
Ablation of Vitamin D Signaling in Cardiomyocytes Leads to Functional Impairment and Stimulation of Pro-Inflammatory and Pro-Fibrotic Gene Regulatory Networks in a Left Ventricular Hypertrophy Model in Mice

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Generation and Characterization of Cardiomyocyte-Specific VDR Knockout Mice
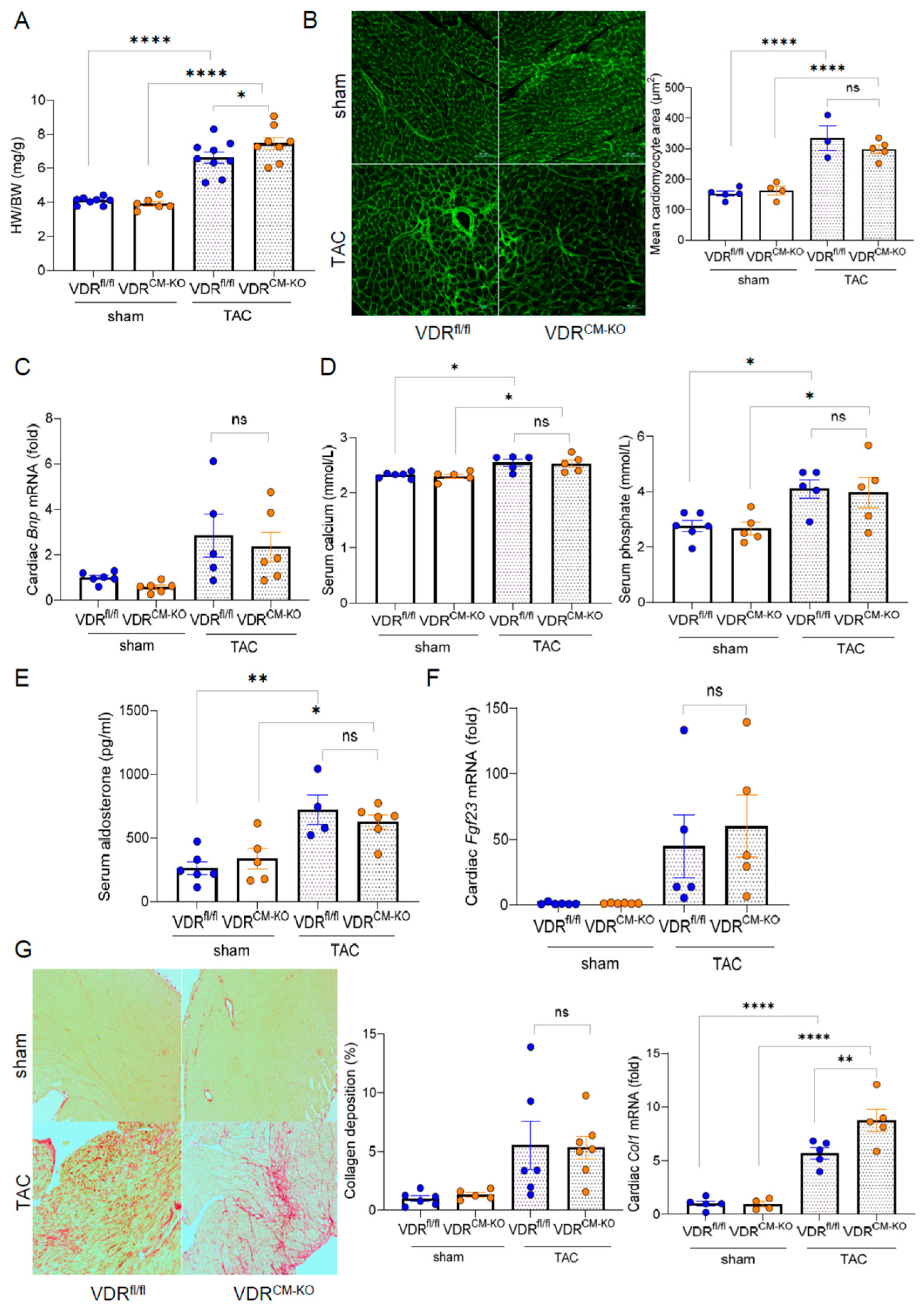
2.2. Development of Afterload-Induced Cardiac Hypertrophy Is Not Modulated by Loss of VDR in Cardiomyocytes
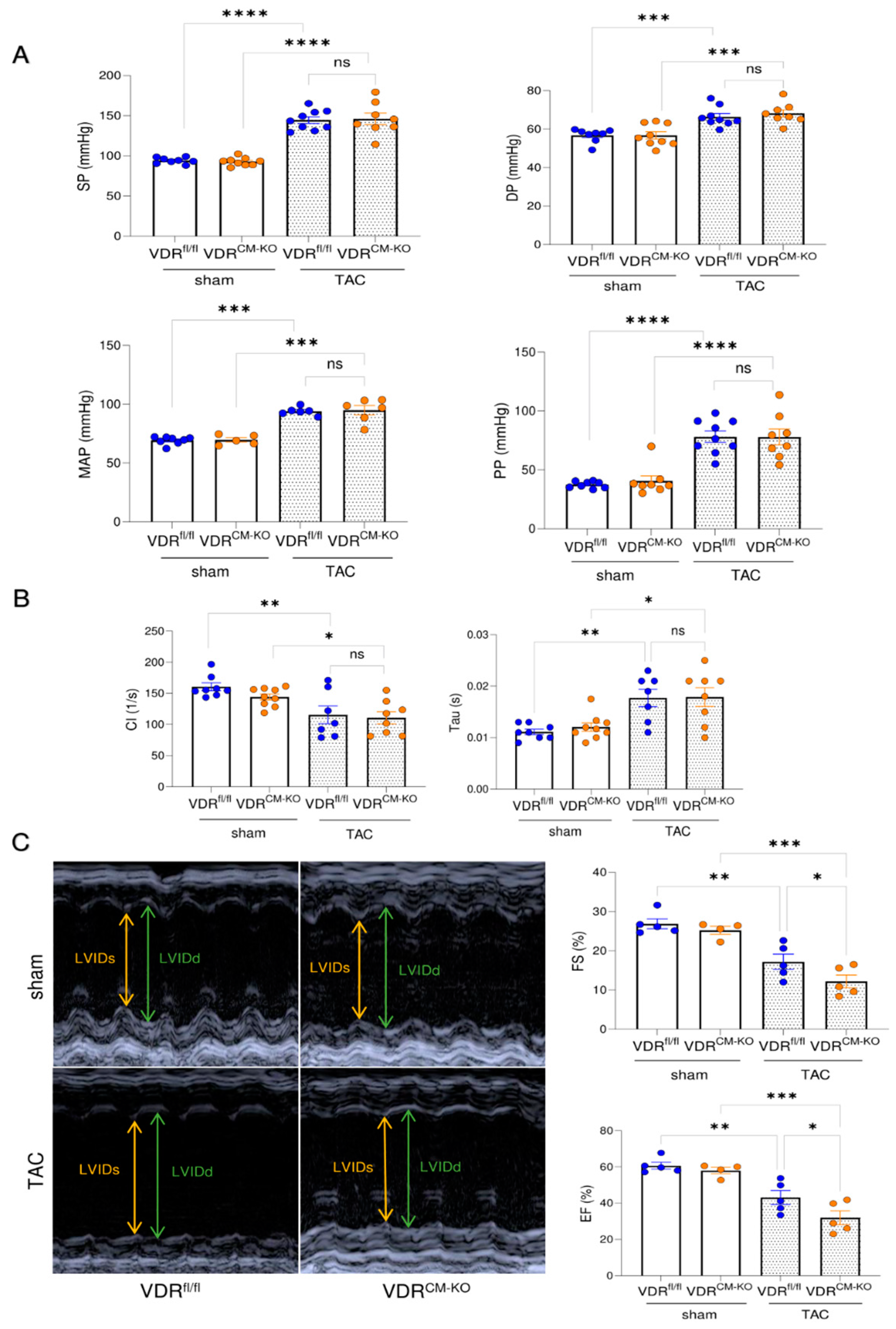
2.3. Lack of VDR Signaling in Cardiomyocytes Aggravates TAC-Induced LV Functional Impairment
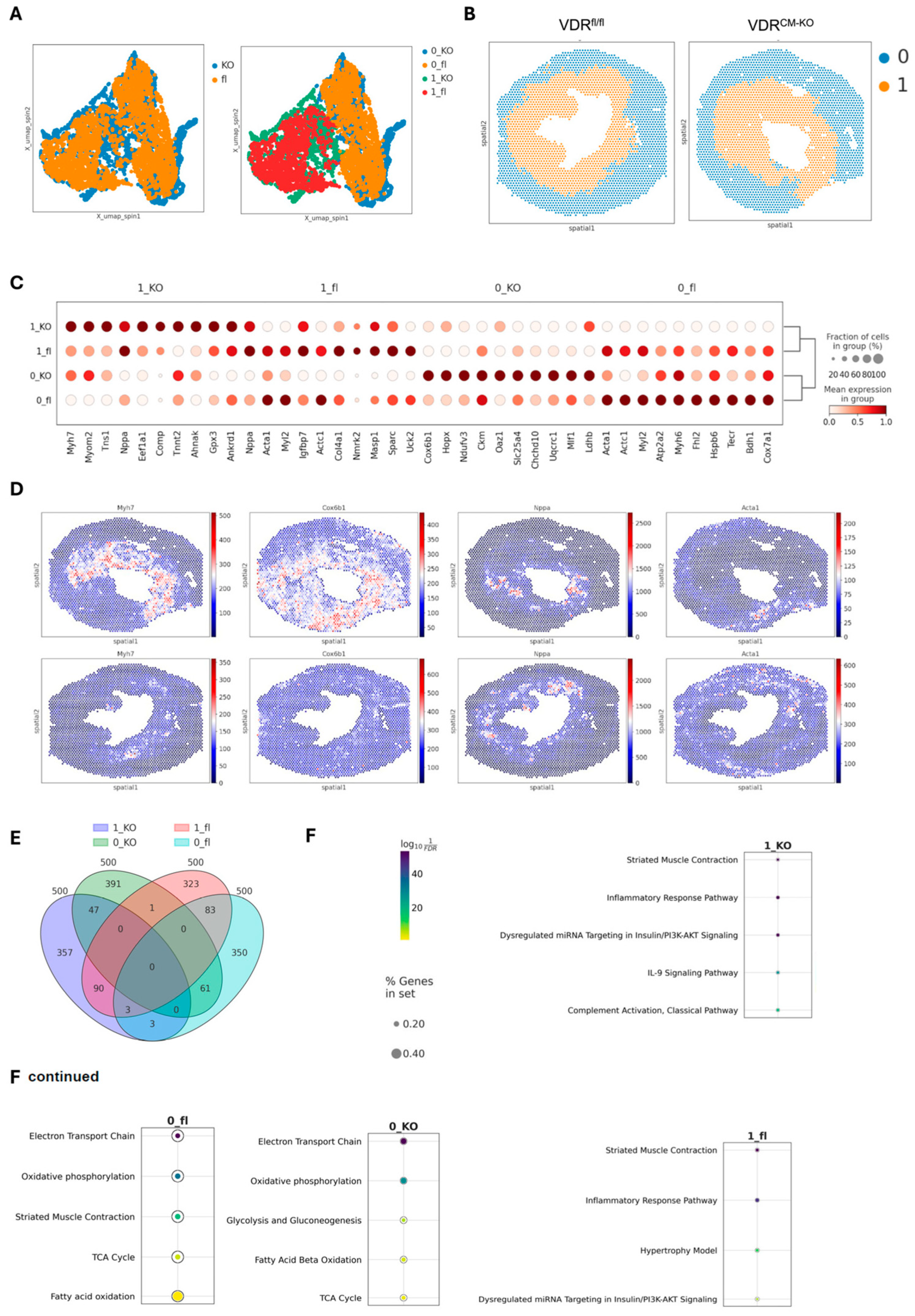
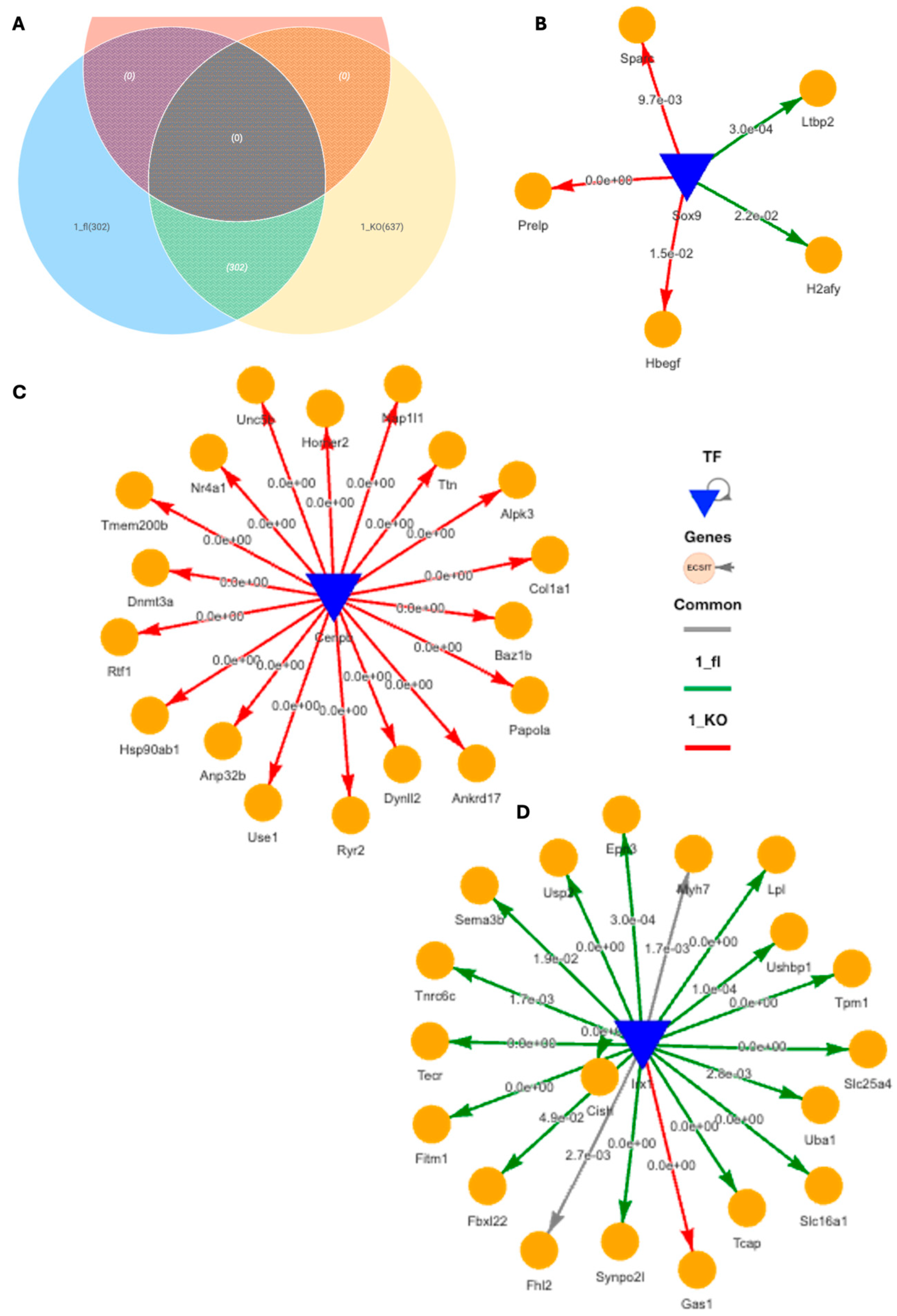
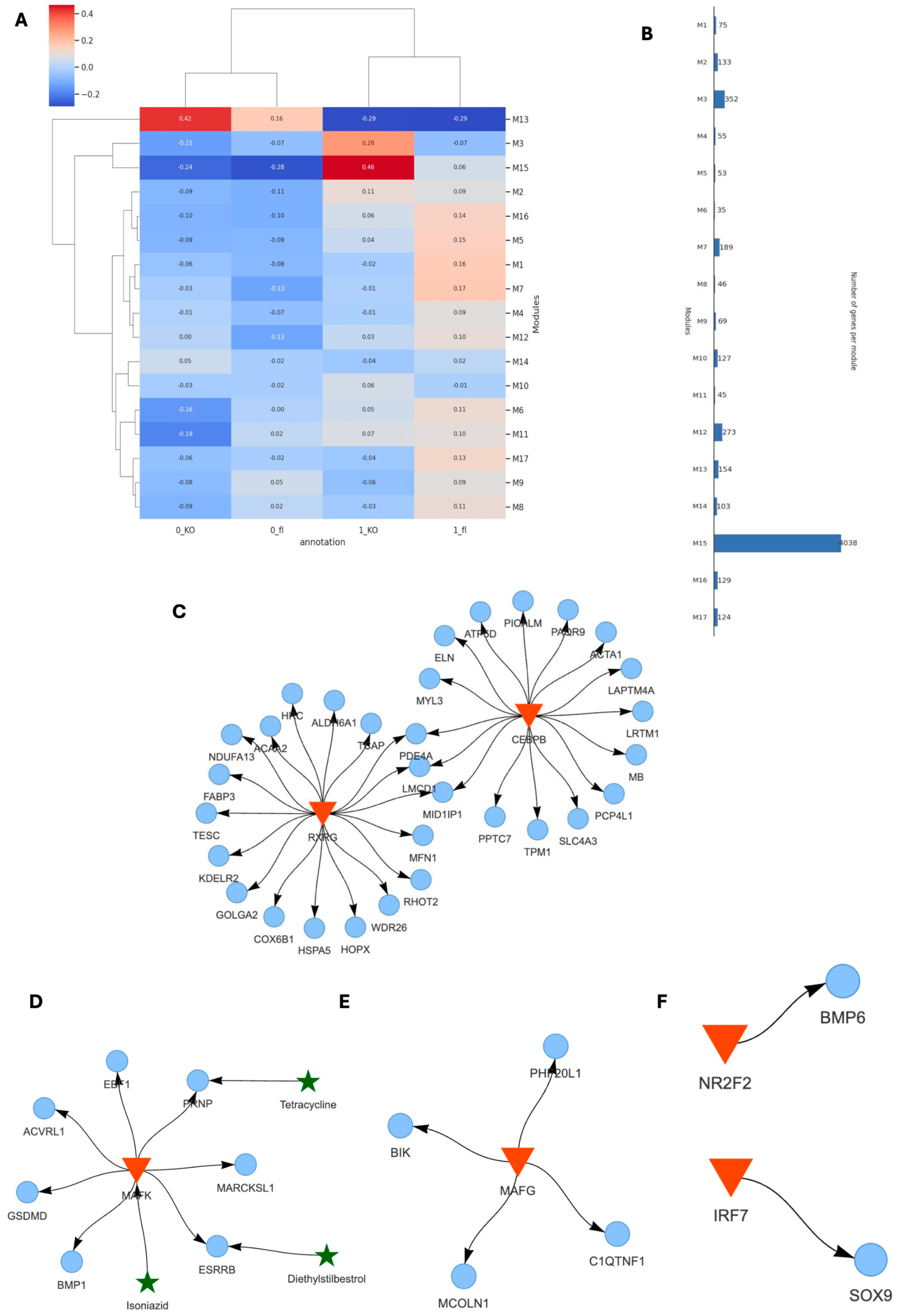
2.4. Spatial Transcriptomics Reveal More Pronounced Pro-Inflammatory and Pro-Fibrotic Gene Regulatory Networks in Hypertrophic Cardiac Tissue Niches of TAC VDRCM-KO Mice
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Transverse Aortic Constriction (TAC)
4.3. Doppler Echocardiography
4.4. Central Arterial and Cardiac Pressure Measurements
4.5. Biochemical Analysis
4.6. Tissue Harvesting and Histological Analysis
4.7. RNA Isolation and Quantitative Real-Time PCR
4.8. Statistical Analysis of Phenotyping Data
4.9. Spatial Transcriptomics and Bioinformatic Analysis
4.9.1. Spatial Transcriptomics Data Preprocessing
4.9.2. Data Integration
4.9.3. Differential Expression and Marker Identification
4.9.4. Functional Enrichment Analysis
4.9.5. Differential Gene Regulatory Network Analysis
4.9.6. Co-Expression Modules and Gene-Regulatory Networks (GRN)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schocken, D.D.; Benjamin, E.J.; Fonarow, G.C.; Krumholz, H.M.; Levy, D.; Mensah, G.A.; Narula, J.; Shor, E.S.; Young, J.B.; Hong, Y. Prevention of heart failure: A scientific statement from the American Heart Association Councils on Epidemiology and Prevention, Clinical Cardiology, Cardiovascular Nursing, and High Blood Pressure Research; Quality of Care and Outcomes Research Interdisciplinary Working Group; and Functional Genomics and Translational Biology Interdisciplinary Working Group. Circulation 2008, 117, 2544–2565. [Google Scholar] [PubMed]
- Cheng, S.; Massaro, J.M.; Fox, C.S.; Larson, M.G.; Keyes, M.J.; McCabe, E.L.; Robins, S.J.; O’Donnell, C.J.; Hoffmann, U.; Jacques, P.F.; et al. Adiposity, cardiometabolic risk, and vitamin D status: The Framingham Heart Study. Diabetes 2010, 59, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Melamed, M.L.; Muntner, P.; Michos, E.D.; Uribarri, J.; Weber, C.; Sharma, J.; Raggi, P. Serum 25-hydroxyvitamin D levels and the prevalence of peripheral arterial disease: Results from NHANES 2001 to 2004. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Meng, X.; Tian, Q.; Cao, W.; Fan, X.; Wu, L.; Song, M.; Meng, Q.; Wang, W.; Wang, Y. Vitamin D and Multiple Health Outcomes: An Umbrella Review of Observational Studies, Randomized Controlled Trials, and Mendelian Randomization Studies. Adv. Nutr. 2022, 13, 1044–1062. [Google Scholar] [CrossRef] [PubMed]
- Scragg, R.; Stewart, A.W.; Waayer, D.; Lawes, C.M.M.; Toop, L.; Sluyter, J.; Murphy, J.; Khaw, K.T.; Camargo, C.A., Jr. Effect of Monthly High-Dose Vitamin D Supplementation on Cardiovascular Disease in the Vitamin D Assessment Study: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Djousse, L.; Cook, N.R.; Kim, E.; Bodar, V.; Walter, J.; Bubes, V.; Luttmann-Gibson, H.; Mora, S.; Joseph, J.; Lee, I.M.; et al. Supplementation With Vitamin D and Omega-3 Fatty Acids and Incidence of Heart Failure Hospitalization: VITAL-Heart Failure. Circulation 2020, 141, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Naughton, D.P. Vitamin D in health and disease: Current perspectives. Nutr. J. 2010, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Cauley, J.A.; Lacroix, A.Z.; Wu, L.; Horwitz, M.; Danielson, M.E.; Bauer, D.C.; Lee, J.S.; Jackson, R.D.; Robbins, J.A.; Wu, C.; et al. Serum 25-hydroxyvitamin D concentrations and risk for hip fractures. Ann. Intern. Med. 2008, 149, 242–250. [Google Scholar] [CrossRef]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef]
- Al Mheid, I.; Patel, R.; Murrow, J.; Morris, A.; Rahman, A.; Fike, L.; Kavtaradze, N.; Uphoff, I.; Hooper, C.; Tangpricha, V.; et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J. Am. Coll. Cardiol. 2011, 58, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, D.X.; Nibbelink, K.A.; Holmberg, K.H.; Dandu, L.; Simpson, R.U. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008, 149, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Glenn, D.J.; Ni, W.; Grigsby, C.L.; Olsen, K.; Nishimoto, M.; Law, C.S.; Gardner, D.G. Expression of the vitamin d receptor is increased in the hypertrophic heart. Hypertension 2008, 52, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.G.; Chen, S.; Glenn, D.J. Vitamin D and the heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R969–R977. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; You, Y.; Swier, V.; Tang, L.; Radwan, M.M.; Pandya, A.N.; Agrawal, D.K. Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2432–2442. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Pan, W.; Kong, J.; Zheng, W.; Szeto, F.L.; Wong, K.E.; Cohen, R.; Klopot, A.; Zhang, Z.; Li, Y.C. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 2007, 282, 29821–29830. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Watts, S.W.; Ng, M.; Chen, S.; Glenn, D.J.; Gardner, D.G. Elimination of vitamin D receptor in vascular endothelial cells alters vascular function. Hypertension 2014, 64, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Riek, A.E.; Darwech, I.; Funai, K.; Shao, J.; Chin, K.; Sierra, O.L.; Carmeliet, G.; Ostlund, R.E., Jr.; Bernal-Mizrachi, C. Deletion of macrophage Vitamin D receptor promotes insulin resistance and monocyte cholesterol transport to accelerate atherosclerosis in mice. Cell Rep. 2015, 10, 1872–1886. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef]
- Andrukhova, O.; Slavic, S.; Zeitz, U.; Riesen, S.C.; Heppelmann, M.S.; Ambrisko, T.D.; Markovic, M.; Kuebler, W.M.; Erben, R.G. Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol. Endocrinol. 2014, 28, 53–64. [Google Scholar] [CrossRef]
- Chen, S.; Law, C.S.; Grigsby, C.L.; Olsen, K.; Hong, T.T.; Zhang, Y.; Yeghiazarians, Y.; Gardner, D.G. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation 2011, 124, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Slavic, S.; Ford, K.; Modert, M.; Becirovic, A.; Handschuh, S.; Baierl, A.; Katica, N.; Zeitz, U.; Erben, R.G.; Andrukhova, O. Genetic Ablation of Fgf23 or Klotho Does not Modulate Experimental Heart Hypertrophy Induced by Pressure Overload. Sci. Rep. 2017, 7, 11298. [Google Scholar] [CrossRef] [PubMed]
- Ford, K.; Latic, N.; Slavic, S.; Zeitz, U.; Dolezal, M.; Andrukhov, O.; Erben, R.G.; Andrukhova, O. Lack of vitamin D signalling per se does not aggravate cardiac functional impairment induced by myocardial infarction in mice. PLoS ONE 2018, 13, e0204803. [Google Scholar] [CrossRef] [PubMed]
- Meir, T.; Levi, R.; Lieben, L.; Libutti, S.; Carmeliet, G.; Bouillon, R.; Silver, J.; Naveh-Many, T. Deletion of the vitamin D receptor specifically in the parathyroid demonstrates a limited role for the receptor in parathyroid physiology. Am. J. Physiol. Renal Physiol. 2009, 297, F1192–F1198. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kubalak, S.W.; Minamisawa, S.; Price, R.L.; Becker, K.D.; Hickey, R.; Ross, J., Jr.; Chien, K.R. Selective requirement of myosin light chain 2v in embryonic heart function. J. Biol. Chem. 1998, 273, 1252–1256. [Google Scholar] [CrossRef] [PubMed]
- Minamisawa, S.; Gu, Y.; Ross, J., Jr.; Chien, K.R.; Chen, J. A post-transcriptional compensatory pathway in heterozygous ventricular myosin light chain 2-deficient mice results in lack of gene dosage effect during normal cardiac growth or hypertrophy. J. Biol. Chem. 1999, 274, 10066–10070. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.; Gil, A.; Gomez, J.; Ruiz, L.; Llano, M.; Garcia, R.; Hurle, M.A.; Nistal, J.F. Experimental modelling of cardiac pressure overload hypertrophy: Modified technique for precise, reproducible, safe and easy aortic arch banding-debanding in mice. Sci. Rep. 2018, 8, 3167. [Google Scholar] [CrossRef] [PubMed]
- Eitner, F.; Richter, B.; Schwanen, S.; Szaroszyk, M.; Vogt, I.; Grund, A.; Thum, T.; Heineke, J.; Haffner, D.; Leifheit-Nestler, M. Comprehensive Expression Analysis of Cardiac Fibroblast Growth Factor 23 in Health and Pressure-induced Cardiac Hypertrophy. Front. Cell Dev. Biol. 2021, 9, 791479. [Google Scholar] [CrossRef]
- Maher, K.; Wu, M.; Zhou, Y.; Huang, J.; Zhang, Q.; Wang, X. Mitigating autocorrelation during spatially resolved transcriptomics data analysis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Olson, T.M.; Michels, V.V.; Thibodeau, S.N.; Tai, Y.S.; Keating, M.T. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998, 280, 750–752. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Wan, J.; Zhang, P.; Pei, F. COX6B1 relieves hypoxia/reoxygenation injury of neonatal rat cardiomyocytes by regulating mitochondrial function. Biotechnol. Lett. 2019, 41, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Brewster, L.M.; Haan, Y.C.; Zwinderman, A.H.; van den Born, B.J.; van Montfrans, G.A. CK (Creatine Kinase) Is Associated With Cardiovascular Hemodynamics: The HELIUS Study. Hypertension 2020, 76, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Nomura, S.; Harada, M.; Yamaguchi, T.; Ko, T.; Sumida, T.; Toko, H.; Naito, A.T.; Takeda, N.; Tobita, T.; et al. High-throughput single-molecule RNA imaging analysis reveals heterogeneous responses of cardiomyocytes to hemodynamic overload. J. Mol. Cell Cardiol. 2019, 128, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yu, P.; Li, D.; Li, Z.; Liao, Y.; Wang, Y.; Zhou, B.; Wang, L. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 2020, 141, 1704–1719. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Nag, S.; Trivedi, D.V.; Sarkar, S.S.; Adhikari, A.S.; Sunitha, M.S.; Sutton, S.; Ruppel, K.M.; Spudich, J.A. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat. Struct. Mol. Biol. 2017, 24, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Barrick, S.K.; Greenberg, M.J. Cardiac myosin contraction and mechanotransduction in health and disease. J. Biol. Chem. 2021, 297, 101297. [Google Scholar] [CrossRef]
- Martens, P.-J.; Gysemans, C.; Verstuyf, A.; Mathieu, C. Vitamin D’s Effect on Immune Function. Nutrients 2020, 12, 1248. [Google Scholar] [CrossRef] [PubMed]
- Oubounyt, M.; Elkjaer, M.L.; Laske, T.; Grønning, A.G.; Moeller, M.J.; Baumbach, J. De-novo reconstruction and identification of transcriptional gene regulatory network modules differentiating single-cell clusters. NAR Genom. Bioinform. 2023, 5, lqad018. [Google Scholar]
- Schauer, A.; Adams, V.; Poitz, D.M.; Barthel, P.; Joachim, D.; Friedrich, J.; Linke, A.; Augstein, A. Loss of Sox9 in cardiomyocytes delays the onset of cardiac hypertrophy and fibrosis. Int. J. Cardiol. 2019, 282, 68–75. [Google Scholar] [CrossRef]
- Zeng, L.; Gu, N.; Chen, J.; Jin, G.; Zheng, Y. IRX1 hypermethylation promotes heart failure by inhibiting CXCL14 expression. Cell Cycle 2019, 18, 3251–3262. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, F.W.; Reischmann, S.; Schwalm, A.; Unger, A.; Ramanujam, D.; Munch, J.; Muller, O.J.; Hengstenberg, C.; Galve, E.; Charron, P.; et al. FHL2 expression and variants in hypertrophic cardiomyopathy. Basic. Res. Cardiol. 2014, 109, 451. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef]
- Hunt, L.C.; Pagala, V.; Stephan, A.; Xie, B.; Kodali, K.; Kavdia, K.; Wang, Y.-D.; Shirinifard, A.; Curley, M.; Graca, F.A.; et al. An adaptive stress response that confers cellular resilience to decreased ubiquitination. Nat. Commun. 2023, 14, 7348. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Li, P.; Hong, J.; Wang, M.; Liu, Y.; Gao, Y.; Dong, J.; Gu, H.; Li, L. Overexpression of Ubiquitin-Specific Protease 2 (USP2) in the Heart Suppressed Pressure Overload-Induced Cardiac Remodeling. Mediat. Inflamm. 2020, 2020, 4121750. [Google Scholar] [CrossRef] [PubMed]
- Erdos, E.; Balint, B.L. NR2F2 Orphan Nuclear Receptor is Involved in Estrogen Receptor Alpha-Mediated Transcriptional Regulation in Luminal A Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 1910. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, V.; Martin, C.; Dorchies, E.; Vallez, E.; Dehondt, H.; Trabelsi, M.S.; Tailleux, A.; Caron, S.; Staels, B. Screening strategy to generate cell specific recombination: A case report with the RIP-Cre mice. Transgenic Res. 2015, 24, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, A.; Ernst, J.B.; Prokop, S.; Fuchs, U.; Dreier, J.; Kuhn, J.; Knabbe, C.; Birschmann, I.; Schulz, U.; Berthold, H.K.; et al. Effect of vitamin D on all-cause mortality in heart failure (EVITA): A 3-year randomized clinical trial with 4000 IU vitamin D daily. Eur. Heart J. 2017, 38, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, O.; Hanel, A.; Carlberg, C. Key Vitamin D Target Genes with Functions in the Immune System. Nutrients 2020, 12, 1140. [Google Scholar] [CrossRef]
- Harries, L.W.; Pilling, L.C.; Hernandez, L.D.; Bradley-Smith, R.; Henley, W.; Singleton, A.B.; Guralnik, J.M.; Bandinelli, S.; Ferrucci, L.; Melzer, D. CCAAT-enhancer-binding protein-beta expression in vivo is associated with muscle strength. Aging Cell 2012, 11, 262–268. [Google Scholar] [CrossRef]
- Latic, N.; Zupcic, A.; Frauenstein, D.; Erben, R.G. Activation of RAAS Signaling Contributes to Hypertension in Aged Hyp Mice. Biomedicines 2022, 10, 1691. [Google Scholar] [CrossRef] [PubMed]
- Latic, N.; Peitzsch, M.; Zupcic, A.; Pietzsch, J.; Erben, R.G. Long-Term Excessive Dietary Phosphate Intake Increases Arterial Blood Pressure, Activates the Renin-Angiotensin-Aldosterone System, and Stimulates Sympathetic Tone in Mice. Biomedicines 2022, 10, 2510. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Liu, X.; Peltz, G. GSEApy: A comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 2022, 39, btac757. [Google Scholar] [CrossRef]
- Oubounyt, M.; Adlung, L.; Patroni, F.; Wenke, N.K.; Maier, A.; Hartung, M.; Baumbach, J.; Elkjaer, M.L. Inference of differential key regulatory networks and mechanistic drug repurposing candidates from scRNA-seq data with SCANet. Bioinformatics 2023, 39, btad644. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zupcic, A.; Latic, N.; Oubounyt, M.; Ramesova, A.; Carmeliet, G.; Baumbach, J.; Elkjaer, M.L.; Erben, R.G. Ablation of Vitamin D Signaling in Cardiomyocytes Leads to Functional Impairment and Stimulation of Pro-Inflammatory and Pro-Fibrotic Gene Regulatory Networks in a Left Ventricular Hypertrophy Model in Mice. Int. J. Mol. Sci. 2024, 25, 5929. https://doi.org/10.3390/ijms25115929
Zupcic A, Latic N, Oubounyt M, Ramesova A, Carmeliet G, Baumbach J, Elkjaer ML, Erben RG. Ablation of Vitamin D Signaling in Cardiomyocytes Leads to Functional Impairment and Stimulation of Pro-Inflammatory and Pro-Fibrotic Gene Regulatory Networks in a Left Ventricular Hypertrophy Model in Mice. International Journal of Molecular Sciences. 2024; 25(11):5929. https://doi.org/10.3390/ijms25115929
Chicago/Turabian StyleZupcic, Ana, Nejla Latic, Mhaned Oubounyt, Alice Ramesova, Geert Carmeliet, Jan Baumbach, Maria L. Elkjaer, and Reinhold G. Erben. 2024. "Ablation of Vitamin D Signaling in Cardiomyocytes Leads to Functional Impairment and Stimulation of Pro-Inflammatory and Pro-Fibrotic Gene Regulatory Networks in a Left Ventricular Hypertrophy Model in Mice" International Journal of Molecular Sciences 25, no. 11: 5929. https://doi.org/10.3390/ijms25115929
APA StyleZupcic, A., Latic, N., Oubounyt, M., Ramesova, A., Carmeliet, G., Baumbach, J., Elkjaer, M. L., & Erben, R. G. (2024). Ablation of Vitamin D Signaling in Cardiomyocytes Leads to Functional Impairment and Stimulation of Pro-Inflammatory and Pro-Fibrotic Gene Regulatory Networks in a Left Ventricular Hypertrophy Model in Mice. International Journal of Molecular Sciences, 25(11), 5929. https://doi.org/10.3390/ijms25115929