
Metabolic Regulation of Endothelial Cells: A New Era for Treating Wet Age-Related Macular Degeneration

Abstract
1. Introduction
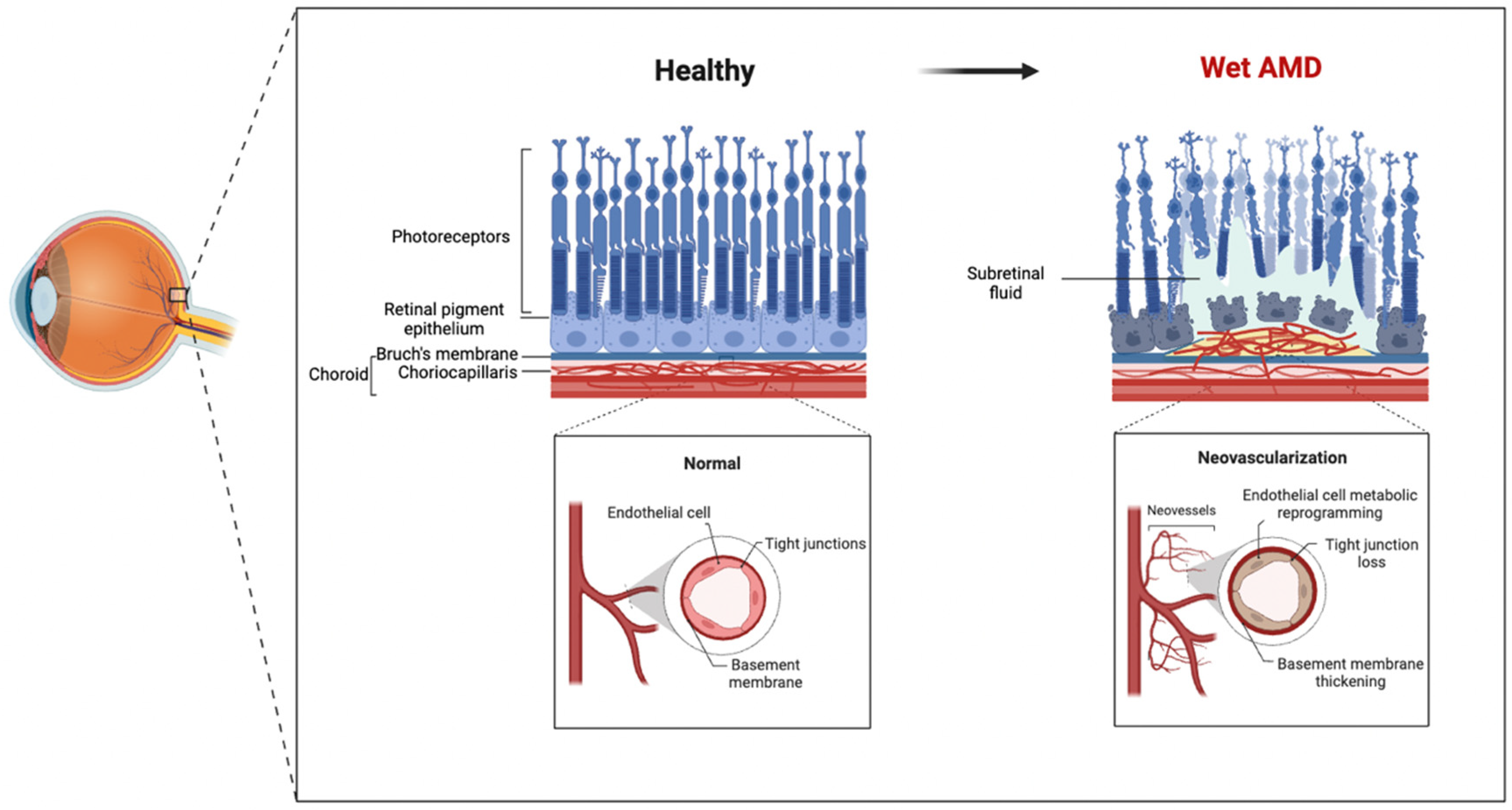
2. The Involvement of ECs in Wet AMD Pathogenesis
2.1. Relationship between EC Dysfunction and Wet AMD
2.2. Evidence for the Involvement of EC Metabolic Dysfunction in the Pathogenic Mechanisms of Wet AMD
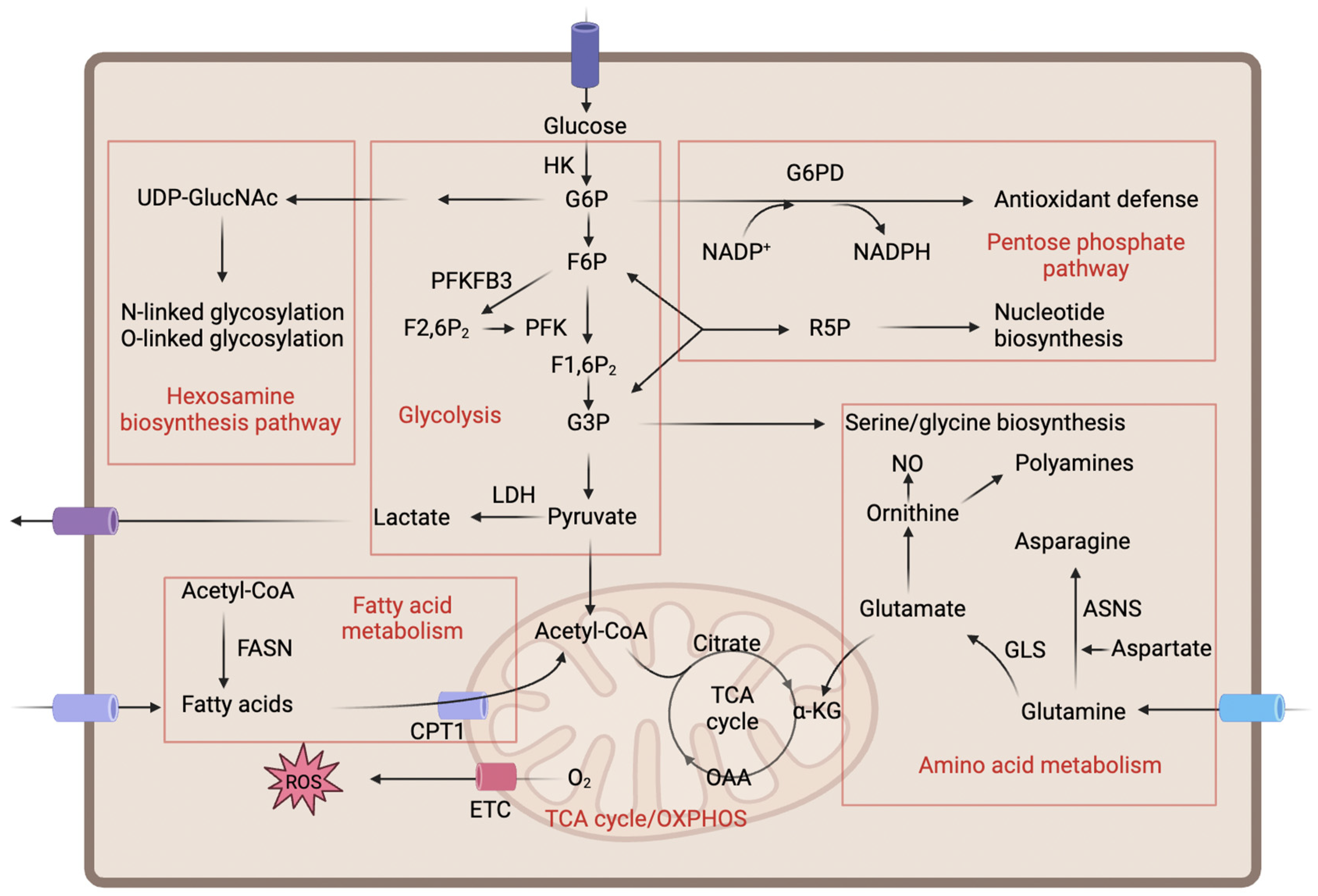
3. Major Metabolic Pathways and Their Regulation in ECs
3.1. Glycolysis and Its Branches
3.2. Mitochondrial Metabolism
3.3. Lipid Metabolism
3.4. Amino Acid Metabolism
4. Metabolic Regulation of ECs and Treatment of Wet AMD
4.1. Targeting Glycolysis for Wet AMD Treatment
4.2. Targeting Mitochondrial Function for Wet AMD Treatment
4.3. Targeting Lipid Metabolism for Wet AMD Treatment
4.4. Targeting Other Metabolic Pathways for Wet AMD Treatment
4.4.1. Pentose Phosphate Pathway
4.4.2. Glycosylation
4.4.3. Glutaminolysis
4.5. Potential Advantages of EC Metabolic Regulation Strategies in Wet AMD Treatment
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Jonas, J.B.; Cheung, C.M.G.; Panda-Jonas, S. Updates on the Epidemiology of Age-Related Macular Degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Colijn, J.M.; Buitendijk, G.H.S.; Prokofyeva, E.; Alves, D.; Cachulo, M.L.; Khawaja, A.P.; Cougnard-Gregoire, A.; Merle, B.M.J.; Korb, C.; Erke, M.G.; et al. Prevalence of Age-Related Macular Degeneration in Europe: The Past and the Future. Ophthalmology 2017, 124, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chen, M.; Liu, Y.; Zhang, D.; Shen, J.; Ni, N.; Tang, Z.; Ju, Y.; Dai, X.; Zhuang, A.; et al. Injectable Anti-Inflammatory Supramolecular Nanofiber Hydrogel to Promote Anti-VEGF Therapy in Age-Related Macular Degeneration Treatment. Adv. Mater. 2023, 35, e2204994. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, R.; Fragiotta, S.; Sarraf, D.; Sadda, S.R.; Freund, K.B.; Parravano, M.; Corradetti, G.; Cabral, D.; Capuano, V.; Miere, A.; et al. Towards a better understanding of non-exudative choroidal and macular neovascularization. Prog. Retin. Eye Res. 2023, 92, 101113. [Google Scholar] [CrossRef]
- Dhoot, D.S.; Kaiser, P.K. Ranibizumab for age-related macular degeneration. Expert. Opin. Biol. Ther. 2012, 12, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Fine, S.L.; Ying, G.S.; Jaffe, G.J.; Grunwald, J.E.; Toth, C.; Redford, M.; Ferris, F.L., 3rd. Ranibizumab and Bevacizumab for Treatment of Neovascular Age-related Macular Degeneration: Two-Year Results. Ophthalmology 2020, 127, S135–S145. [Google Scholar] [CrossRef]
- Sarwar, S.; Clearfield, E.; Soliman, M.K.; Sadiq, M.A.; Baldwin, A.J.; Hanout, M.; Agarwal, A.; Sepah, Y.J.; Do, D.V.; Nguyen, Q.D. Aflibercept for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2016, 2, Cd011346. [Google Scholar] [CrossRef]
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to Anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 2021, 82, 100906. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, J.; Sun, X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: A comprehensive review. Drug Des. Dev. Ther. 2016, 10, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Bierhansl, L.; Conradi, L.C.; Treps, L.; Dewerchin, M.; Carmeliet, P. Central Role of Metabolism in Endothelial Cell Function and Vascular Disease. Physiology 2017, 32, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Carmeliet, P. The Link between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquière, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zou, R.; Zhang, X.; Shen, M.; Chen, X.; Wang, J.; Niu, W.; Yuan, Y.; Yuan, F. YAP promotes ocular neovascularization by modifying PFKFB3-driven endothelial glycolysis. Angiogenesis 2021, 24, 489–504. [Google Scholar] [CrossRef]
- Kim, B.; Arany, Z. Endothelial Lipid Metabolism. Cold Spring Harb. Perspect. Med. 2022, 12, a041162. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef] [PubMed]
- Chirco, K.R.; Sohn, E.H.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Structural and molecular changes in the aging choroid: Implications for age-related macular degeneration. Eye 2017, 31, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Machalinska, A.; Safranow, K.; Dziedziejko, V.; Mozolewska-Piotrowska, K.; Paczkowska, E.; Klos, P.; Pius, E.; Grymula, K.; Wiszniewska, B.; Karczewicz, D.; et al. Different populations of circulating endothelial cells in patients with age-related macular degeneration: A novel insight into pathogenesis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Totan, Y.; Koca, C.; Erdurmuş, M.; Keskin, U.; Yiğitoğlu, R. Endothelin-1 and Nitric Oxide Levels in Exudative Age-Related Macular Degeneration. J. Ophthalmic Vis. Res. 2015, 10, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.A.; Baba, T.; Merges, C.; McLeod, D.S.; Lutty, G.A. Low nitric oxide synthases (NOSs) in eyes with age-related macular degeneration (AMD). Exp. Eye Res. 2010, 90, 155–167. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef]
- Wang, H.; Han, X.; Wittchen, E.S.; Hartnett, M.E. TNF-α mediates choroidal neovascularization by upregulating VEGF expression in RPE through ROS-dependent β-catenin activation. Mol. Vis. 2016, 22, 116–128. [Google Scholar]
- Machalińska, A.; Kawa, M.P.; Marlicz, W.; Machaliński, B. Complement system activation and endothelial dysfunction in patients with age-related macular degeneration (AMD): Possible relationship between AMD and atherosclerosis. Acta Ophthalmol. 2012, 90, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374. [Google Scholar] [CrossRef]
- Marneros, A.G. NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Cell Rep. 2013, 4, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Goveia, J.; García-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e814. [Google Scholar] [CrossRef]
- Yeo, N.J.Y.; Wazny, V.; Nguyen, N.L.U.; Ng, C.Y.; Wu, K.X.; Fan, Q.; Cheung, C.M.G.; Cheung, C. Single-Cell Transcriptome of Wet AMD Patient-Derived Endothelial Cells in Angiogenic Sprouting. Int. J. Mol. Sci. 2022, 23, 12549. [Google Scholar] [CrossRef]
- Song, J.; Lee, K.; Park, S.W.; Chung, H.; Jung, D.; Na, Y.R.; Quan, H.; Cho, C.S.; Che, J.H.; Kim, J.H.; et al. Lactic Acid Upregulates VEGF Expression in Macrophages and Facilitates Choroidal Neovascularization. Invest. Ophthalmol. Vis. Sci. 2018, 59, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, J.; Wang, Y.; Guo, X.; Sun, Y. Integrative metabolome and lipidome analyses of plasma in neovascular macular degeneration. Heliyon 2023, 9, e20329. [Google Scholar] [CrossRef] [PubMed]
- Laíns, I.; Kelly, R.S.; Miller, J.B.; Silva, R.; Vavvas, D.G.; Kim, I.K.; Murta, J.N.; Lasky-Su, J.; Miller, J.W.; Husain, D. Human Plasma Metabolomics Study across All Stages of Age-Related Macular Degeneration Identifies Potential Lipid Biomarkers. Ophthalmology 2018, 125, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Wei, P.; He, M.; Teng, H. Glucose Metabolic Characterization of Human Aqueous Humor in Relation to Wet Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2020, 61, 49. [Google Scholar] [CrossRef] [PubMed]
- Yumnamcha, T.; Guerra, M.; Singh, L.P.; Ibrahim, A.S. Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy. Antioxidants 2020, 9, 1244. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Nakajima, K.; Korekane, H.; Takamatsu, S.; Gao, C.; Angata, T.; Ohtsubo, K.; Taniguchi, N. Hypoxic regulation of glycosylation via the N-acetylglucosamine cycle. J. Clin. Biochem. Nutr. 2011, 48, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell 2014, 54, 820–831. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Vizán, P.; Sánchez-Tena, S.; Alcarraz-Vizán, G.; Soler, M.; Messeguer, R.; Pujol, M.D.; Lee, W.N.; Cascante, M. Characterization of the metabolic changes underlying growth factor angiogenic activation: Identification of new potential therapeutic targets. Carcinogenesis 2009, 30, 946–952. [Google Scholar] [CrossRef]
- Jongkind, J.F.; Verkerk, A.; Baggen, R.G. Glutathione metabolism of human vascular endothelial cells under peroxidative stress. Free Radic. Biol. Med. 1989, 7, 507–512. [Google Scholar] [CrossRef]
- Ghesquière, B.; Wong, B.W.; Kuchnio, A.; Carmeliet, P. Metabolism of stromal and immune cells in health and disease. Nature 2014, 511, 167–176. [Google Scholar] [CrossRef]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Vosseller, K.; Hart, G.W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001, 291, 2376–2378. [Google Scholar] [CrossRef]
- Bousseau, S.; Vergori, L.; Soleti, R.; Lenaers, G.; Martinez, M.C.; Andriantsitohaina, R. Glycosylation as new pharmacological strategies for diseases associated with excessive angiogenesis. Pharmacol. Ther. 2018, 191, 92–122. [Google Scholar] [CrossRef]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Cerliani, J.P.; Dalotto-Moreno, T.; Méndez-Huergo, S.P.; Mascanfroni, I.D.; Dergan-Dylon, S.; Toscano, M.A.; Caramelo, J.J.; García-Vallejo, J.J.; Ouyang, J.; et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef]
- Chandler, K.B.; Leon, D.R.; Kuang, J.; Meyer, R.D.; Rahimi, N.; Costello, C.E. N-Glycosylation regulates ligand-dependent activation and signaling of vascular endothelial growth factor receptor 2 (VEGFR2). J. Biol. Chem. 2019, 294, 13117–13130. [Google Scholar] [CrossRef]
- Chandler, K.B.; Leon, D.R.; Meyer, R.D.; Rahimi, N.; Costello, C.E. Site-Specific N-Glycosylation of Endothelial Cell Receptor Tyrosine Kinase VEGFR-2. J. Proteome Res. 2017, 16, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Blouin, A.; Bolender, R.P.; Weibel, E.R. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J. Cell Biol. 1977, 72, 441–455. [Google Scholar] [CrossRef]
- Dranka, B.P.; Hill, B.G.; Darley-Usmar, V.M. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic. Biol. Med. 2010, 48, 905–914. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Yao, J.; Wang, Z.; Xu, J. Mitochondria in endothelial cells angiogenesis and function: Current understanding and future perspectives. J. Transl. Med. 2023, 21, 441. [Google Scholar] [CrossRef] [PubMed]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Harjes, U.; Bridges, E.; McIntyre, A.; Fielding, B.A.; Harris, A.L. Fatty acid-binding protein 4, a point of convergence for angiogenic and metabolic signaling pathways in endothelial cells. J. Biol. Chem. 2014, 289, 23168–23176. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Bochem, A.E.; Yvan-Charvet, L.; Murphy, A.J.; Wang, N.; Tall, A.R. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ. Res. 2014, 114, 157–170. [Google Scholar] [CrossRef]
- Terasaka, N.; Yu, S.; Yvan-Charvet, L.; Wang, N.; Mzhavia, N.; Langlois, R.; Pagler, T.; Li, R.; Welch, C.L.; Goldberg, I.J.; et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J. Clin. Investig. 2008, 118, 3701–3713. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Eelen, G.; Dubois, C.; Cantelmo, A.R.; Goveia, J.; Brüning, U.; DeRan, M.; Jarugumilli, G.; van Rijssel, J.; Saladino, G.; Comitani, F.; et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic. Biol. Med. 2017, 109, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Yang, K.; Qiu, T.; Zhou, J.; Gong, X.; Zhang, X.; Lan, Y.; Zhang, Z.; Ji, Y. Blockage of glycolysis by targeting PFKFB3 suppresses the development of infantile hemangioma. J. Transl. Med. 2023, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Thirusangu, P.; Ray, U.; Sarkar Bhattacharya, S.; Oien, D.B.; Jin, L.; Staub, J.; Kannan, N.; Molina, J.R.; Shridhar, V. PFKFB3 regulates cancer stemness through the hippo pathway in small cell lung carcinoma. Oncogene 2022, 41, 4003–4017. [Google Scholar] [CrossRef]
- Poels, K.; Schnitzler, J.G.; Waissi, F.; Levels, J.H.M.; Stroes, E.S.G.; Daemen, M.; Lutgens, E.; Pennekamp, A.M.; De Kleijn, D.P.V.; Seijkens, T.T.P.; et al. Inhibition of PFKFB3 Hampers the Progression of Atherosclerosis and Promotes Plaque Stability. Front. Cell Dev. Biol. 2020, 8, 581641. [Google Scholar] [CrossRef]
- Merchan, J.R.; Kovács, K.; Railsback, J.W.; Kurtoglu, M.; Jing, Y.; Piña, Y.; Gao, N.; Murray, T.G.; Lehrman, M.A.; Lampidis, T.J. Antiangiogenic activity of 2-deoxy-D-glucose. PLoS ONE 2010, 5, e13699. [Google Scholar] [CrossRef]
- Singh, S.; Pandey, S.; Chawla, A.S.; Bhatt, A.N.; Roy, B.G.; Saluja, D.; Dwarakanath, B.S. Dietary 2-deoxy-D-glucose impairs tumour growth and metastasis by inhibiting angiogenesis. Eur. J. Cancer 2019, 123, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Escudero, J.; Clemente, C.; García-Weber, D.; Acín-Pérez, R.; Millán, J.; Enríquez, J.A.; Bentley, K.; Carmeliet, P.; Arroyo, A.G. PKM2 regulates endothelial cell junction dynamics and angiogenesis via ATP production. Sci. Rep. 2019, 9, 15022. [Google Scholar] [CrossRef]
- Cho, Y.S.; Jung, H.J.; Seok, S.H.; Payumo, A.Y.; Chen, J.K.; Kwon, H.J. Functional inhibition of UQCRB suppresses angiogenesis in zebrafish. Biochem. Biophys. Res. Commun. 2013, 433, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Kim, Y.; Chang, J.; Kang, S.W.; Kim, J.H.; Kwon, H.J. Mitochondrial UQCRB regulates VEGFR2 signaling in endothelial cells. J. Mol. Med. 2013, 91, 1117–1128. [Google Scholar] [CrossRef]
- Jung, H.J.; Shim, J.S.; Lee, J.; Song, Y.M.; Park, K.C.; Choi, S.H.; Kim, N.D.; Yoon, J.H.; Mungai, P.T.; Schumacker, P.T.; et al. Terpestacin inhibits tumor angiogenesis by targeting UQCRB of mitochondrial complex III and suppressing hypoxia-induced reactive oxygen species production and cellular oxygen sensing. J. Biol. Chem. 2010, 285, 11584–11595. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yoon, N.G.; Im, J.Y.; Lee, J.H.; Kim, J.; Jeon, Y.; Choi, Y.J.; Lee, J.H.; Uemura, A.; Park, D.H.; et al. Targeting the Mitochondrial Chaperone TRAP1 Alleviates Vascular Pathologies in Ischemic Retinopathy. Adv. Sci. 2024, 11, e2302776. [Google Scholar] [CrossRef]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e15. [Google Scholar] [CrossRef]
- Seguin, F.; Carvalho, M.A.; Bastos, D.C.; Agostini, M.; Zecchin, K.G.; Alvarez-Flores, M.P.; Chudzinski-Tavassi, A.M.; Coletta, R.D.; Graner, E. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br. J. Cancer 2012, 107, 977–987. [Google Scholar] [CrossRef]
- Sagara, N.; Kawaji, T.; Takano, A.; Inomata, Y.; Inatani, M.; Fukushima, M.; Tanihara, H. Effect of pitavastatin on experimental choroidal neovascularization in rats. Exp. Eye Res. 2007, 84, 1074–1080. [Google Scholar] [CrossRef]
- Zambarakji, H.J.; Nakazawa, T.; Connolly, E.; Lane, A.M.; Mallemadugula, S.; Kaplan, M.; Michaud, N.; Hafezi-Moghadam, A.; Gragoudas, E.S.; Miller, J.W. Dose-dependent effect of pitavastatin on VEGF and angiogenesis in a mouse model of choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2623–2631. [Google Scholar] [CrossRef]
- Leopold, J.A.; Walker, J.; Scribner, A.W.; Voetsch, B.; Zhang, Y.Y.; Loscalzo, A.J.; Stanton, R.C.; Loscalzo, J. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J. Biol. Chem. 2003, 278, 32100–32106. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, F.; Zhang, Y.; Lin, Z.; Yang, J.; Han, X.; Feng, Y.; Pei, X.; Li, F.; Liu, Q.; et al. Targeting glucose-6-phosphate dehydrogenase by 6-AN induces ROS-mediated autophagic cell death in breast cancer. FEBS J. 2023, 290, 763–779. [Google Scholar] [CrossRef]
- Chong, C.R.; Xu, J.; Lu, J.; Bhat, S.; Sullivan, D.J., Jr.; Liu, J.O. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem. Biol. 2007, 2, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Del Carratore, R.; Carpi, A.; Beffy, P.; Lubrano, V.; Giorgetti, L.; Maserti, B.E.; Carluccio, M.A.; Simili, M.; Iervasi, G.; Balzan, S. Itraconazole inhibits HMEC-1 angiogenesis. Biomed. Pharmacother. 2012, 66, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.H.; Hwang, A.R.; Kim, C.Y.; Yu, H.G.; Koh, H.J.; Yang, W.I.; Chang, H.R.; Lee, S.C. Intravitreal itraconazole inhibits laser-induced choroidal neovascularization in rats. PLoS ONE 2017, 12, e0180482. [Google Scholar] [CrossRef] [PubMed]
- Peyton, K.J.; Liu, X.M.; Yu, Y.; Yates, B.; Behnammanesh, G.; Durante, W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem. Pharmacol. 2018, 156, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Wik, J.A.; Lundbäck, P.; la Cour Poulsen, L.; Haraldsen, G.; Skålhegg, B.S.; Hol, J. 3PO inhibits inflammatory NFκB and stress-activated kinase signaling in primary human endothelial cells independently of its target PFKFB3. PLoS ONE 2020, 15, e0229395. [Google Scholar] [CrossRef]
- Zhu, W.; Ye, L.; Zhang, J.; Yu, P.; Wang, H.; Ye, Z.; Tian, J. PFK15, a Small Molecule Inhibitor of PFKFB3, Induces Cell Cycle Arrest, Apoptosis and Inhibits Invasion in Gastric Cancer. PLoS ONE 2016, 11, e0163768. [Google Scholar] [CrossRef]
- Matsumoto, K.; Noda, T.; Kobayashi, S.; Sakano, Y.; Yokota, Y.; Iwagami, Y.; Yamada, D.; Tomimaru, Y.; Akita, H.; Gotoh, K.; et al. Inhibition of glycolytic activator PFKFB3 suppresses tumor growth and induces tumor vessel normalization in hepatocellular carcinoma. Cancer Lett. 2021, 500, 29–40. [Google Scholar] [CrossRef]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [PubMed]
- Baier, D.; Schoenhacker-Alte, B.; Rusz, M.; Pirker, C.; Mohr, T.; Mendrina, T.; Kirchhofer, D.; Meier-Menches, S.M.; Hohenwallner, K.; Schaier, M.; et al. The Anticancer Ruthenium Compound BOLD-100 Targets Glycolysis and Generates a Metabolic Vulnerability towards Glucose Deprivation. Pharmaceutics 2022, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Wang, S.Y.; Lin, L.L.; Wang, P.W.; Chen, T.Y.; Hsu, W.M.; Lin, T.K.; Liou, C.W.; Chuang, J.H. Glycolytic inhibitor 2-deoxyglucose simultaneously targets cancer and endothelial cells to suppress neuroblastoma growth in mice. Dis. Model. Mech. 2015, 8, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Aghaee, F.; Pirayesh Islamian, J.; Baradaran, B. Enhanced radiosensitivity and chemosensitivity of breast cancer cells by 2-deoxy-d-glucose in combination therapy. J. Breast Cancer 2012, 15, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; He, J.; Song, X.; Tan, L.; Wang, M.; Jiang, P.; Li, Y.; Cao, Z.; Peng, C. Pharmacological properties and derivatives of shikonin-A review in recent years. Pharmacol. Res. 2019, 149, 104463. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, L.; Wang, J.; Wang, Q.; Sun, C.; Li, Y.; Jia, K.; Wang, J.; Xu, T.; Ming, R.; et al. Anti-angiogenic effect of Shikonin in rheumatoid arthritis by downregulating PI3K/AKT and MAPKs signaling pathways. J. Ethnopharmacol. 2020, 260, 113039. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, X.; Gao, X.; Chen, H.; Geng, L. Shikonin suppresses IL-17-induced VEGF expression via blockage of JAK2/STAT3 pathway. Int. Immunopharmacol. 2014, 19, 327–333. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, L.; Zhu, M.; Guo, Y.; Tu, Y.; Zhou, Y.; Zeng, J.; Zhu, L.; Du, S.; Wang, Z.; et al. Shikonin alleviates choroidal neovascularization by inhibiting proangiogenic factor production from infiltrating macrophages. Exp. Eye Res. 2021, 213, 108823. [Google Scholar] [CrossRef]
- Jung, H.J.; Lee, H.B.; Kim, C.J.; Rho, J.R.; Shin, J.; Kwon, H.J. Anti-angiogenic activity of terpestacin, a bicyclo sesterterpene from Embellisia chlamydospora. J. Antibiot. 2003, 56, 492–496. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Y.; Zhang, H.; Chen, R.; Lv, F.; Li, Z.; Jiang, T.; Lin, D.; Zhang, H.; Yang, L.; et al. The Antioxidant MitoQ Protects Against CSE-Induced Endothelial Barrier Injury and Inflammation by Inhibiting ROS and Autophagy in Human Umbilical Vein Endothelial Cells. Int. J. Biol. Sci. 2019, 15, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Missiaen, R.; Morales-Rodriguez, F.; Eelen, G.; Carmeliet, P. Targeting endothelial metabolism for anti-angiogenesis therapy: A pharmacological perspective. Vascul Pharmacol. 2017, 90, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Browne, C.D.; Hindmarsh, E.J.; Smith, J.W. Inhibition of endothelial cell proliferation and angiogenesis by orlistat, a fatty acid synthase inhibitor. FASEB J. 2006, 20, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jin, G.; Mi, R.; Zhang, J.; Zhang, J.; Xu, H.; Cheng, S.; Zhang, Y.; Song, W.; Liu, F. Inhibition of fatty acid synthase suppresses neovascularization via regulating the expression of VEGF-A in glioma. J. Cancer Res. Clin. Oncol. 2016, 142, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Elliott, V.A.; Rychahou, P.; Mustain, W.C.; Kim, J.T.; Valentino, J.; Gao, T.; O’Connor, K.L.; Neltner, J.M.; Lee, E.Y.; et al. Cancer cell-associated fatty acid synthase activates endothelial cells and promotes angiogenesis in colorectal cancer. Carcinogenesis 2014, 35, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Vincent, L.; Chen, W.; Hong, L.; Mirshahi, F.; Mishal, Z.; Mirshahi-Khorassani, T.; Vannier, J.P.; Soria, J.; Soria, C. Inhibition of endothelial cell migration by cerivastatin, an HMG-CoA reductase inhibitor: Contribution to its anti-angiogenic effect. FEBS Lett. 2001, 495, 159–166. [Google Scholar] [CrossRef]
- Vincent, L.; Soria, C.; Mirshahi, F.; Opolon, P.; Mishal, Z.; Vannier, J.P.; Soria, J.; Hong, L. Cerivastatin, an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme a reductase, inhibits endothelial cell proliferation induced by angiogenic factors in vitro and angiogenesis in in vivo models. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Roizenblatt, M.; Naranjit, N.; Maia, M.; Gehlbach, P.L. The Question of a Role for Statins in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2018, 19, 3688. [Google Scholar] [CrossRef]
- Pan, S.; World, C.J.; Kovacs, C.J.; Berk, B.C. Glucose 6-phosphate dehydrogenase is regulated through c-Src-mediated tyrosine phosphorylation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 895–901. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF and Intraocular Neovascularization: From Discovery to Therapy. Transl. Vis. Sci. Technol. 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Grassi, P.; Dell, A.; Haslam, S.M.; Liu, J.O. The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells. J. Biol. Chem. 2011, 286, 44045–44056. [Google Scholar] [CrossRef] [PubMed]
- Aftab, B.T.; Dobromilskaya, I.; Liu, J.O.; Rudin, C.M. Itraconazole inhibits angiogenesis and tumor growth in non-small cell lung cancer. Cancer Res. 2011, 71, 6764–6772. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- Tang, Z.; Fan, X.; Chen, Y.; Gu, P. Ocular Nanomedicine. Adv. Sci. 2022, 9, e2003699. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Metabolic Pathways | Targets | Compounds | Effects | References |
|---|---|---|---|---|
| Glycolysis | PFKFB3 | 3PO | Inhibition of pathological angiogenesis in CNV and ROP models | [18] |
| PFK-15 | Inhibition of infantile hemangioma angiogenesis | [77] | ||
| PFK-158 | Anti-cancer and anti-atherosclerosis effects | [78,79] | ||
| HK2 | 2-DG | Reduced HUVEC angiogenesis and anti-tumor angiogenesis | [80,81] | |
| PKM2 | Shikonin | Instability of EC junctions and weakened angiogenesis | [82] | |
| Mitochondrial Metabolism | Mitochondrial respiratory chain | Terpestacin | Inhibition of angiogenesis in zebrafish and anti-tumor angiogenesis | [83,84,85] |
| oxidative stress | MitoQ | Improved vascular lesions in OIR and STZ mouse models | [86] | |
| Lipid metabolism | FASN | Orlistat | Reduced vascular tuft formation in ROP model and anti-tumor angiogenesis | [87,88] |
| CPT1 | Etomoxir | Inhibition of retinal EC proliferation and pathological angiogenesis in ROP model | [43] | |
| HMG-CoA | Pitavastatin | Anti-angiogenic effects on CNV in rats and mice | [89,90] | |
| Pentose phosphate pathway | G6PD | 6-AN | Inhibition of angiogenic response and anti-tumor effects | [91,92] |
| Glycosylation | VEGFR2 | Itraconazole | Anti-angiogenic effects on various ECs of different origins in vitro, and inhibition of CNV development in rats | [93,94,95] |
| Glutaminolysis | GLS | DON, BPTES, CB839 | Inhibition of angiogenesis in various EC sources and reduced vascular tuft formation in ROP model | [21,96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Xu, Y.; Ju, Y.; Gu, P. Metabolic Regulation of Endothelial Cells: A New Era for Treating Wet Age-Related Macular Degeneration. Int. J. Mol. Sci. 2024, 25, 5926. https://doi.org/10.3390/ijms25115926
Chen X, Xu Y, Ju Y, Gu P. Metabolic Regulation of Endothelial Cells: A New Era for Treating Wet Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2024; 25(11):5926. https://doi.org/10.3390/ijms25115926
Chicago/Turabian StyleChen, Xirui, Yang Xu, Yahan Ju, and Ping Gu. 2024. "Metabolic Regulation of Endothelial Cells: A New Era for Treating Wet Age-Related Macular Degeneration" International Journal of Molecular Sciences 25, no. 11: 5926. https://doi.org/10.3390/ijms25115926
APA StyleChen, X., Xu, Y., Ju, Y., & Gu, P. (2024). Metabolic Regulation of Endothelial Cells: A New Era for Treating Wet Age-Related Macular Degeneration. International Journal of Molecular Sciences, 25(11), 5926. https://doi.org/10.3390/ijms25115926