
Acute Kidney Injury in Sepsis

Abstract
:1. Introduction
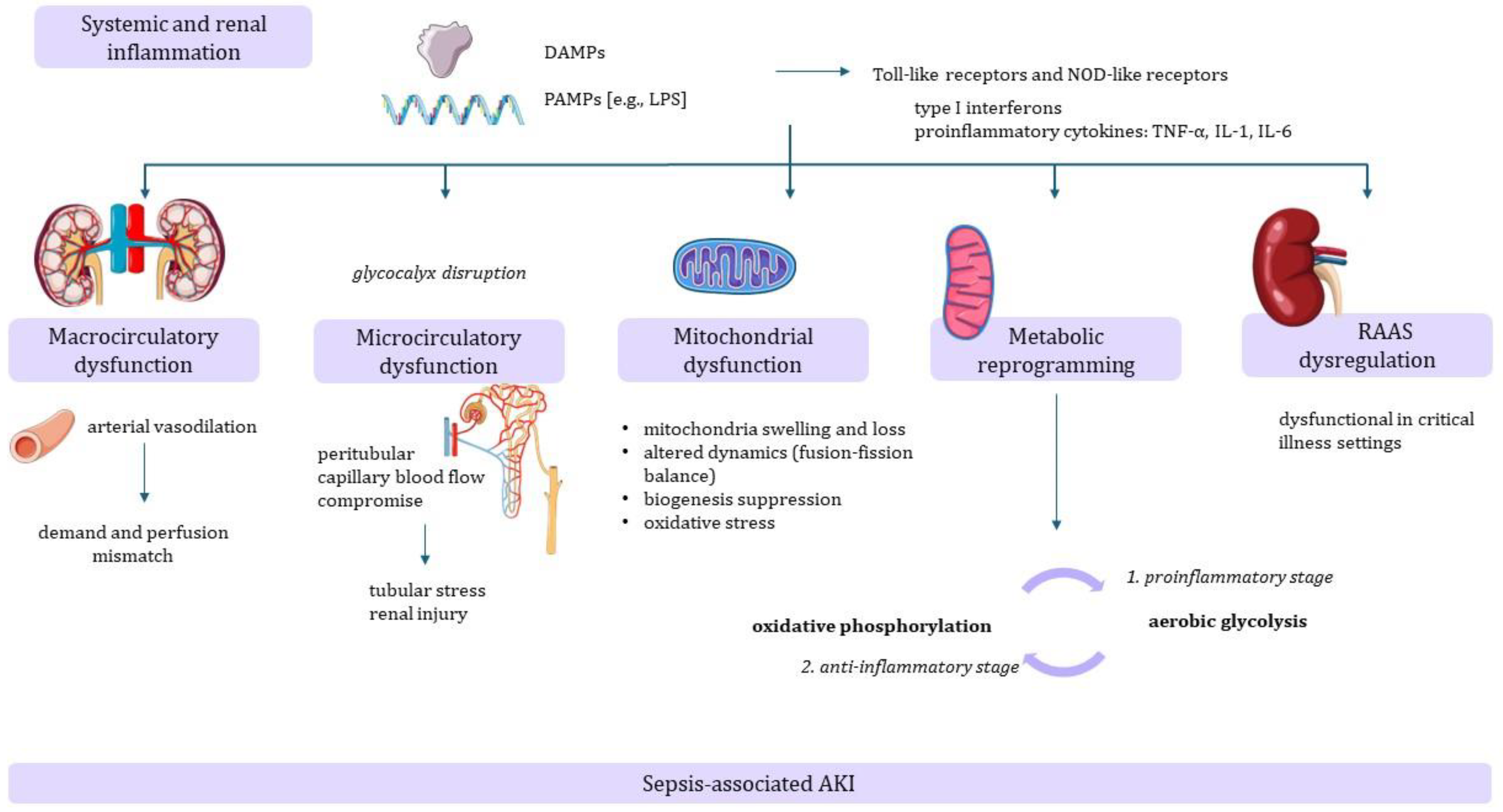
2. Pathophysiology
3. Treatment
3.1. Antimicrobial Administration
3.2. Fluid Resuscitation
3.3. Vasoactive Drugs
3.4. Renal Replacement Therapy
3.5. Extracorporeal Therapies
3.6. Mechanical Ventilation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; Mcintyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock 2021. Crit. Care Med. 2021, 49, E1063–E1143. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA—J. Am. Med. Assoc. 2016, 315, 801–810. [Google Scholar] [CrossRef]
- KDIGO Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. Suppl. 2012, 2, 1–138. [Google Scholar] [CrossRef]
- Zarbock, A.; Nadim, M.K.; Pickkers, P.; Gomez, H.; Bell, S.; Joannidis, M.; Kashani, K.; Koyner, J.L.; Pannu, N.; Meersch, M.; et al. Sepsis-associated acute kidney injury: Consensus report of the 28th Acute Disease Quality Initiative workgroup. Nat. Rev. Nephrol. 2023, 19, 401–417. [Google Scholar] [CrossRef]
- Bagshaw, S.M.; George, C.; Bellomo, R. Early acute kidney injury and sepsis: A multicentre evaluation. Crit. Care 2008, 12, R47. [Google Scholar] [CrossRef]
- Liu, J.; Xie, H.; Ye, Z.; Li, F.; Wang, L. Rates, predictors, and mortality of sepsis-associated acute kidney injury: A systematic review and meta-analysis. BMC Nephrol. 2020, 21, 318. [Google Scholar] [CrossRef]
- Hoste, E.A.J.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gómez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Bagshaw, S.M.; Uchino, S.; Bellomo, R.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; Gibney, N.; et al. Septic acute kidney injury in critically ill patients: Clinical characteristics and outcomes. Clin. J. Am. Soc. Nephrol. 2007, 2, 431–439. [Google Scholar] [CrossRef]
- Shum, H.P.; Yan, W.W.; Chan, T.M. Recent knowledge on the pathophysiology of septic acute kidney injury: A narrative review. J. Crit. Care 2016, 31, 82–89. [Google Scholar] [CrossRef]
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, i1585. [Google Scholar] [CrossRef]
- Kuwabara, S.; Goggins, E.; Okusa, M.D. The Pathophysiology of Sepsis-Associated AKI. Clin. J. Am. Soc. Nephrol. 2022, 17, 1050–1069. [Google Scholar] [CrossRef]
- Kounatidis, D.; Vallianou, N.G.; Psallida, S.; Panagopoulos, F.; Margellou, E.; Tsilingiris, D.; Karampela, I.; Stratigou, T.; Dalamaga, M. Sepsis-Associated Acute Kidney Injury: Where Are We Now? Medicina 2024, 60, 434. [Google Scholar] [CrossRef]
- Sun, S.; Chen, R.; Dou, X.; Dai, M.; Long, J.; Wu, Y.; Lin, Y. Immunoregulatory mechanism of acute kidney injury in sepsis: A Narrative Review. Biomed. Pharmacother. 2023, 159, 114202. [Google Scholar] [CrossRef]
- Oliveira, F.R.M.B.; Assreuy, J.; Sordi, R. The role of nitric oxide in sepsis-associated kidney injury. Biosci. Rep. 2022, 42, BSR20220093. [Google Scholar] [CrossRef]
- Schrier, R.W.; Wang, W. Acute renal failure and sepsis. N. Engl. J. Med. 2004, 351, 159–169. [Google Scholar] [CrossRef]
- Zafrani, L.; Payen, D.; Azoulay, E.; Ince, C. The Microcirculation of the Septic Kidney. Semin. Nephrol. 2015, 35, 75–84. [Google Scholar] [CrossRef]
- Wu, L.; Tiwari, M.M.; Messer, K.J.; Holthoff, J.H.; Gokden, N.; Brock, R.W.; Mayeux, P.R. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. Am. J. Physiol. Renal Physiol. 2007, 292, F261–F268. [Google Scholar] [CrossRef]
- Sun, N.; Zheng, S.; Rosin, D.L.; Poudel, N.; Yao, J.; Perry, H.M.; Cao, R.; Okusa, M.D.; Hu, S. Development of a photoacoustic microscopy technique to assess peritubular capillary function and oxygen metabolism in the mouse kidney. Kidney Int. 2021, 100, 613–620. [Google Scholar] [CrossRef]
- Morrell, E.D.; Kellum, J.A.; Pastor-Soler, N.M.; Hallows, K.R. Septic acute kidney injury: Molecular mechanisms and the importance of stratification and targeting therapy. Crit. Care 2014, 18, 501. [Google Scholar] [CrossRef]
- Zhang, X.; Agborbesong, E.; Li, X. The role of mitochondria in acute kidney injury and chronic kidney disease and its therapeutic potential. Int. J. Mol. Sci. 2021, 22, 11253. [Google Scholar] [CrossRef] [PubMed]
- Gómez, H.; Kellum, J.A.; Ronco, C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat. Rev. Nephrol. 2017, 13, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Flannery, A.H.; Ortiz-Soriano, V.; Li, X.; Gianella, F.G.; Toto, R.D.; Moe, O.W.; Devarajan, P.; Goldstein, S.L.; Neyra, J.A. Serum renin and major adverse kidney events in critically ill patients: A multicenter prospective study. Crit. Care 2021, 25, 294. [Google Scholar] [CrossRef] [PubMed]
- Ehrmann, S.; Helms, J.; Joret, A.; Martin-Lefevre, L.; Quenot, J.P.; Herbrecht, J.E.; Benzekri-Lefevre, D.; Robert, R.; Desachy, A.; Bellec, F.; et al. Nephrotoxic drug burden among 1001 critically ill patients: Impact on acute kidney injury. Ann. Intensive Care 2019, 9, 106. [Google Scholar] [CrossRef]
- Marshall, J.C.; al Naqbi, A. Principles of Source Control in the Management of Sepsis. Crit. Care Nurs. Clin. N. Am. 2011, 23, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Lagunes, L.; Encina, B.; Ramirez-Estrada, S. Current understanding in source control management in septic shock patients: A review. Ann. Transl. Med. 2016, 4, 330. [Google Scholar] [CrossRef] [PubMed]
- Payen, D.; de Pont, A.C.; Sakr, Y.; Spies, C.; Reinhart, K.; Vincent, J.L. A positive fluid balance is associated with a worse outcome in patients with acute renal failure. Crit. Care 2008, 12, R74. [Google Scholar] [CrossRef] [PubMed]
- Grams, M.E.; Estrella, M.M.; Coresh, J.; Brower, R.G.; Liu, K.D. Fluid balance, diuretic use, and mortality in acute kidney injury. Clin. J. Am. Soc. Nephrol. 2011, 6, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.P. Fluid overload and acute kidney injury. Indian J. Crit. Care Med. 2020, 24, S94–S97. [Google Scholar] [CrossRef]
- Zampieri, F.G.; Machado, F.R.; Biondi, R.S.; Freitas, F.G.R.; Veiga, V.C.; Figueiredo, R.C.; Lovato, W.J.; Serpa-Neto, A.; Paranhos, J.L.R.; Guedes, M.A.V.; et al. Effect of Intravenous Fluid Treatment with a Balanced Solution vs. 0.9% Saline Solution on Mortality in Critically Ill Patients: The BaSICS Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2021, 326, 818–829. [Google Scholar] [CrossRef]
- Finfer, S.; Micallef, S.; Hammond, N.; Navarra, L.; Bellomo, R.; Billot, L.; Delaney, A.; Gallagher, M.; Gattas, D.; Li, Q.; et al. Balanced Multielectrolyte Solution versus Saline in Critically Ill Adults. N. Engl. J. Med. 2022, 386, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Yealy, D.M.; Kellum, J.A.; Huang, D.T.; Barnato, A.E.; Weissfeld, L.A.; Pike, F.; Terndrup, T.; Wang, H.E.; Hou, P.C.; LoVecchio, F.; et al. A Randomized Trial of Protocol-Based Care for Early Septic Shock. N. Engl. J. Med. 2014, 370, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- The ARISE Investigators and the ANZICS Clinical Trials Group. Goal-Directed Resuscitation for Patients with Early Septic Shock. N. Engl. J. Med. 2014, 371, 1496–1506. [Google Scholar] [CrossRef]
- Mouncey, P.R.; Osborn, T.M.; Power, G.S.; Harrison, D.A.; Sadique, M.Z.; Grieve, R.D.; Jahan, R.; Harvey, S.E.; Bell, D.; Bion, J.F.; et al. Trial of Early, Goal-Directed Resuscitation for Septic Shock. N. Engl. J. Med. 2015, 372, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- The PRISM Investigators. Early, Goal-Directed Therapy for Septic Shock—A Patient-Level Meta-Analysis. N. Engl. J. Med. 2017, 376, 2223–2234. [Google Scholar] [CrossRef]
- Liu, Z.-M.; Chen, J.; Kou, Q.; Lin, Q.; Huang, X.; Tang, Z.; Kang, Y.; Li, K.; Zhou, L.; Song, Q.; et al. Terlipressin versus norepinephrine as infusion in patients with septic shock: A multicentre, randomised, double-blinded trial. Intensive Care Med. 2018, 44, 1816–1825. [Google Scholar] [CrossRef]
- Shi, R.; Hamzaoui, O.; De Vita, N.; Monnet, X.; Teboul, J.-L. Vasopressors in septic shock: Which, when, and how much? Ann. Transl. Med. 2020, 8, 794. [Google Scholar] [CrossRef]
- Gordon, A.C.; Mason, A.J.; Thirunavukkarasu, N.; Perkins, G.D.; Cecconi, M.; Cepkova, M.; Pogson, D.G.; Aya, H.D.; Anjum, A.; Frazier, G.J.; et al. Effect of early vasopressin vs norepinephrine on kidney failure in patients with septic shock: The VANISH randomized clinical trial. JAMA—J. Am. Med. Assoc. 2016, 316, 509–518. [Google Scholar] [CrossRef]
- Tumlin, J.A.; Murugan, R.; Deane, A.M.; Ostermann, M.; Busse, L.W.; Ham, K.R.; Kashani, K.; Szerlip, H.M.; Prowle, J.R.; Bihorac, A.; et al. Outcomes in patients with vasodilatory shock and renal replacement therapy treated with intravenous angiotensin II. Crit. Care Med. 2018, 46, 949–957. [Google Scholar] [CrossRef]
- Myburgh, J.A.; Higgins, A.; Jovanovska, A.; Lipman, J.; Ramakrishnan, N.; Santamaria, J. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med. 2008, 34, 2226–2234. [Google Scholar] [CrossRef]
- De Backer, D.; Biston, P.; Devriendt, J.; Madl, C.; Chochrad, D.; Aldecoa, C.; Brasseur, A.; Defrance, P.; Gottignies, P.; Vincent, J.-L. Comparison of Dopamine and Norepinephrine in the Treatment of Shock. N. Engl. J. Med. 2010, 362, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Asfar, P.; Radermacher, P.; Vincent, J.L.; Martin, C. Optimizing mean arterial pressure in septic shock: A critical reappraisal of the literature. Crit. Care 2015, 19, 101. [Google Scholar] [CrossRef] [PubMed]
- Strandgaard, S.; Olesen, J.; Skinh, E.; Lassen, N.A. Autoregulation of Brain Circulation in Severe Arterial Hypertension. Br. Med. J. 1973, 1, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, M.; De Backer, D.; Antonelli, M.; Beale, R.; Bakker, J.; Hofer, C.; Jaeschke, R.; Mebazaa, A.; Pinsky, M.R.; Teboul, J.L.; et al. Consensus on circulatory shock and hemodynamic monitoring. Task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2014, 40, 1795–1815. [Google Scholar] [CrossRef] [PubMed]
- Asfar, P.; Meziani, F.; Hamel, J.-F.; Grelon, F.; Megarbane, B.; Anguel, N.; Mira, J.-P.; Dequin, P.-F.; Gergaud, S.; Weiss, N.; et al. High versus Low Blood-Pressure Target in Patients with Septic Shock. N. Engl. J. Med. 2014, 370, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, H.; Fukui, S.; Higashio, K.; Endo, A.; Takasu, A.; Yamakawa, K. Optimal target blood pressure in critically ill adult patients with vasodilatory shock: A systematic review and meta-analysis. Front. Physiol. 2022, 13, 962670. [Google Scholar] [CrossRef] [PubMed]
- Karvellas, C.J.; Farhat, M.R.; Sajjad, I.; Mogensen, S.S.; Leung, A.A.; Wald, R.; Bagshaw, S.M. A comparison of early versus late initiation of renal replacement therapy in critically ill patients with acute kidney injury: A systematic review and meta-analysis. Crit. Care 2011, 15, R72. [Google Scholar] [CrossRef] [PubMed]
- Seabra, V.F.; Balk, E.M.; Liangos, O.; Sosa, M.A.; Cendoroglo, M.; Jaber, B.L. Timing of Renal Replacement Therapy Initiation in Acute Renal Failure: A Meta-analysis. Am. J. Kidney Dis. 2008, 52, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Kellum, J.A.; Schmidt, C.; Van Aken, H.; Wempe, C.; Pavenstädt, H.; Boanta, A.; Gerß, J.; Meersch, M. Effect of early vs delayed initiation of renal replacement therapy on mortality in critically ill patients with acute kidney injury: The ELAIN randomized clinical trial. JAMA—J. Am. Med. Assoc. 2016, 315, 2190–2199. [Google Scholar] [CrossRef]
- Gaudry, S.; Hajage, D.; Schortgen, F.; Martin-Lefevre, L.; Pons, B.; Boulet, E.; Boyer, A.; Chevrel, G.; Lerolle, N.; Carpentier, D.; et al. Initiation Strategies for Renal-Replacement Therapy in the Intensive Care Unit. N. Engl. J. Med. 2016, 375, 122–133. [Google Scholar] [CrossRef]
- Barbar, S.D.; Clere-Jehl, R.; Bourredjem, A.; Hernu, R.; Montini, F.; Bruyère, R.; Lebert, C.; Bohé, J.; Badie, J.; Eraldi, J.-P.; et al. Timing of Renal-Replacement Therapy in Patients with Acute Kidney Injury and Sepsis. N. Engl. J. Med. 2018, 379, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- STARRT-AKI Investigators. Timing of Initiation of Renal-Replacement Therapy in Acute Kidney Injury. N. Engl. J. Med. 2020, 383, 240–251. [Google Scholar] [CrossRef]
- Fayad, A.I.I.; Buamscha, D.G.; Ciapponi, A. Timing of renal replacement therapy initiation for acute kidney injury. Cochrane Database Syst. Rev. 2018, 2018, CD010612. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, Z. When to start renal replacement therapy in acute kidney injury: What are we waiting for? J. Intensive Med. 2024, in press. [Google Scholar] [CrossRef]
- Chen, J.-J.; Lee, C.-C.; Kuo, G.; Fan, P.-C.; Lin, C.-Y.; Chang, S.-W.; Tian, Y.-C.; Chen, Y.-C.; Chang, C.-H. Comparison between watchful waiting strategy and early initiation of renal replacement therapy in the critically ill acute kidney injury population: An updated systematic review and meta-analysis. Ann. Intensive Care 2020, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Palevsky, P.M.; Zhang, J.H.; Star, R.A.; Smith, M.W. Design of the VA/NIH Acute Renal Failure Trial Network (ATN) Study: Intensive versus Conventional Renal Support in Acute Renal Failure. Clin. Trials 2005, 2, 423–435. [Google Scholar] [CrossRef]
- Investigators RRTS. Intensity of continuous renal-replacement therapy in critically ill patients. N. Engl. J. Med. 2009, 361, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Lee, H.; Kee, Y.K.; Park, S.; Oh, H.J.; Han, S.H.; Joo, K.W.; Lim, C.-S.; Kim, Y.S.; Kang, S.-W.; et al. High-Dose Versus Conventional-Dose Continuous Venovenous Hemodiafiltration and Patient and Kidney Survival and Cytokine Removal in Sepsis-Associated Acute Kidney Injury: A Randomized Controlled Trial. Am. J. Kidney Dis. 2016, 68, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Coates, E.C.; Smith, D.J., Jr.; Karlnoski, R.A.; Hickerson, W.L.; Arnold-Ross, A.L.; Mosier, M.J.; Halerz, M.; Sprague, A.M.; Mullins, R.F.; et al. High-volume hemofiltration in adult burn patients with septic shock and acute kidney injury: A multicenter randomized controlled trial. Crit. Care 2017, 21, 289. [Google Scholar] [CrossRef] [PubMed]
- Joannes-Boyau, O.; Honoré, P.M.; Perez, P.; Bagshaw, S.M.; Grand, H.; Canivet, J.-L.; Dewitte, A.; Flamens, C.; Pujol, W.; Grandoulier, A.-S.; et al. High-volume versus standard-volume haemofiltration for septic shock patients with acute kidney injury (IVOIRE study): A multicentre randomized controlled trial. Intensive Care Med. 2013, 39, 1535–1546. [Google Scholar] [CrossRef]
- Schefold, J.C.; Haehling, S.; Pschowski, R.; Bender, T.O.; Berkmann, C.; Briegel, S.; Hasper, D.; Jörres, A. The effect of continuous versus intermittent renal replacement therapy on the outcome of critically ill patients with acute renal failure (CONVINT): A prospective randomized controlled trial. Crit. Care 2014, 18, R11. [Google Scholar] [CrossRef]
- Poston, J.T.; Koyner, J.L. Sepsis associated acute kidney injury. BMJ 2019, 364, k4891. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Koyner, J.L.; Gomez, H.; Pickkers, P.; Forni, L. Sepsis-associated acute kidney injury—Treatment standard. Nephrol. Dial. Transplant. 2023, 39, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Bagshaw, S.M.; Antonelli, M.; Foster, D.M.; Klein, D.J.; Marshall, J.C.; Palevsky, P.M.; Weisberg, L.S.; Schorr, C.A.; Trzeciak, S.; et al. Effect of Targeted Polymyxin B Hemoperfusion on 28-Day Mortality in Patients with Septic Shock and Elevated Endotoxin Level: The EUPHRATES Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2018, 320, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Payen, D.M.; Guilhot, J.; Launey, Y.; Lukaszewicz, A.C.; Kaaki, M.; Veber, B.; Pottecher, J.; Joannes-Boyau, O.; Martin-Lefevre, L.; Jabaudon, M.; et al. Early use of polymyxin B hemoperfusion in patients with septic shock due to peritonitis: A multicenter randomized control trial. Intensive Care Med. 2015, 41, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Chawla, L.; Husain-Syed, F.; Kellum, J.A. Rationale for sequential extracorporeal therapy (SET) in sepsis. Crit. Care 2023, 27, 50. [Google Scholar] [CrossRef]
- Hepokoski, M.L.; Malhotra, A.; Singh, P.; Alexander, L.E.C. Ventilator-Induced Kidney Injury: Are Novel Biomarkers the Key to Prevention? Nephron 2018, 140, 90–93. [Google Scholar] [CrossRef]
- van den Akker, J.P.C.; Egal, M.; Groeneveld, J.A.B. Invasive mechanical ventilation as a risk factor for acute kidney injury in the critically ill: A systematic review and meta-analysis. Crit. Care 2013, 17, R98. [Google Scholar] [CrossRef] [PubMed]
- Heimann, J.C.; Hedley-Whyte, J. Mechanisms of Renal Dysfunction During Pressure Ventilation Positive. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1981, 50, 643–649. [Google Scholar]
- Hepokoski, M.; Englert, J.A.; Baron, R.M.; Crotty-Alexander, L.E.; Fuster, M.M.; Beitler, J.R.; Malhotra, A.; Singh, P. Ventilator-induced lung injury increases expression of endothelial inflammatory mediators in the kidney. Am. J. Physiol. Renal Physiol. 2017, 312, F654–F660. [Google Scholar] [CrossRef]
- Imai, Y.; Parodo, J.; Kajikawa, O.; de Perrot, M.; Fischer, S.; Edwards, V.; Cutz, E.; Liu, M.; Keshavjee, S.; Martin, T.R.; et al. Injurious Mechanical Ventilation and End-Organ Epithelial Cell Apoptosis and Organ Dysfunction in an Experimental Model of Acute Respiratory Distress Syndrome. JAMA 2003, 289, 2104–2112. [Google Scholar] [CrossRef]

{kind=link}
| Serum Creatinine Criteria | Urine Output | |
|---|---|---|
| Stage 1 | SCr 1.5 to 1.9 times the baseline value OR SCr ≥0.3 mg/dL (≥26.5 µmol/L) | <0.5 mL/kg/h for 6–12 h |
| Stage 2 | SCr 2.0 to 2.9 times baseline | <0.5 mL/kg/h for ≥12 h |
| Stage 3 | SCr rises to 3.0 times baseline OR Increase in SCr to ≥4.0 mg/dL (≥354 µmol/L) OR Need for initiation of renal replacement therapy | <0.3 mL/kg/h for ≥24 h OR Anuria for ≥12 h |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pais, T.; Jorge, S.; Lopes, J.A. Acute Kidney Injury in Sepsis. Int. J. Mol. Sci. 2024, 25, 5924. https://doi.org/10.3390/ijms25115924
Pais T, Jorge S, Lopes JA. Acute Kidney Injury in Sepsis. International Journal of Molecular Sciences. 2024; 25(11):5924. https://doi.org/10.3390/ijms25115924
Chicago/Turabian StylePais, Telma, Sofia Jorge, and José António Lopes. 2024. "Acute Kidney Injury in Sepsis" International Journal of Molecular Sciences 25, no. 11: 5924. https://doi.org/10.3390/ijms25115924