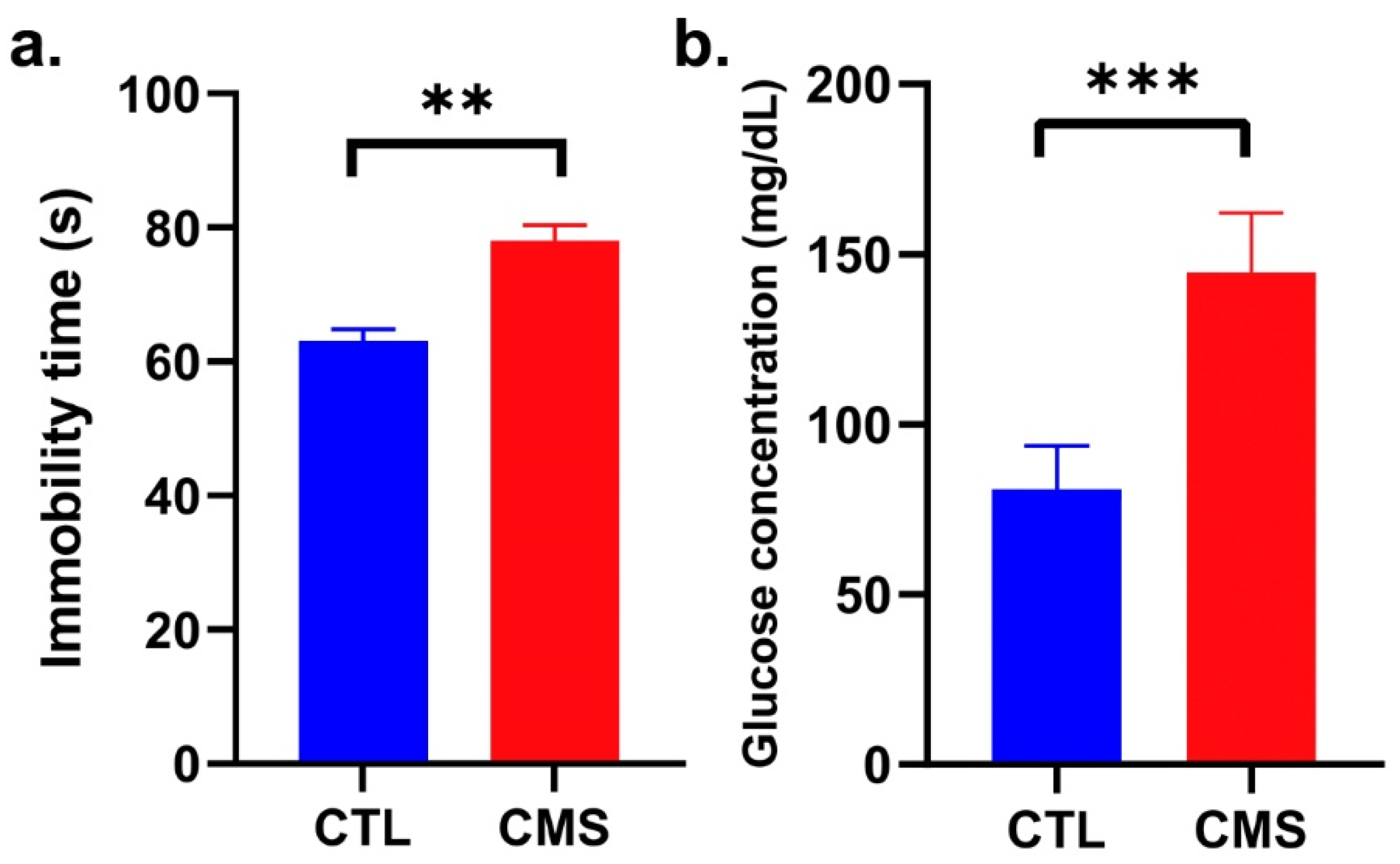
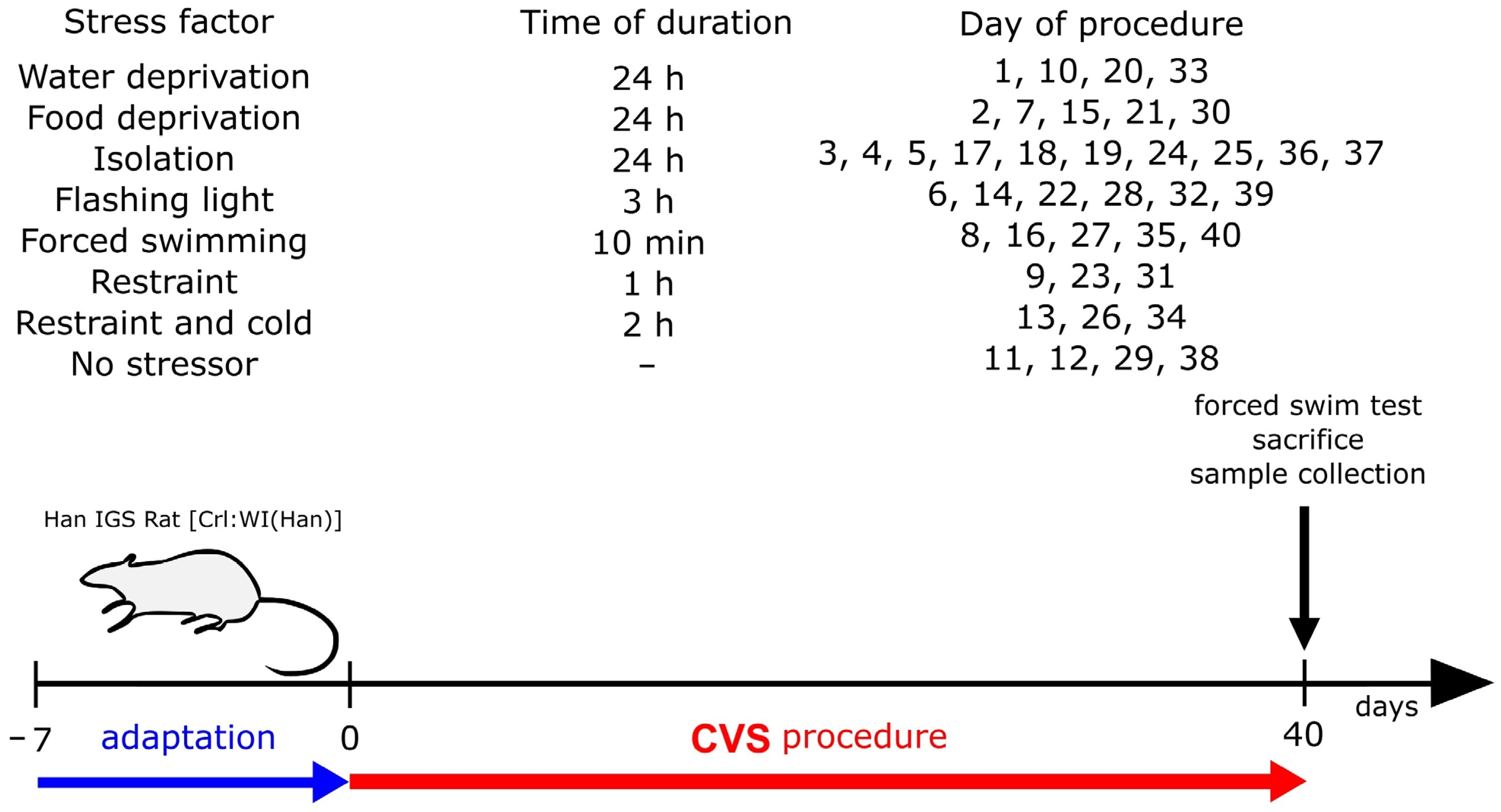
2.1. Establishment of the CVS Model
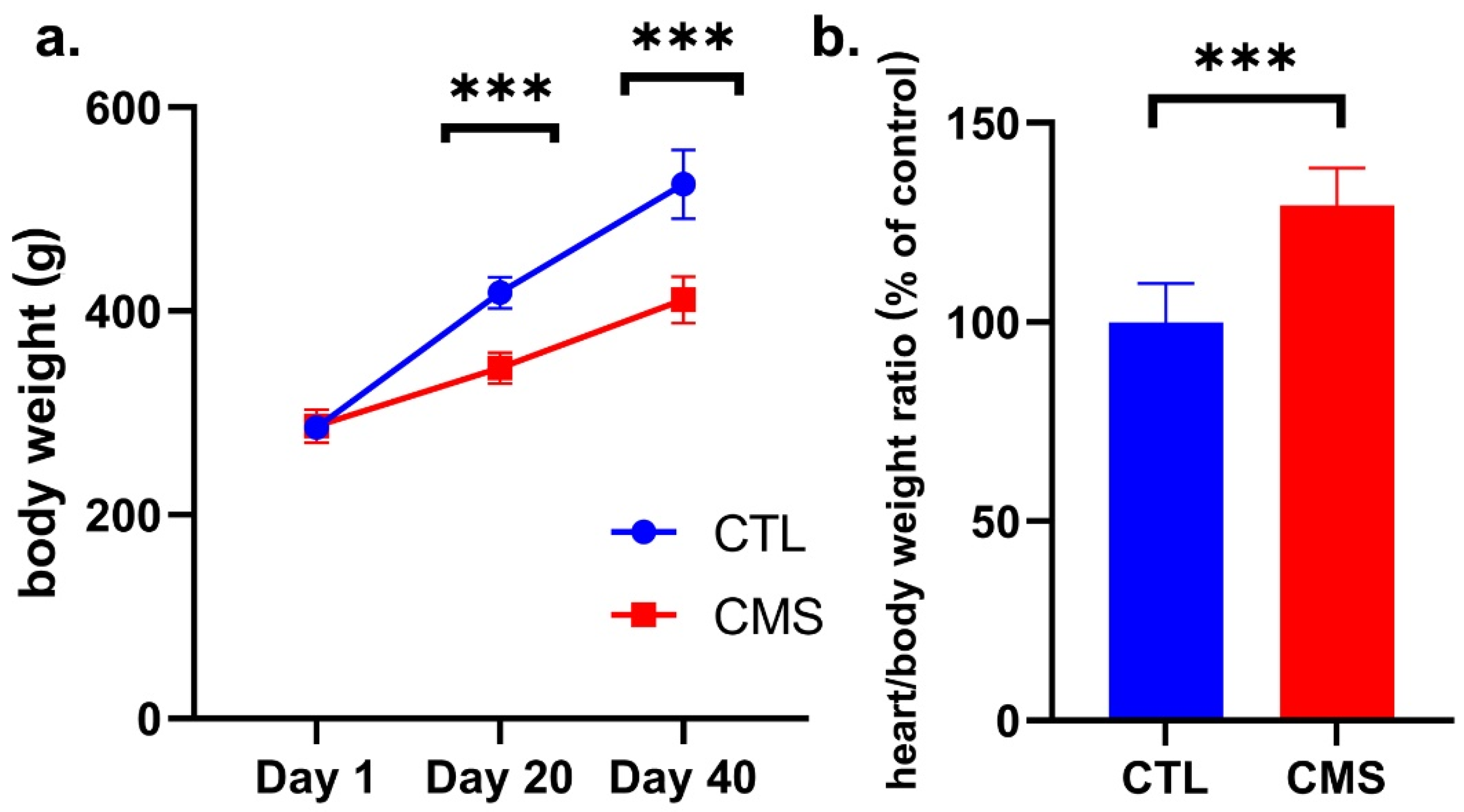
2.2. Body Weight Assessment
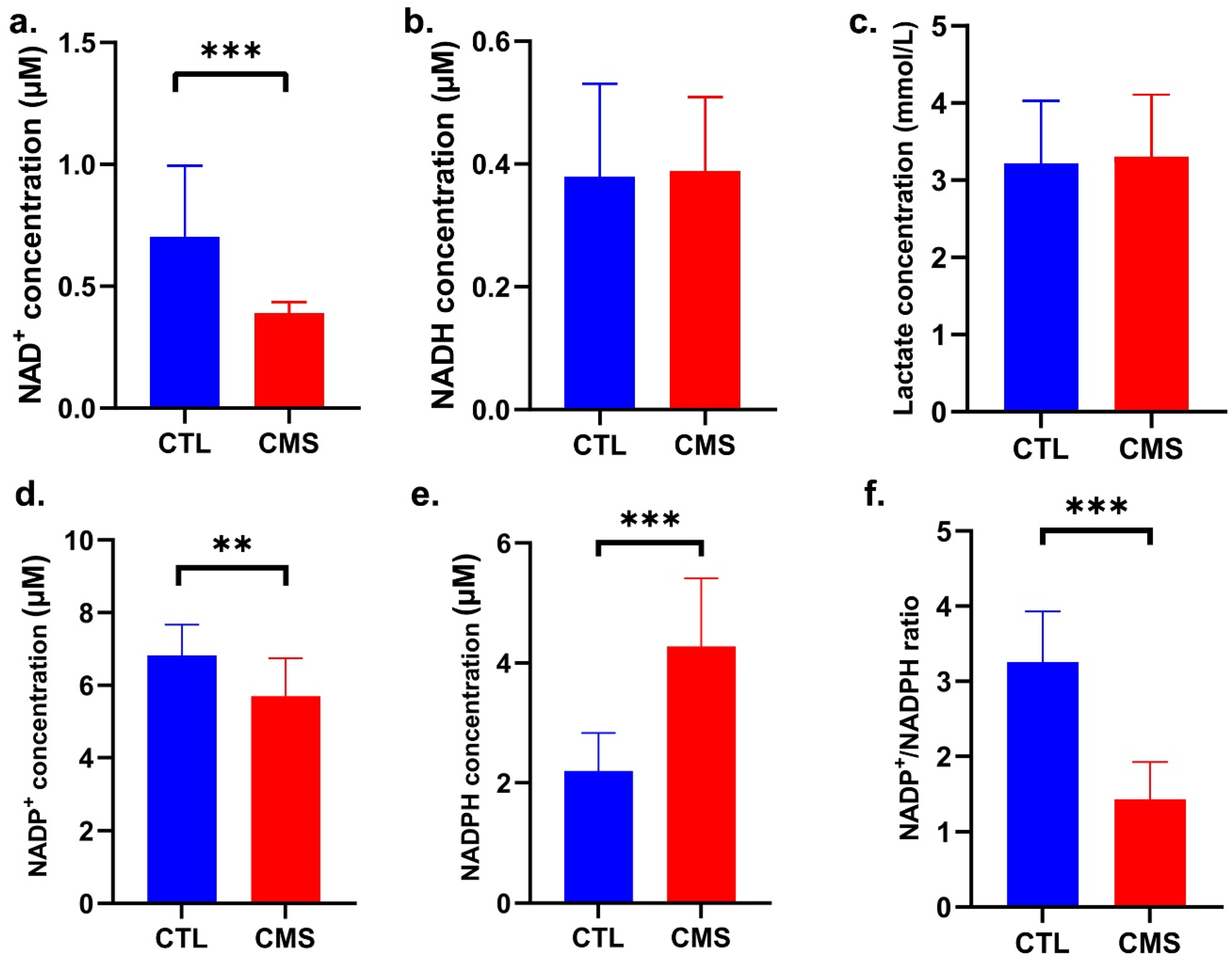
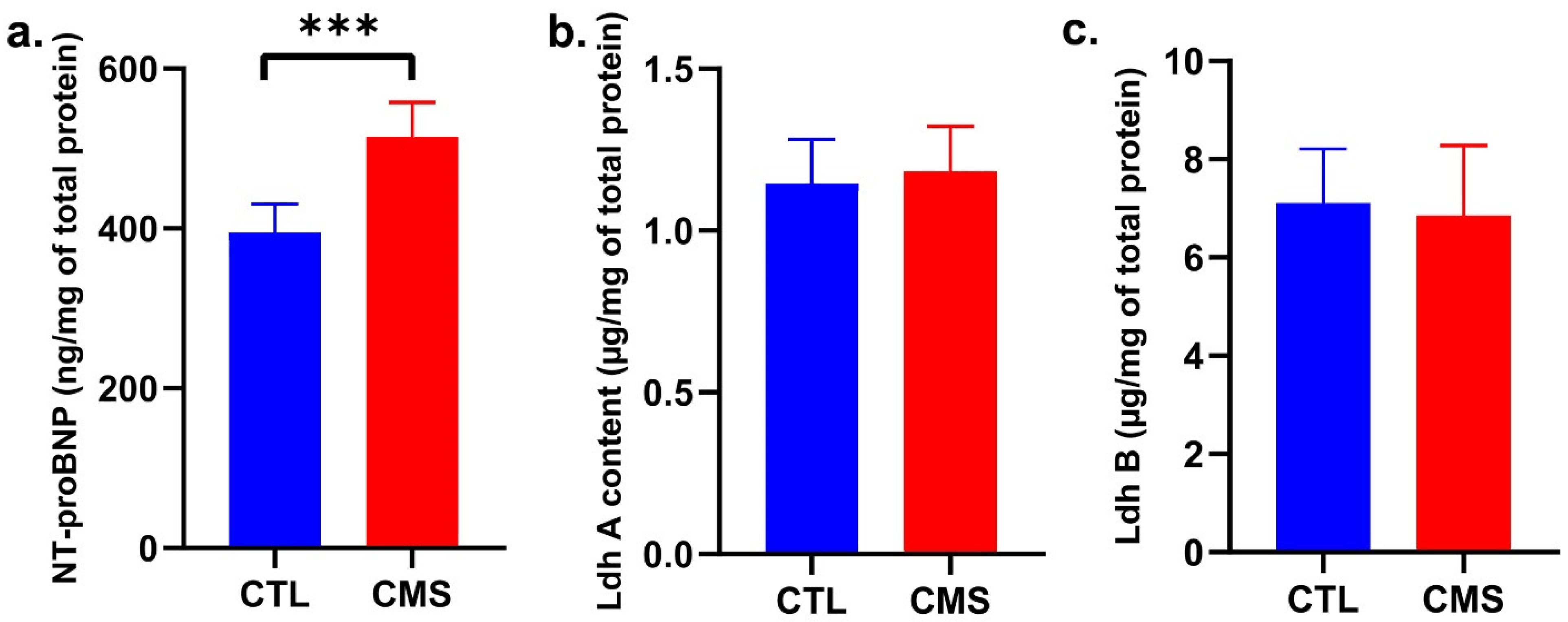
2.3. Biochemical Analysis
2.4. Free Amino Acid Analysis
2.5. Oxidative DNA Damage
2.6. Molecular Analysis
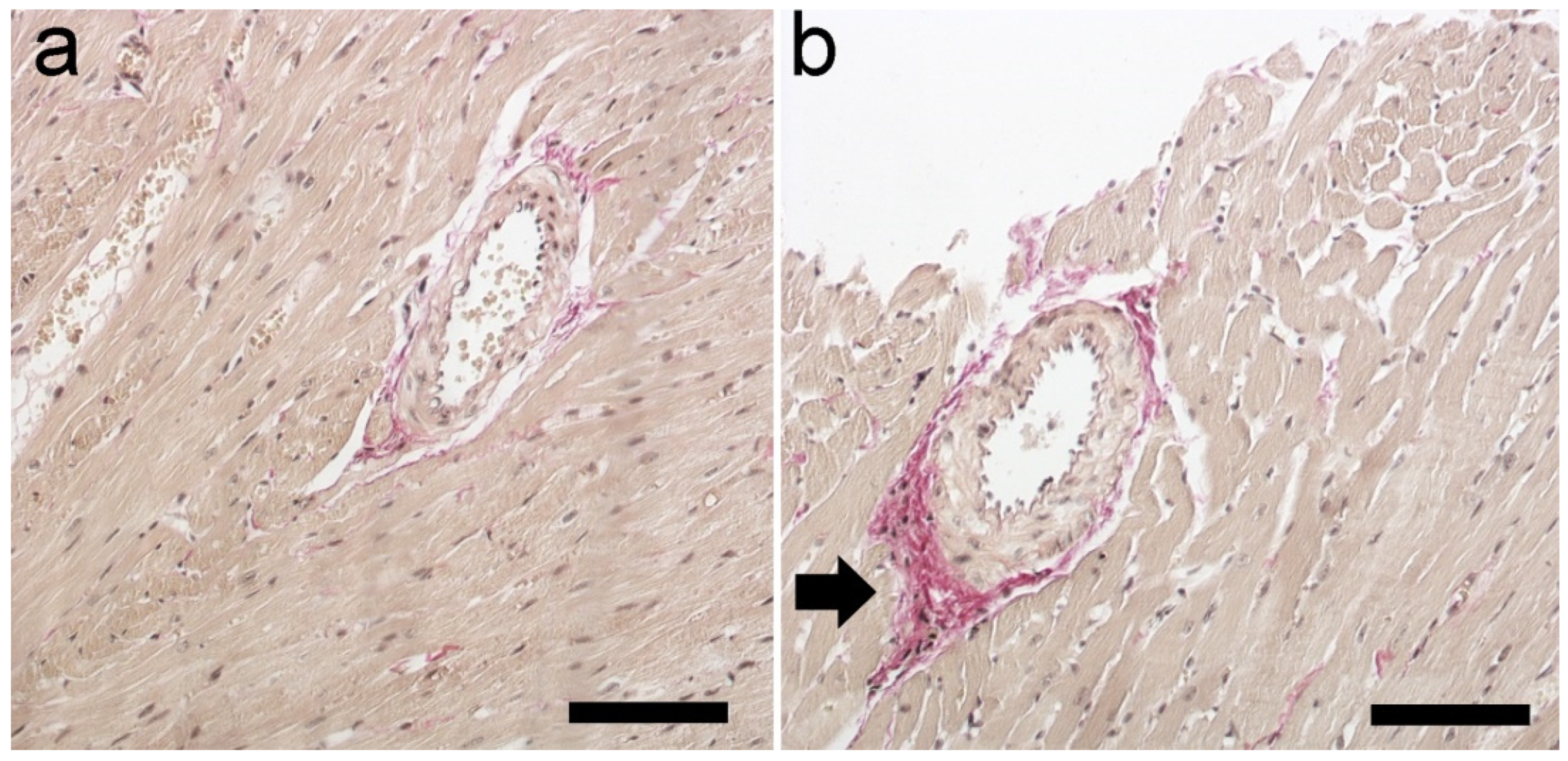
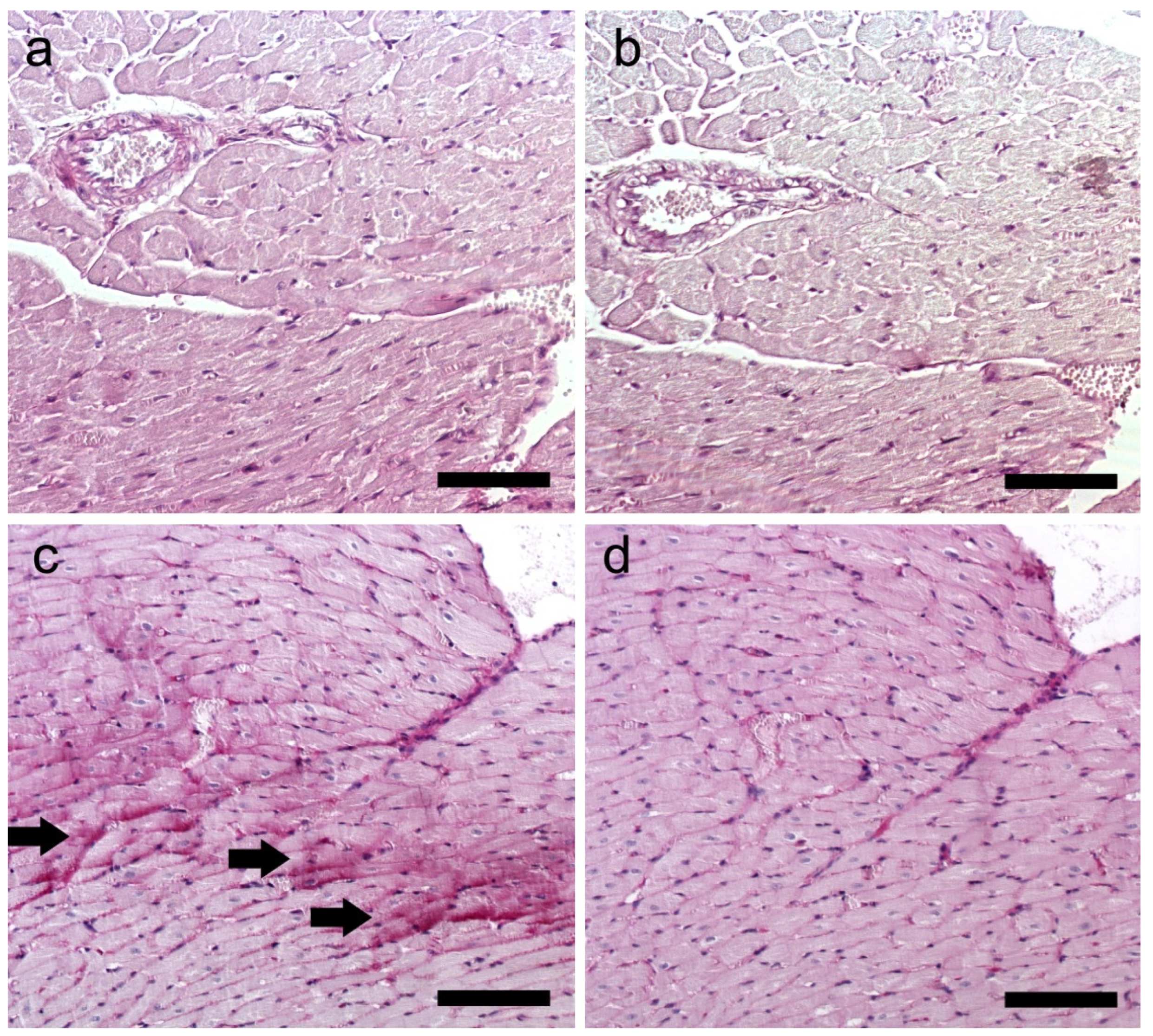
2.7. Histological Staining
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. CVS Procedure
4.3. Behavior
4.4. Biochemical Assays
4.5. Free Amino Acid Analysis
4.6. Oxidative DNA Damage
4.7. Molecular Analysis
4.8. Histological Staining
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gamaro, G.D.; Manoli, L.P.; Torres, I.L.S.; Silveira, R.; Dalmaz, C. Effects of Chronic Variate Stress on Feeding Behavior and on Monoamine Levels in Different Rat Brain Structures. Neurochem. Int. 2003, 42, 107–114. [Google Scholar] [CrossRef]
- Willner, P. The Chronic Mild Stress (CMS) Model of Depression: History, Evaluation and Usage. Neurobiol. Stress 2017, 6, 78–93. [Google Scholar] [CrossRef]
- Katz, R.J.; Roth, K.A.; Carroll, B.J. Acute and Chronic Stress Effects on Open Field Activity in the Rat: Implications for a Model of Depression. Neurosci. Biobehav. Rev. 1981, 5, 247–251. [Google Scholar] [CrossRef]
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 22 May 2024).
- Steptoe, A.; Kivimäki, M. Stress and Cardiovascular Disease: An Update on Current Knowledge. Annu. Rev. Public Health 2013, 34, 337–354. [Google Scholar] [CrossRef]
- Nicholson, A.; Kuper, H.; Hemingway, H. Depression as an Aetiologic and Prognostic Factor in Coronary Heart Disease: A Meta-Analysis of 6362 Events among 146 538 Participants in 54 Observational Studies. Eur. Heart J. 2006, 27, 2763–2774. [Google Scholar] [CrossRef]
- Chaturvedi, K.; Vishwakarma, D.K.; Singh, N. COVID-19 and Its Impact on Education, Social Life and Mental Health of Students: A Survey. Child. Youth Serv. Rev. 2021, 121, 105866. [Google Scholar] [CrossRef]
- Santomauro, D.F.; Herrera, A.M.M.; Shadid, J.; Zheng, P.; Ashbaugh, C.; Pigott, D.M.; Abbafati, C.; Adolph, C.; Amlag, J.O.; Aravkin, A.Y.; et al. Global Prevalence and Burden of Depressive and Anxiety Disorders in 204 Countries and Territories in 2020 Due to the COVID-19 Pandemic. Lancet 2021, 398, 1700–1712. [Google Scholar] [CrossRef]
- Rugulies, R. Depression as a Predictor for Coronary Heart Disease. a Review and Meta-Analysis. Am. J. Prev. Med. 2002, 23, 51–61. [Google Scholar] [CrossRef]
- Sullivan, M.; Simon, G.; Spertus, J.; Russo, J. Depression-Related Costs in Heart Failure Care. Arch. Intern. Med. 2002, 162, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Lesman-Leegte, I.; Lundgren, J.; Hillege, H.L.; Hoes, A.; Sanderman, R.; van Veldhuisen, D.J.; Jaarsma, T. Time-Course of Depressive Symptoms in Patients with Heart Failure. J. Psychosom. Res. 2013, 74, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.L.; Toukhsati, S.R.; Johansson, P.; Jaarsma, T. Depression and Cardiovascular Disease: A Clinical Review. Eur. Heart J. 2014, 35, 1365–1372. [Google Scholar] [CrossRef]
- Cudnoch-Jedrzejewska, A.; Szczepanska-Sadowska, E.; Dobruch, J.; Gomolka, R.; Puchalska, L. Brain Vasopressin V(1) Receptors Contribute to Enhanced Cardiovascular Responses to Acute Stress in Chronically Stressed Rats and Rats with Myocardial Infarcton. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R672–R680. [Google Scholar] [CrossRef]
- Grippo, A.J.; Moffitt, J.A.; Sgoifo, A.; Jepson, A.J.; Bates, S.L.; Chandler, D.L.; McNeal, N.; Preihs, K. The Integration of Depressive Behaviors and Cardiac Dysfunction during an Operational Measure of Depression: Investigating the Role of Negative Social Experiences Using an Animal Model. Psychosom. Med. 2012, 74, 612–619. [Google Scholar] [CrossRef]
- Park, S.E.; Park, D.; Song, K.-I.; Seong, J.-K.; Chung, S.; Youn, I. Differential Heart Rate Variability and Physiological Responses Associated with Accumulated Short- and Long-Term Stress in Rodents. Physiol. Behav. 2017, 171, 21–31. [Google Scholar] [CrossRef]
- O’Neil, A.; Fisher, A.J.; Kibbey, K.J.; Jacka, F.N.; Kotowicz, M.A.; Williams, L.J.; Stuart, A.L.; Berk, M.; Lewandowski, P.A.; Taylor, C.B.; et al. Depression Is a Risk Factor for Incident Coronary Heart Disease in Women: An 18-Year Longitudinal Study. J. Affect. Disord. 2016, 196, 117–124. [Google Scholar] [CrossRef]
- Vaccarino, V.; Sullivan, S.; Hammadah, M.; Wilmot, K.; Al Mheid, I.; Ramadan, R.; Elon, L.; Pimple, P.M.; Garcia, E.V.; Nye, J.; et al. Mental Stress Induced-Myocardial Ischemia in Young Patients with Recent Myocardial Infarction: Sex Differences and Mechanisms. Circulation 2018, 137, 794–805. [Google Scholar] [CrossRef]
- Vaccarino, V.; Badimon, L.; Bremner, J.D.; Cenko, E.; Cubedo, J.; Dorobantu, M.; Duncker, D.J.; Koller, A.; Manfrini, O.; Milicic, D.; et al. Depression and Coronary Heart Disease: 2018 Position Paper of the ESC Working Group on Coronary Pathophysiology and Microcirculation. Eur. Heart J. 2020, 41, 1687–1696. [Google Scholar] [CrossRef]
- Scherrer, J.F.; Garfield, L.D.; Lustman, P.J.; Hauptman, P.J.; Chrusciel, T.; Zeringue, A.; Carney, R.M.; Freedland, K.E.; Bucholz, K.K.; Owen, R.; et al. Antidepressant Drug Compliance: Reduced Risk of MI and Mortality in Depressed Patients. Am. J. Med. 2011, 124, 318–324. [Google Scholar] [CrossRef]
- Jiang, W.; Davidson, J.R.T. Antidepressant Therapy in Patients with Ischemic Heart Disease. Am. Heart J. 2005, 150, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, D.; Sato, H.; Sakata, Y.; Shiotani, I.; Kinjo, K.; Mizuno, H.; Shimizu, M.; Ito, H.; Koretsune, Y.; Hirayama, A.; et al. Influence of Serotonin Transporter Gene Polymorphism on Depressive Symptoms and New Cardiac Events after Acute Myocardial Infarction. Am. Heart J. 2005, 150, 652–658. [Google Scholar] [CrossRef] [PubMed]
- AbuRuz, M.E.; Alaloul, F.; Al-Dweik, G. Depressive Symptoms Are Associated with In-Hospital Complications Following Acute Myocardial Infarction. Appl. Nurs. Res. ANR 2018, 39, 65–70. [Google Scholar] [CrossRef]
- Pivonello, R.; Simeoli, C.; De Martino, M.C.; Cozzolino, A.; De Leo, M.; Iacuaniello, D.; Pivonello, C.; Negri, M.; Pellecchia, M.T.; Iasevoli, F.; et al. Neuropsychiatric Disorders in Cushing’s Syndrome. Front. Neurosci. 2015, 9, 129. [Google Scholar] [CrossRef]
- Shukla, L. Cushing’s Syndrome and Treatment-Resistant Depression. Indian J. Psychol. Med. 2017, 39, 213–214. [Google Scholar] [CrossRef]
- Vaseghi, M.; Shivkumar, K. The Role of the Autonomic Nervous System in Sudden Cardiac Death. Prog. Cardiovasc. Dis. 2008, 50, 404–419. [Google Scholar] [CrossRef]
- Li, J.; Duan, R.; Zhang, Y.; Zhao, X.; Cheng, Y.; Chen, Y.; Yuan, J.; Li, H.; Zhang, J.; Chu, L.; et al. Beta-Adrenergic Activation Induces Cardiac Collapse by Aggravating Cardiomyocyte Contractile Dysfunction in Bupivacaine Intoxication. PLoS ONE 2018, 13, e0203602. [Google Scholar] [CrossRef]
- Su, Z.; Liu, Y.; Zhang, H. Adaptive Cardiac Metabolism Under Chronic Hypoxia: Mechanism and Clinical Implications. Front. Cell Dev. Biol. 2021, 9, 625524. [Google Scholar] [CrossRef]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef]
- Pettersen, I.K.N.; Tusubira, D.; Ashrafi, H.; Dyrstad, S.E.; Hansen, L.; Liu, X.-Z.; Nilsson, L.I.H.; Løvsletten, N.G.; Berge, K.; Wergedahl, H.; et al. Upregulated PDK4 Expression Is a Sensitive Marker of Increased Fatty Acid Oxidation. Mitochondrion 2019, 49, 97–110. [Google Scholar] [CrossRef]
- Lundsgaard, A.-M.; Fritzen, A.M.; Kiens, B. Chapter 36—Exercise Physiology in Men and Women. In Principles of Gender-Specific Medicine, 3rd ed.; Legato, M.J., Ed.; Academic Press: San Diego, CA, USA, 2017; pp. 525–542. ISBN 978-0-12-803506-1. [Google Scholar]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Geng, C.; Guo, Y.; Wang, C.; Liao, D.; Han, W.; Zhang, J.; Jiang, P. Systematic Impacts of Chronic Unpredictable Mild Stress on Metabolomics in Rats. Sci. Rep. 2020, 10, 700. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J.; Whitfield, J.; Janzen, N.R.; Belhaj, M.R.; Galic, S.; Murray-Segal, L.; Smiles, W.J.; Ling, N.X.Y.; Dite, T.A.; Scott, J.W.; et al. Genetic Loss of AMPK-Glycogen Binding Destabilises AMPK and Disrupts Metabolism. Mol. Metab. 2020, 41, 101048. [Google Scholar] [CrossRef]
- Kow, Y.W.; Dare, A. Detection of Abasic Sites and Oxidative DNA Base Damage Using an ELISA-like Assay. Methods 2000, 22, 164–169. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef]
- Aimo, A.; Castiglione, V.; Borrelli, C.; Saccaro, L.F.; Franzini, M.; Masi, S.; Emdin, M.; Giannoni, A. Oxidative Stress and Inflammation in the Evolution of Heart Failure: From Pathophysiology to Therapeutic Strategies. Eur. J. Prev. Cardiol. 2020, 27, 494–510. [Google Scholar] [CrossRef]
- Higo, T.; Naito, A.T.; Sumida, T.; Shibamoto, M.; Okada, K.; Nomura, S.; Nakagawa, A.; Yamaguchi, T.; Sakai, T.; Hashimoto, A.; et al. DNA Single-Strand Break-Induced DNA Damage Response Causes Heart Failure. Nat. Commun. 2017, 8, 15104. [Google Scholar] [CrossRef]
- van Bilsen, M.; Smeets, P.J.H.; Gilde, A.J.; van der Vusse, G.J. Metabolic Remodelling of the Failing Heart: The Cardiac Burn-out Syndrome? Cardiovasc. Res. 2004, 61, 218–226. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial Energy Metabolism in Heart Failure: A Question of Balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxid. Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; McLaughlin, D.; Robinson, E.; Harvey, A.P.; Hookham, M.B.; Shah, A.M.; McDermott, B.J.; Grieve, D.J. Nox2 NADPH Oxidase Promotes Pathologic Cardiac Remodeling Associated with Doxorubicin Chemotherapy. Cancer Res. 2010, 70, 9287–9297. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines 2020, 8, 206. [Google Scholar] [CrossRef]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative Stress Regulates Collagen Synthesis and Matrix Metalloproteinase Activity in Cardiac Fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280, C53–C60. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Phang, J.M.; Liu, W.; Zabirnyk, O. Proline Metabolism and Microenvironmental Stress. Annu. Rev. Nutr. 2010, 30, 441–463. [Google Scholar] [CrossRef]
- Ben Rejeb, K.; Abdelly, C.; Savouré, A. How Reactive Oxygen Species and Proline Face Stress Together. Plant Physiol. Biochem. 2014, 80, 278–284. [Google Scholar] [CrossRef]
- Vale, S. Psychosocial Stress and Cardiovascular Diseases. Postgrad. Med. J. 2005, 81, 429–435. [Google Scholar] [CrossRef]
- Al, L.-L.; Hb, J.; Mdc, E.V.; Mb, P.; Gv, P.; Fja, A. Chronic Unpredictable Mild Stress Generates Oxidative Stress and Systemic Inflammation in Rats. Physiol. Behav. 2016, 161, 15–23. [Google Scholar] [CrossRef]
- Greaney, J.L.; Saunders, E.F.H.; Santhanam, L.; Alexander, L.M. Oxidative Stress Contributes to Microvascular Endothelial Dysfunction in Men and Women with Major Depressive Disorder. Circ. Res. 2019, 124, 564–574. [Google Scholar] [CrossRef]
- Carley, A.N.; Taegtmeyer, H.; Lewandowski, E.D. Mechanisms Linking Energy Substrate Metabolism to the Function of the Heart. Circ. Res. 2014, 114, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Chabowski, A.; Coort, S.L.M.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. The Subcellular Compartmentation of Fatty Acid Transporters Is Regulated Differently by Insulin and by AICAR. FEBS Lett. 2005, 579, 2428–2432. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Lopaschuk, G.D. Cardiac Fatty Acid Oxidation in Heart Failure Associated with Obesity and Diabetes. Biochim. Biophys. Acta 2016, 1861, 1525–1534. [Google Scholar] [CrossRef]
- Buckley, D.; Duke, G.; Heuer, T.S.; O’Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty Acid Synthase—Modern Tumor Cell Biology Insights into a Classical Oncology Target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.M.; Wertz, S.L.; Pollard, C.M.; Desimine, V.L.; Maning, J.; McCrink, K.A.; Lymperopoulos, A. Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. Int. J. Mol. Sci. 2018, 19, 3764. [Google Scholar] [CrossRef]
- Ngwenyama, N.; Carrillo-Salinas, F.; Aronovitz, M.; Alcaide, P. Self-Antigen-Specific T Cell Receptor Activation of CD4+ T Cells in the Heart Is Required for Maladaptive Cardiac Remodeling in Pressure Overload Induced Heart Failure. FASEB J. 2019, 33, 374.1. [Google Scholar] [CrossRef]
- Razmara, E.; Garshasbi, M. Whole-Exome Sequencing Identifies R1279X of MYH6 Gene to Be Associated with Congenital Heart Disease. BMC Cardiovasc. Disord. 2018, 18, 137. [Google Scholar] [CrossRef]
- Ching, Y.-H.; Ghosh, T.K.; Cross, S.J.; Packham, E.A.; Honeyman, L.; Loughna, S.; Robinson, T.E.; Dearlove, A.M.; Ribas, G.; Bonser, A.J.; et al. Mutation in Myosin Heavy Chain 6 Causes Atrial Septal Defect. Nat. Genet. 2005, 37, 423–428. [Google Scholar] [CrossRef]
- James, S.K.; Lindahl, B.; Siegbahn, A.; Stridsberg, M.; Venge, P.; Armstrong, P.; Barnathan, E.S.; Califf, R.; Topol, E.J.; Simoons, M.L.; et al. N-Terminal pro-Brain Natriuretic Peptide and Other Risk Markers for the Separate Prediction of Mortality and Subsequent Myocardial Infarction in Patients with Unstable Coronary Artery Disease: A Global Utilization of Strategies To Open Occluded Arteries (GUSTO)-IV Substudy. Circulation 2003, 108, 275–281. [Google Scholar] [CrossRef]
- Rutledge, T.; Reis, V.A.; Linke, S.E.; Greenberg, B.H.; Mills, P.J. Depression in Heart Failure a Meta-Analytic Review of Prevalence, Intervention Effects, and Associations with Clinical Outcomes. J. Am. Coll. Cardiol. 2006, 48, 1527–1537. [Google Scholar] [CrossRef]
- Zuzarte, P.; Scola, G.; Duong, A.; Kostiw, K.; Figueira, M.L.; Costa-Vitali, A. NT-ProBNP Is a Potential Mediator between Reduced Ejection Fraction and Depression in Patients with Heart Failure. J. Psychiatr. Res. 2018, 104, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Strekalova, T.; Liu, Y.; Kiselev, D.; Khairuddin, S.; Chiu, J.L.Y.; Lam, J.; Chan, Y.-S.; Pavlov, D.; Proshin, A.; Lesch, K.-P.; et al. Chronic Mild Stress Paradigm as a Rat Model of Depression: Facts, Artifacts, and Future Perspectives. Psychopharmacology 2022, 239, 663–693. [Google Scholar] [CrossRef] [PubMed]
- Close, B.; Banister, K.; Baumans, V.; Bernoth, E.M.; Bromage, N.; Bunyan, J.; Erhardt, W.; Flecknell, P.; Gregory, N.; Hackbarth, H.; et al. Recommendations for Euthanasia of Experimental Animals: Part 1. DGXI of the European Commission. Lab. Anim. 1996, 30, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Close, B.; Banister, K.; Baumans, V.; Bernoth, E.M.; Bromage, N.; Bunyan, J.; Erhardt, W.; Flecknell, P.; Gregory, N.; Hackbarth, H.; et al. Recommendations for Euthanasia of Experimental Animals: Part 2. DGXT of the European Commission. Lab. Anim. 1997, 31, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Porsolt, R.D.; Le Pichon, M.; Jalfre, M. Depression: A New Animal Model Sensitive to Antidepressant Treatments. Nature 1977, 266, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Brattelid, T.; Winer, L.H.; Levy, F.O.; Liestøl, K.; Sejersted, O.M.; Andersson, K.B. Reference Gene Alternatives to Gapdh in Rodent and Human Heart Failure Gene Expression Studies. BMC Mol. Biol. 2010, 11, 22. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Biochemical Name | CTL | CMS | Fold Change | p-Value |
|---|---|---|---|---|---|
| Median Concentration ± SD [µmol/mL] | |||||
| Glycine, serine, and threonine metabolism | glycine | 2.852 ± 0.863 | 3.527 ± 0.582 | 1.237 | 0.135 |
| serine | 3.265 ± 1.147 | 2.644 ± 0.652 | 0.810 | 0.285 | |
| phosphoserine | 0.734 ± 0.043 | 0.663 ± 0.059 | 0.903 | 0.031 | |
| threonine | 18.774 ± 6.903 | 22.289 ± 5.210 | 1.187 | 0.362 | |
| Alanine and aspartate metabolism | alanine | 16.282 ± 2.097 | 17.534 ± 3.611 | 1.077 | 0.388 |
| asparagine | 4.360 ± 1.851 | 7.842 ± 2.160 | 1.799 | 0.011 | |
| Glutamate metabolism | glutamine | 42.344 ± 10.766 | 38.485 ± 8.400 | 0.909 | 0.506 |
| glutamic acid | 68.252 ± 13.054 | 80.088 ± 14.123 | 1.173 | 0.095 | |
| gamma-aminobutyrate (GABA) | 0.074 ± 0.021 | 0.060 ± 0.022 | 0.811 | 0.302 | |
| Histidine metabolism | histidine | 0.832 ± 0.084 | 0.901 ± 0.215 | 1.083 | 0.615 |
| 1-methylhistidine | 0.194 ± 0.068 | 0.202 ± 0.032 | 1.041 | 0.615 | |
| Lysine metabolism | lysine | 1.728 ± 0.141 | 1.641 ± 0.324 | 0.950 | 0.468 |
| Phenylalanine and tyrosine metabolism | phenylalanine | 0.836 ± 0.137 | 0.882 ± 0.129 | 1.055 | 0.621 |
| tyrosine | 0.660 ± 0.061 | 0.593 ± 0.064 | 0.898 | 0.082 | |
| Valine, leucine, and isoleucine metabolism | isoleucine | 0.632 ± 0.136 | 0.738 ± 0.089 | 1.168 | 0.074 |
| leucine | 1.369 ± 0.197 | 1.406 ± 0.150 | 1.027 | 0.799 | |
| valine | 1.081 ± 0.264 | 1.559 ± 0.191 | 1.442 | 0.003 | |
| Cysteine, methionine, SAM, and taurine metabolism | cysteine | 0.069 ± 0.013 | 0.079 ± 0.010 | 1.145 | 0.151 |
| taurine | 111.129 ± 5.822 | 106.945 ± 7.418 | 0.962 | 0.271 | |
| methionine | 0.520 ± 0.053 | 0.663 ± 0.078 | 1.275 | 0.003 | |
| Urea cycle; arginine and proline metabolism | arginine | 1.780 ± 0.243 | 1.380 ± 0.274 | 0.775 | 0.013 |
| ornithine | 0.036 ± 0.016 | 0.038 ± 0.008 | 1.056 | 0.799 | |
| urea | 28.327 ± 3.093 | 28.470 ± 3.375 | 1.005 | 0.815 | |
| proline | 0.161 ± 0.025 | 0.248 ± 0.102 | 1.540 | 0.008 | |
| citrulline | 0.361 ± 0.089 | 0.302 ± 0.056 | 0.837 | 0.146 | |
| Glycerolipid metabolism | ethanolamine | 0.249 ± 0.028 | 0.191 ± 0.035 | 0.767 | 0.006 |
| Pathway | Gene Symbol | CTL | CMS | p-Value | ||
|---|---|---|---|---|---|---|
| Me RQ ± SD | Me RQ ± SD | |||||
| Glucose transport | Slc2a1 | 1.010 ± 0.145 | 0.993 ± 0.160 | 0.776 | Scale (RQ) | |
| Slc2a4 | 1.008 ± 0.131 | 1.193 ± 0.146 | <0.001 | >1.6 | ||
| Glucose metabolism | Hk2 | 1.017 ± 0.182 | 1.417 ± 0.220 | <0.001 | 1.21–1.59 | |
| Pfkl | 1.005 ± 0.097 | 1.036 ± 0.176 | 0.538 | 0.8–1.20 | ||
| Tpi1 | 1.008 ± 0.130 | 1.182 ± 0.148 | <0.001 | 0.5–0.79 | ||
| Got2 | 1.011 ± 0.155 | 1.117 ± 0.203 | 0.006 | 0–0.49 | ||
| Gluconeogenesis | Fbp1 | 1.015 ± 0.174 | 0.441 ± 0.151 | <0.001 | ||
| Fbp2 | 1.009 ± 0.136 | 2.116 ± 1.521 | <0.001 | |||
| G6pc | 1.008 ± 0.131 | 1.642 ± 0.558 | <0.001 | |||
| Pc | 1.012 ± 0.156 | 1.341 ± 0.730 | 0.714 | |||
| Glutaminolysis | Slc1a5 | 1.011 ± 0.149 | 1.441 ± 0.245 | <0.001 | ||
| Gls | 1.019 ± 0.200 | 0.954 ± 0.138 | 0.208 | |||
| Pyruvate metabolism | Me1 | 1.012 ± 0.158 | 1.161 ± 0.279 | 0.021 | ||
| Ldha | 1.005 ± 0.104 | 1.273 ± 0.120 | <0.001 | |||
| Pdk2 | 1.006 ± 0.117 | 1.010 ± 0.146 | 0.823 | |||
| Pdk4 | 1.014 ± 0.170 | 4.523 ± 1.753 | <0.001 | |||
| Mpc1 | 1.005 ± 0.107 | 1.038 ± 0.160 | 0.396 | |||
| Mpc2 | 1.007 ± 0.118 | 1.085 ± 0.202 | 0.155 | |||
| Monocarboxylate transport | Slc16a1 | 1.006 ± 0.113 | 1.471 ± 0.231 | <0.001 | ||
| Urea cycle | Cs | 1.007 ± 0.122 | 1.237 ± 0.186 | <0.001 | ||
| Sdhc | 1.005 ± 0.106 | 1.045 ± 0.094 | 0.118 | |||
| Mdh1 | 1.006 ± 0.114 | 1.178 ± 0.208 | 0.001 | |||
| Idh2 | 1.010 ± 0.150 | 1.152 ± 0.144 | 0.001 | |||
| Lipid transport | Cd36 | 1.004 ± 0.093 | 1.319 ± 0.108 | <0.001 | ||
| Slc27a1 | 1.011 ± 0.145 | 1.231 ± 0.205 | <0.001 | |||
| Cpt2 | 1.011 ± 0.154 | 1.005 ± 0.184 | 0.762 | |||
| Lipid synthesis | Acaca | 1.014 ± 0.172 | 1.058 ± 0.243 | 0.653 | ||
| Dgat1 | 1.005 ± 0.100 | 1.255 ± 0.323 | 0.004 | |||
| Acly | 1.016 ± 0.185 | 1.286 ± 0.180 | <0.001 | |||
| Prkaa2 | 1.012 ± 0.162 | 1.555 ± 0.249 | <0.001 | |||
| Mlycd | 1.005 ± 0.107 | 1.101 ± 0.236 | 0.304 | |||
| β-oxidation | Cpt1a | 1.007 ± 0.123 | 1.581 ± 0.0256 | <0.001 | ||
| Cpt1b | 1.014 ± 0.171 | 1.030 ± 0.170 | 0.890 | |||
| Ppargc1a | 1.012 ± 0.161 | 1.942 ± 0.324 | <0.001 | |||
| Ppara | 1.008 ± 0.130 | 1.002 ± 0.256 | 0.728 | |||
| Acsl1 | 1.011 ± 0.155 | 1.218 ± 0.230 | 0.001 | |||
| Acadm | 1.014 ± 0.174 | 1.202 ± 0.228 | 0.002 | |||
| Oxidative stress | Cat | 1.013 ± 0.164 | 1.216 ± 0.241 | 0.002 | ||
| Sod2 | 1.009 ± 0.135 | 1.200 ± 0.181 | <0.001 | |||
| Heart remodeling | Myh6 | 1.012 ± 0.160 | 1.663 ± 0.316 | <0.001 | ||
| Myh7 | 1.017 ± 0.192 | 0.628 ± 0.203 | <0.001 | |||
| Transcription factors | Myc | 1.016 ± 0.186 | 1.904 ± 0.244 | <0.001 | ||
| Hif1a | 1.008 ± 0.128 | 1.150 ± 0.200 | 0.005 | |||
| Srebp1c | 1.008 ± 0.129 | 0.975 ± 0.164 | 0.313 | |||
| Morphological Feature | CTL | CMS |
|---|---|---|
| Mononuclear cell infiltration | − (8) | + (8) |
| Wavy course of fibers (necrosis) | − (9) | + (9) |
| Increased acidophilus (necrosis) | − (10) | + (9) |
| Collagen deposition | − (9) | + (10) |
| Glycogen deposition | − (10) | ++ (10) |
| Pathway/Function | Gene Symbol | RefSeq | Sequence (5′ → 3′) | |
|---|---|---|---|---|
| Glucose transport | Slc2a1 | NM_138827.1 | F: gcc tga gac cag ttg aaa gc | R: gag tgt ccg tgt ctt cag ca |
| Slc2a4 | NM_012751.1 | F: gct tct gtt gcc ctt ctg tc | R: tgg acg ctc tct ttc caa ct | |
| Glucose metabolism | Hk2 | NM_012735.2 | F: ctc cat ccc aca gga ggt ta | R: tga gga gga tgc tct ggt ct |
| Pfkl | NM_031715.1 | F: ggc ttt gag gct tac aca gg | R: cag act gct tga ttc ggt ca | |
| Tpi1 | NM_022922.2 | F: atg tca cca aga tgg ctt cc | R: aca cat cca gta ggg ctt gg | |
| Got2 | NM_013177.2 | F: acc atc cac tgc cgt ctt ac | R: tct tga agg ctt cgg tca ct | |
| Gluconeogenesis | Fbp1 | NM_012558.3 | F: tta tag gct ccc cca gga ct | R: cag ggt gct gat atc cgt tt |
| Fbp2 | NM_053716.1 | F: cag ggg atg agg tga aga aa | R: agt ggg tca aag caa acc ac | |
| G6pc | NM_013098.2 | F: acc ctg gta gcc ctg tct tt | R: ggg ctt tct ctt ctg tgt cg | |
| Pc | NM_012744.2 | F: gag att gcc atc cga gtg tt | R: ctc ctt ggc cac ctt aat ga | |
| Glutaminolysis | Slc1a5 | NM_175758.3 | F: ttc tat gcc ctg gtg acc tc | R: tgc cat ctc ctt gaa tag gg |
| Gls | NM_012569.2 | F: cac aca cac gga ttt ctt gg | R: gcc gaa gct gac ttt gaa ac | |
| Pyruvate transport | Mpc1 | NM_133561.1 | F: act ttc gcc ctc tgt tgc ta | R: gca ctg tcc ctt tca aga gc |
| Mpc2 | NM_001077643.2 | F: ttt tct ggg ctc cga taa tg | R: gat ccg aaa cag ctg aga gg | |
| Pyruvate metabolism | Me1 | NM_012600.2 | F: gcc ctg aat atg atg cgt tt | R: cct gga aca gca ctg tct ga |
| Ldha | NM_017025.1 | F: ggt ggt tga cag tgc ata cg | R: agg ata cat ggg acg ctg ag | |
| Pdk2 | NM_030872.1 | F: agg aag tca atg cca cca ac | R: ttt tga tgg gag gga gag tg | |
| Pdk4 | NM_053551.1 | F: cct ttg gct ggt ttt ggt ta | R: cac cag tca tca gcc tca ga | |
| Monocarboxylate transport | Slc16a1 | NM_012716.2 | F: tat gcc gga ggt cct atc ag | R: agt tga aag caa gcc caa ga |
| Urea cycle | Cs | NM_130755.1 | F: aag gct aaa ggt ggg gaa ga | R: gtg cag att ggt cgg aaa at |
| Sdhc | NM_001005534.1 | F: ctt tgt gct gct tct tgc tg | R: cca cgc tgc tct tta tct cc | |
| Mdh1 | NM_033235.2 | F: gaa gcc ctc aaa gac gac ag | R: cga cag gga acg agt aga gc | |
| Idh2 | NM_001014161.1 | F: cag tct gac atc ctg gct ca | R: aga tgc tgg caa tag ggt tg | |
| Lipid transport | Cd36 | NM_031561.2 | F: gca aca aca agg cca ggt at | R: aag agc tag gca gca tgg aa |
| Slc27a1 | NM_053580.2 | F: cct cac atc aca gca gga ga | R: gct ctg tcc aca ccc ttc at | |
| Cpt2 | NM_012930.1 | F: tcc tcg atc aag atg gga ac | R: gat cct tca tcg gga agt ca | |
| Lipid synthesis | Acaca | NM_022193.1 | F: tac aac gca ggc atc aga ag | R: tgt gct gca gga aga ttg ac |
| Dgat1 | NM_053437.1 | F: ctg ctg ccc aga aca ctg ta | R: aag ctg ggt gaa aaa gag ca | |
| Acly | NM_016987.2 | F: ctc aca cgg aag ctc atc aa | R: atg gca aca ccc tcg tag ac | |
| Prkaa2 | NM_023991.1 | F: agc tcg cag tgg ctt atc at | R: ggg gct gtc tgc tat gag ag | |
| Mlycd | NM_053477.1 | F: gcc tgg tac ctt tac ggt ga | R: gct acc agg ctg agg atc tg | |
| β-oxidation | Cpt1a | NM_031559.2 | F: atg acg gct atg gtg tct cc | R: gtg agg cca aac aag gtg at |
| Cpt1b | NM_013200.1 | F: gca aac tgg acc gag aag ag | R: cct tga aga agc gac ctt tg | |
| Ppargc1a | NM_031347.1 | F: atg tgt cgc ctt ctt gct ct | R: atc tac tgc ctg ggg acc tt | |
| Ppara | NM_013196.1 | F: tca cac aat gca atc cgt tt | R: ggc ctt gac ctt gtt cat gt | |
| Acsl1 | NM_012820.1 | F: aac gat gta cga tgg ctt cc | R: ggt cac cca ctc agg tct gt | |
| Acadm | NM_016986.2 | F: caa gag agc ctg gga act tg | R: ccc caa aga att tgc ttc aa | |
| Oxidative stress | Cat | NM_012520.2 | F: aca tgg tct ggg act tct gg | R: caa gtt ttt gat gcc ctg gt |
| Sod2 | NM_017051.2 | F: cac tgt ggc tga gct gtt gt | R: tcc aag caa ttc aag cct ct | |
| Heart remodeling | Myh6 | NM_017239.2 | F: tga tga ctc cga gga gct tt | R: tga cac aga ccc ttg agc ag |
| Myh7 | NM_017240.2 | F: cct cgc aat atc aag gga aa | R: tac agg tgc atc agc tcc ag | |
| Transcription factors | Myc | NM_012603.2 | F: cga gct gaa gcg tag ctt tt | R: ctc gcc gtt tcc tca gta ag |
| Hif1a | NM_024359.1 | F: tca agt cag caa cgt gga ag | R: tat cga ggc tgt gtc gac tg | |
| Srebp1c | NM_001276707.1 | F: gtg gtc ttc cag agg ctg ag | R: ggg tga gag cct tga gac ag | |
| Reference genes | Rpl32 | NM_013226 | F: aga ttc aag ggc cag atc ct | R: cga tgg ctt ttc ggt tct ta |
| Polr2a | XM_343922 | F: cgt atc cgc atc atg aac agt ga | R: tca tcc atc tta tcc acc acc tct t | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostrowska-Leśko, M.; Herbet, M.; Pawłowski, K.; Korga-Plewko, A.; Poleszak, E.; Dudka, J. Pathological Changes and Metabolic Adaptation in the Myocardium of Rats in Response to Chronic Variable Mild Stress. Int. J. Mol. Sci. 2024, 25, 5899. https://doi.org/10.3390/ijms25115899
Ostrowska-Leśko M, Herbet M, Pawłowski K, Korga-Plewko A, Poleszak E, Dudka J. Pathological Changes and Metabolic Adaptation in the Myocardium of Rats in Response to Chronic Variable Mild Stress. International Journal of Molecular Sciences. 2024; 25(11):5899. https://doi.org/10.3390/ijms25115899
Chicago/Turabian StyleOstrowska-Leśko, Marta, Mariola Herbet, Kamil Pawłowski, Agnieszka Korga-Plewko, Ewa Poleszak, and Jarosław Dudka. 2024. "Pathological Changes and Metabolic Adaptation in the Myocardium of Rats in Response to Chronic Variable Mild Stress" International Journal of Molecular Sciences 25, no. 11: 5899. https://doi.org/10.3390/ijms25115899
APA StyleOstrowska-Leśko, M., Herbet, M., Pawłowski, K., Korga-Plewko, A., Poleszak, E., & Dudka, J. (2024). Pathological Changes and Metabolic Adaptation in the Myocardium of Rats in Response to Chronic Variable Mild Stress. International Journal of Molecular Sciences, 25(11), 5899. https://doi.org/10.3390/ijms25115899