
Osteoarthritis as a Systemic Disease Promoted Prostate Cancer In Vivo and In Vitro
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
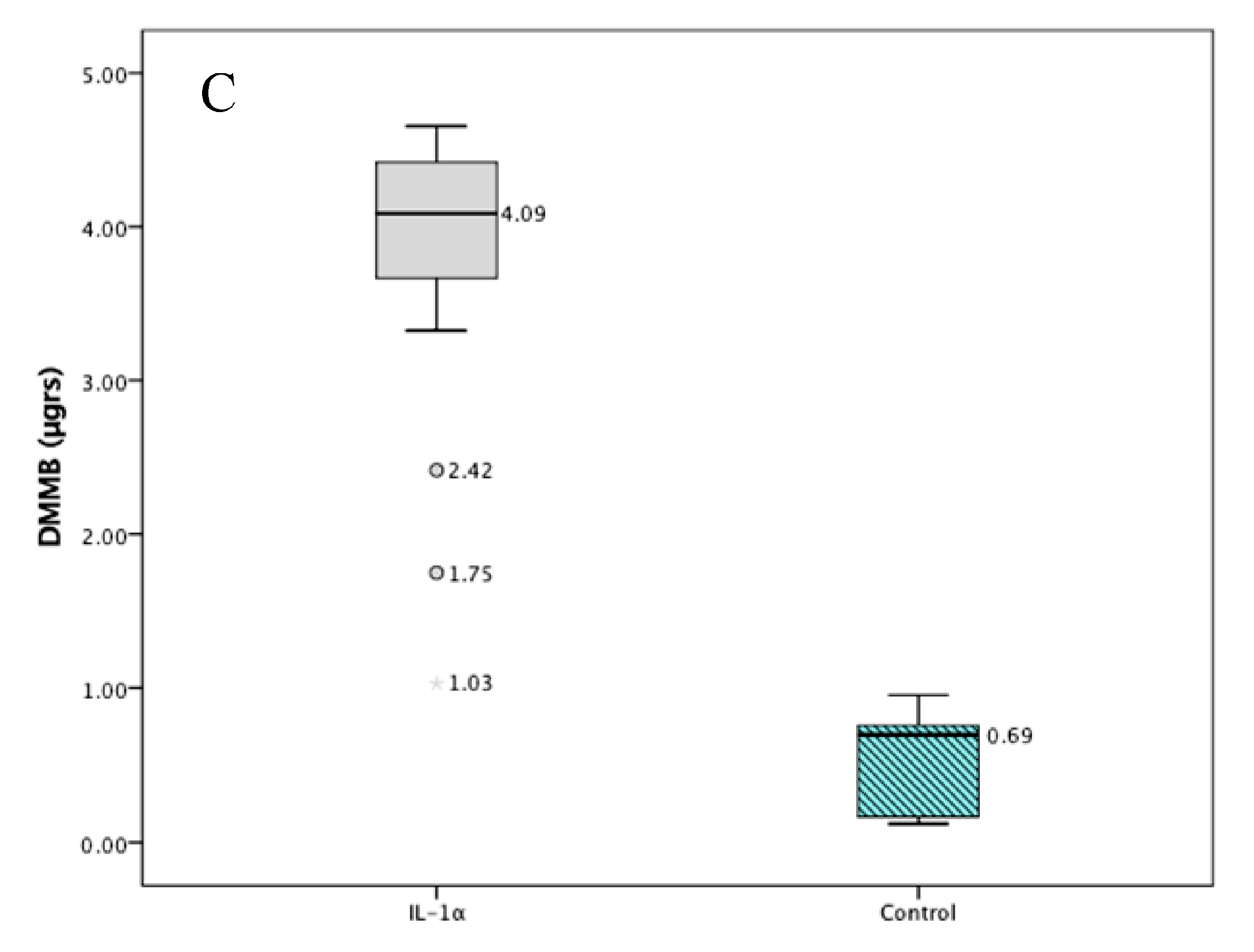
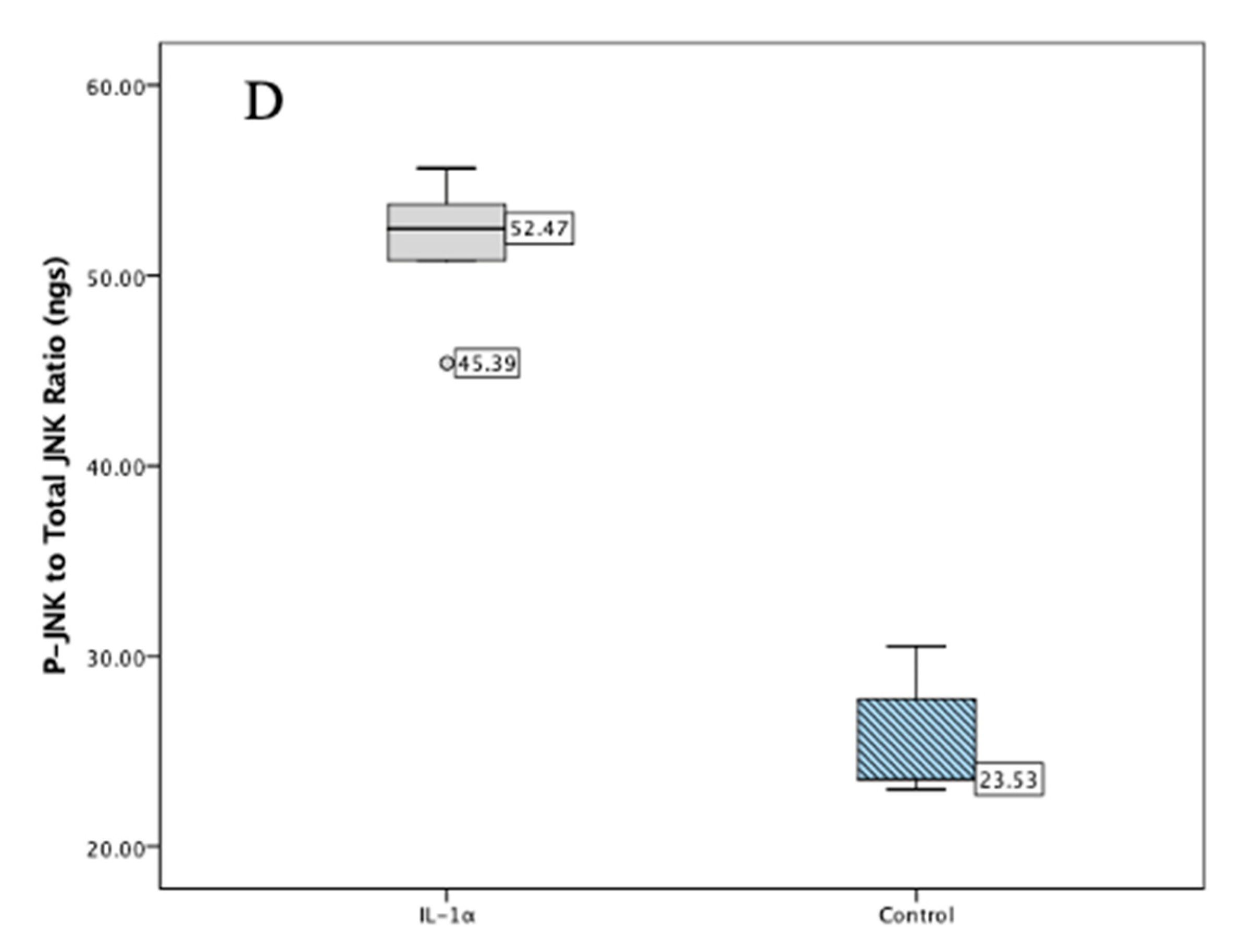
2.1. Validation Studies
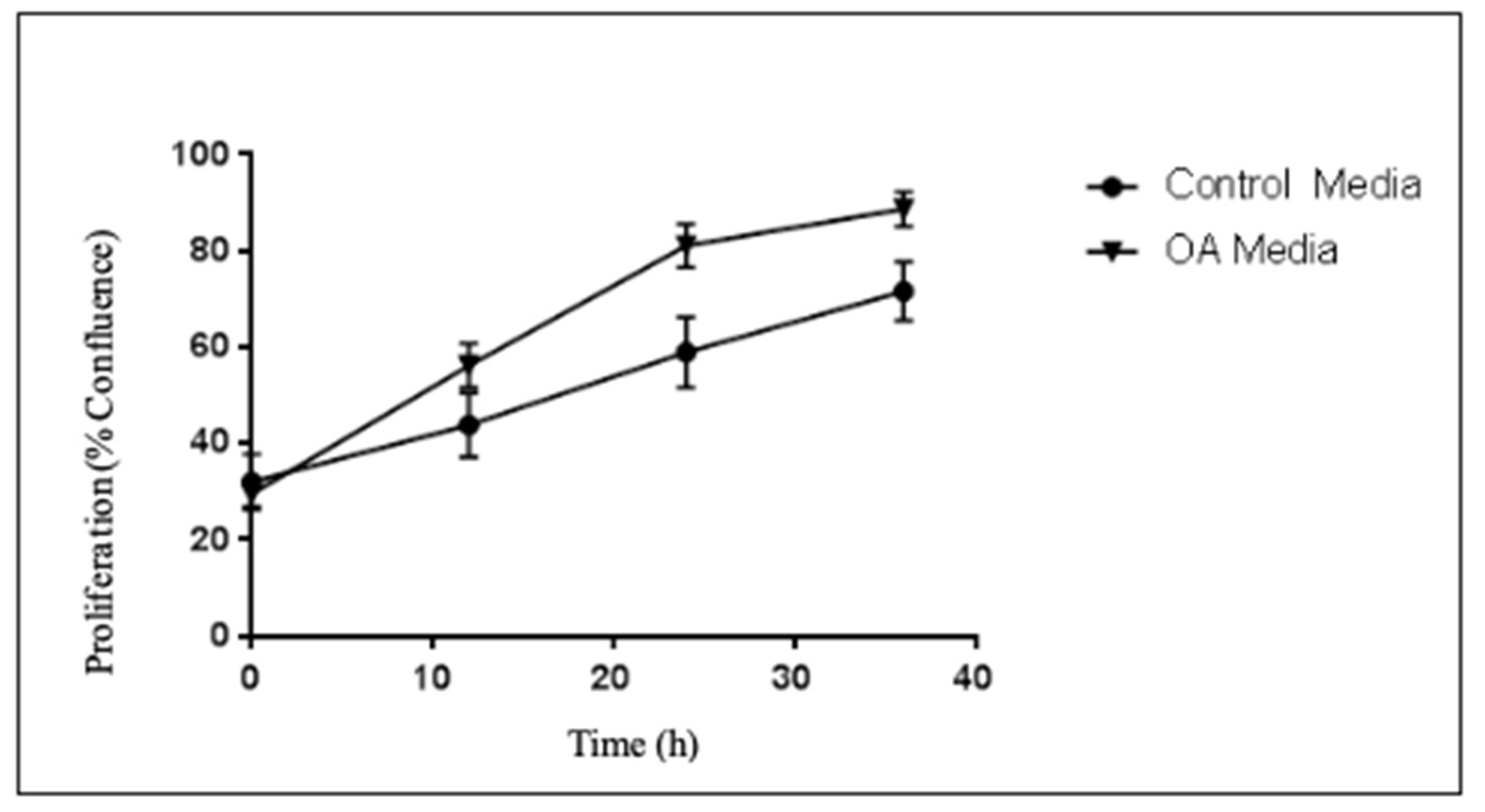
2.2. Proliferation Studies
2.3. Migration Studies
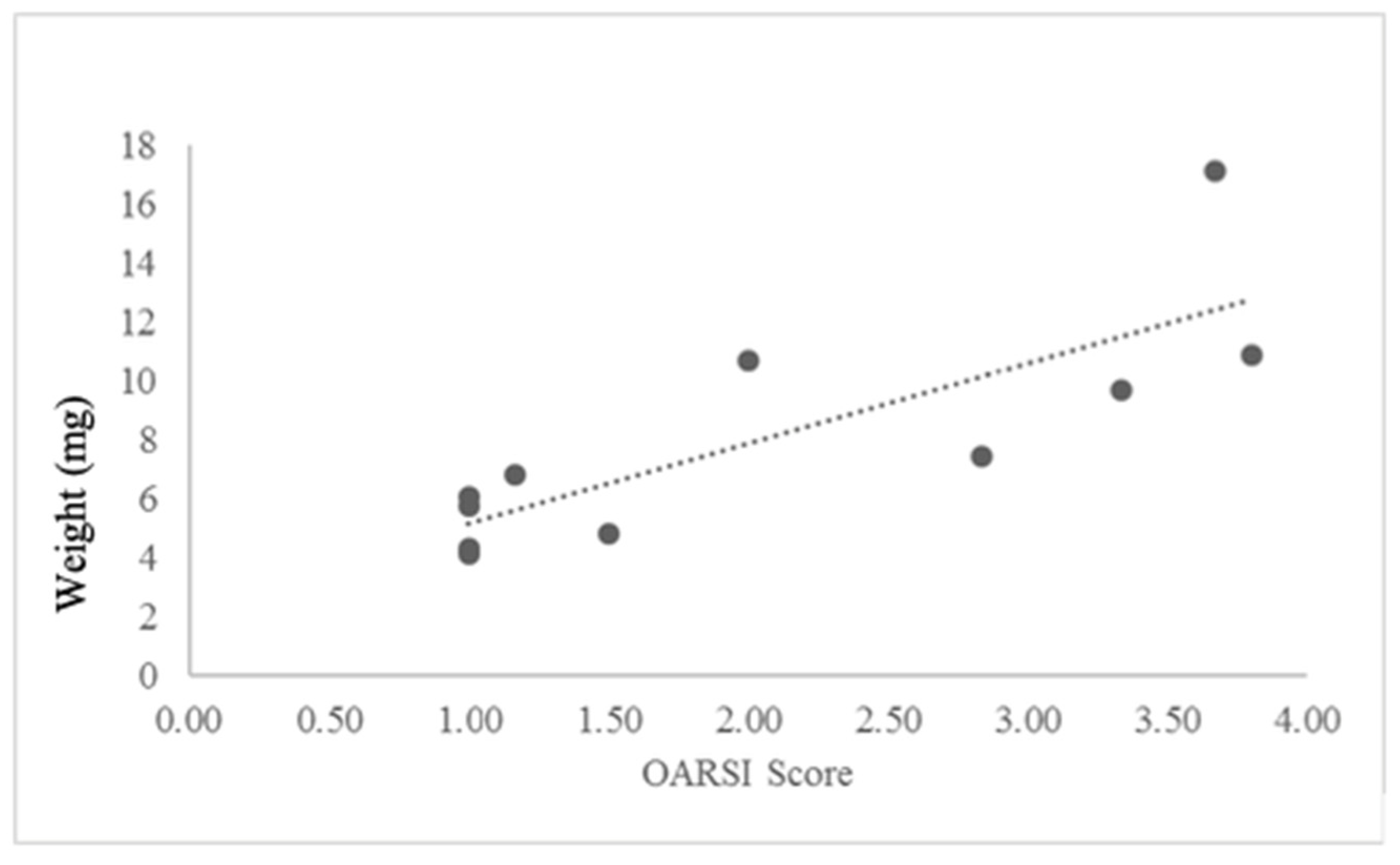
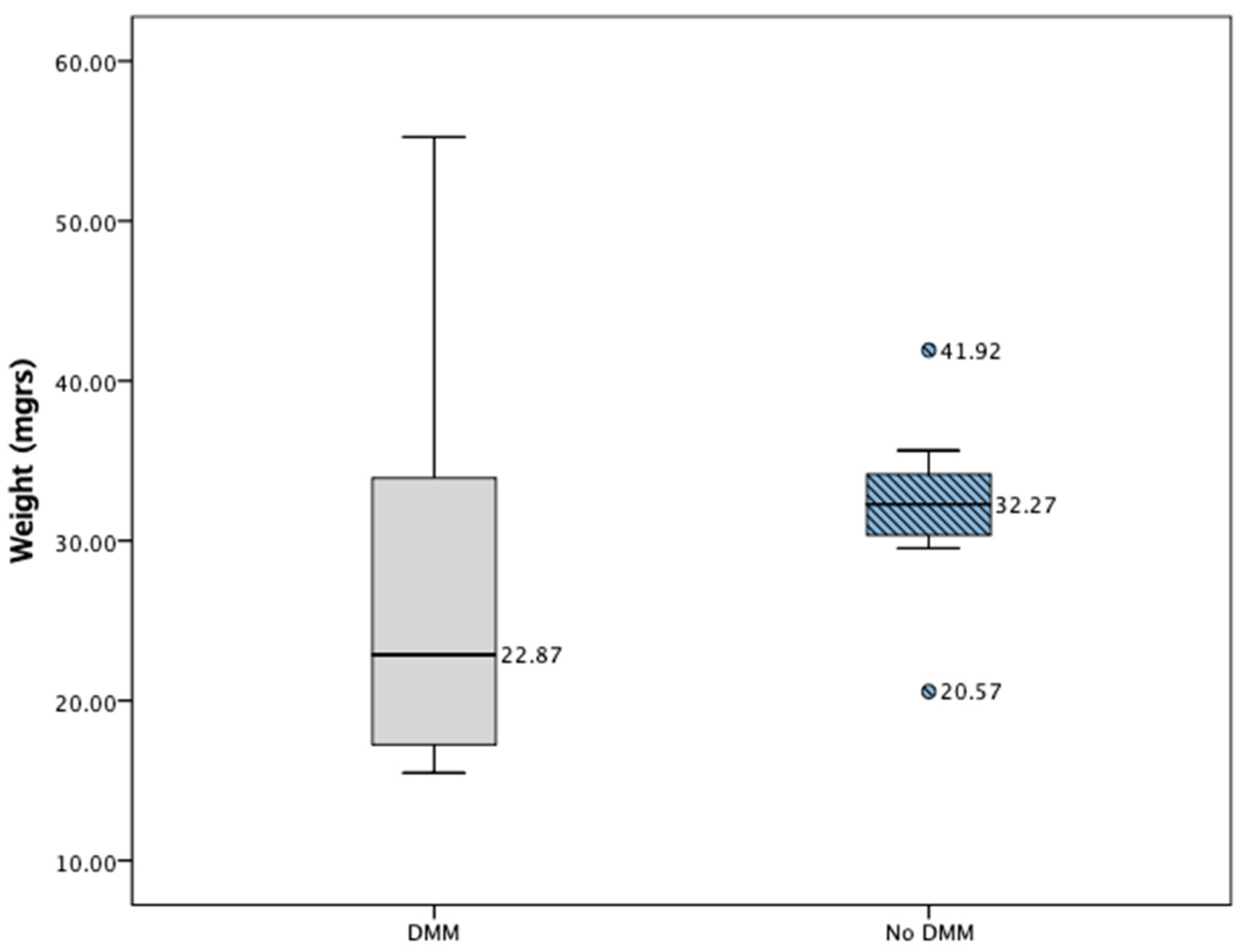
2.4. In Vivo Results
3. Discussion
Limitations
4. Materials and Methods
4.1. In Vitro
4.1.1. Prostate Cancer Cells
4.1.2. Reagents and Antibodies
4.1.3. Proliferation Studies
4.1.4. Migration Studies
4.1.5. In Vivo Model
4.1.6. Other Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roche, M.; Law, T.Y.; Kurowicki, J.; Rosas, S.; Rush, A.J., 3rd. Effect of Obesity on Total Knee Arthroplasty Costs and Revision Rate. J. Knee Surg. 2018, 31, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Rosas, S.; Luo, T.D.; Jinnah, A.H.; Marquez-Lara, A.; Roche, M.W.; Emory, C.L. Previous History of Breast Cancer Increases Rates of Pulmonary Embolism and Costs after Total Knee Arthroplasty: An Evaluation of 185,114 Matched Patients. J. Knee Surg. 2019, 32, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Rosas, S.; Marquez-Lara, A.; Bracey, D.N.; Kurowicki, J.; Roche, M.W.; Emory, C.L. History of Breast Cancer Increases 90-Day Pulmonary Embolism Rates and Reimbursements After Total Hip Arthroplasty: A National Matched-Pair Analysis. J. Arthroplast. 2018, 33, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.P.; Plate, J.F.; Goltz, D.E.; Attarian, D.E.; Wellman, S.S.; Seyler, T.M.; Jiranek, W.A. Should Medical Severity-Diagnosis Related Group Classification Be Utilized for Reimbursement? An Analysis of Elixhauser Comorbidities and Cost of Care. J. Arthroplast. 2019, 34, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Peisch, S.F.; Van Blarigan, E.L.; Chan, J.M.; Stampfer, M.J.; Kenfield, S.A. Prostate cancer progression and mortality: A review of diet and lifestyle factors. World J. Urol. 2017, 35, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Rosas, S.; Hughes, R.T.; Farris, M.; Lee, H.; McTyre, E.R.; Plate, J.F.; Shi, L.; Emory, C.L.; Blackstock, A.W.; Kerr, B.A.; et al. Cartilage oligomeric matrix protein in patients with osteoarthritis is independently associated with metastatic disease in prostate cancer. Oncotarget 2019, 10, 4776–4785. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.L.; Hecht, J.T. The role of cartilage oligomeric matrix protein (COMP) in skeletal disease. Curr. Drug Targets 2008, 9, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Englund, E.; Canesin, G.; Papadakos, K.S.; Vishnu, N.; Persson, E.; Reitsma, B.; Anand, A.; Jacobsson, L.; Helczynski, L.; Mulder, H.; et al. Cartilage oligomeric matrix protein promotes prostate cancer progression by enhancing invasion and disrupting intracellular calcium homeostasis. Oncotarget 2017, 8, 98298–98311. [Google Scholar] [CrossRef]
- Liu, T.T.; Liu, X.S.; Zhang, M.; Liu, X.N.; Zhu, F.X.; Zhu, F.M.; Ouyang, S.W.; Li, S.B.; Song, C.L.; Sun, H.M.; et al. Cartilage oligomeric matrix protein is a prognostic factor and biomarker of colon cancer and promotes cell proliferation by activating the Akt pathway. J. Cancer Res. Clin. Oncol. 2018, 144, 1049–1063. [Google Scholar] [CrossRef]
- Englund, E.; Bartoschek, M.; Reitsma, B.; Jacobsson, L.; Escudero-Esparza, A.; Orimo, A.; Leandersson, K.; Hagerling, C.; Aspberg, A.; Storm, P.; et al. Cartilage oligomeric matrix protein contributes to the development and metastasis of breast cancer. Oncogene 2016, 35, 5585–5596. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Wang, Y.; Liu, W.; Zhang, S. Cartilage oligomeric matrix protein acts as a molecular biomarker in multiple cancer types. Clin. Transl. Oncol. 2023, 25, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Kleeff, J.; Guo, J.; Gazdhar, A.; Liao, Q.; Di Cesare, P.E.; Büchler, M.W.; Friess, H. Cartilage oligomeric matrix protein expression in hepatocellular carcinoma and the cirrhotic liver. J. Gastroenterol. Hepatol. 2004, 19, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Attur, M.; Krasnokutsky-Samuels, S.; Samuels, J.; Abramson, S.B. Prognostic biomarkers in osteoarthritis. Curr. Opin. Rheumatol. 2013, 25, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Pu, H.; Kyprianou, N. Targeting TGF-beta in prostate cancer: Therapeutic possibilities during tumor progression. Expert. Opin. Ther. Targets 2009, 13, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.R.; Chay, C.H.; Pienta, K.J. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia 2002, 4, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Loberg, R.D.; Day, L.L.; Harwood, J.; Ying, C.; St John, L.N.; Giles, R.; Neeley, C.K.; Pienta, K.J. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia 2006, 8, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Allin, K.H.; Nordestgaard, B.G. Elevated C-reactive protein in the diagnosis, prognosis, and cause of cancer. Crit. Rev. Clin. Lab. Sci. 2011, 48, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Glimcher, L.H.; Aliprantis, A.O. Osteoimmunology at the nexus of arthritis, osteoporosis, cancer, and infection. J. Clin. Investig. 2011, 121, 2534–2542. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Stewart, L.M.D.; Ajayi, L.; Gray, I.C.; Gray, N.E.; Roberts, K.G.; Watson, G.J.; Kaisary, A.V.; Snary, D. The expression profile for the tumour suppressor gene PTEN and associated polymorphic markers. Br. J. Cancer 2000, 82, 1671–1676. [Google Scholar]
- Isaacs, W.B.; Carter, B.S.; Ewing, C.M. Wild-Type Suppresses Growth of Human Prostate Cancer Cells Containing Mutant Alleles. Cancer Res. 1991, 51, 4716–4720. [Google Scholar] [PubMed]
- Carroll, A.G.; Voeller, H.J.; Sugars, L.; Gelmann, E.P. p53 oncogene mutations in three human prostate cancer cell lines. Prostate 1993, 23, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Rock, M.J.; Holden, P.; Horton, W.A.; Cohn, D.H. Cartilage oligomeric matrix protein promotes cell attachment via two independent mechanisms involving CD47 and alphaVbeta3 integrin. Mol. Cell. Biochem. 2010, 338, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Das Roy, L.; Pathangey, L.B.; Tinder, T.L.; Schettini, J.L.; Gruber, H.E.; Mukherjee, P. Breast-cancer-associated metastasis is significantly increased in a model of autoimmune arthritis. Breast Cancer Res. 2009, 11, R56. [Google Scholar] [CrossRef] [PubMed]
- Perruccio, A.V.; Chandran, V.; Power, J.D.; Kapoor, M.; Mahomed, N.N.; Gandhi, R. Systemic inflammation and painful joint burden in osteoarthritis: A matter of sex? Osteoarthr. Cartil. 2017, 25, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Nasi, S.; Ea, H.K.; So, A.; Busso, N. Revisiting the Role of Interleukin-1 Pathway in Osteoarthritis: Interleukin-1α and -1β, and NLRP3 Inflammasome Are Not Involved in the Pathological Features of the Murine Menisectomy Model of Osteoarthritis. Front. Pharmacol. 2017, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Bay-Jensen, A.C.; van Spil, W.E.; Larkin, J.; Levesque, M.C. Osteoarthritis Year in Review 2016: Biomarkers (biochemical markers). Osteoarthr. Cartil. 2017, 25, 199–208. [Google Scholar] [CrossRef]
- Smitten, A.L.; Simon, T.A.; Hochberg, M.C.; Suissa, S. A meta-analysis of the incidence of malignancy in adult patients with rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R45. [Google Scholar] [CrossRef]
- Hemminki, K.; Li, X.; Sundquist, K.; Sundquist, J. Cancer risk in hospitalized rheumatoid arthritis patients. Rheumatology 2008, 47, 698–701. [Google Scholar] [CrossRef]
- Gridley, G.; McLaughlin, J.K.; Ekbom, A.; Klareskog, L.; Adami, H.O.; Hacker, D.G.; Hoover, R.; Fraumeni, J.F., Jr. Incidence of cancer among patients with rheumatoid arthritis. J. Natl. Cancer Inst. 1993, 85, 307–311. [Google Scholar] [CrossRef]
- Shen, K.; Xu, G.; Wu, Q.; Zhou, D.; Li, J. Risk of multiple myeloma in rheumatoid arthritis: A meta-analysis of case-control and cohort studies. PLoS ONE 2014, 9, e91461. [Google Scholar] [CrossRef] [PubMed]
- Dixon, W.G.; Watson, K.D.; Lunt, M.; Mercer, L.K.; Hyrich, K.L.; Symmons, D.P. Influence of anti-tumor necrosis factor therapy on cancer incidence in patients with rheumatoid arthritis who have had a prior malignancy: Results from the British Society for Rheumatology Biologics Register. Arthritis Care Res. 2010, 62, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.A.; Shi, L.; Jinnah, A.H.; Willey, J.S.; Lennon, D.P.; Caplan, A.I.; Byzova, T.V. Kindlin-3 Mutation in Mesenchymal Stem Cells Results in Enhanced Chondrogenesis. bioRxiv 2019, 399, 578690. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, A.A.; Foster, B.A.; Allison, J.P.; Greenberg, N.M.; Kwon, E.D. The TRAMP mouse as a model for prostate cancer. Curr. Protoc. Immunol. 2001, 45, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Gelman, I.H. How the TRAMP Model Revolutionized the Study of Prostate Cancer Progression. Cancer Res. 2016, 76, 6137–6139. [Google Scholar] [CrossRef]
- McCabe, N.P.; Kerr, B.A.; Madajka, M.; Vasanji, A.; Byzova, T.V. Augmented osteolysis in SPARC-deficient mice with bone-residing prostate cancer. Neoplasia 2011, 13, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, A.H.; Zacks, B.C.; Gwam, C.U.; Kerr, B.A. Emerging and Established Models of Bone Metastasis. Cancers 2018, 10, 176. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ribeiro, B.; Li, H.; Myer, L.; Chase, P.; Surti, N.; Lippy, J.; Zhang, L.; Cvijic, M.E. Leveraging the IncuCyte Technology for Higher-Throughput and Automated Chemotaxis Assays for Target Validation and Compound Characterization. SLAS Discov. Adv. Life Sci. R D 2018, 23, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.T.; Shah, E.T.; Chopin, L.K.; Sean McElwain, D.L.; Simpson, M.J. Estimating cell diffusivity and cell proliferation rate by interpreting IncuCyte ZOOM assay data using the Fisher-Kolmogorov model. BMC Syst. Biol. 2015, 9, 38. [Google Scholar] [CrossRef]
- Yammani, R.R.; Carlson, C.S.; Bresnick, A.R.; Loeser, R.F. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: Role of the receptor for advanced glycation end products. Arthritis Rheum. 2006, 54, 2901–2911. [Google Scholar] [CrossRef]
- Lindburg, C.A.; Willey, J.S.; Dean, D. Effects of low dose X-ray irradiation on porcine articular cartilage explants. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2013, 31, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- O’Clair, L.; Roddy, M.; Tikhonenko, M.; Syzbut, C.; Bevan, N.; Schroeder, K.; Appledorn, D.M. Quantification of cell migration and invasion using the IncuCyte™ chemotaxis assay. Essen BioScience 2015, 1, 1–5. [Google Scholar]
- Maini, P.K.; McElwain, D.S.; Leavesley, D.I. Traveling wave model to interpret a wound-healing cell migration assay for human peritoneal mesothelial cells. Tissue Eng. 2004, 10, 475–482. [Google Scholar] [CrossRef]
- Ghassemi, P.; Harris, K.S.; Ren, X.; Foster, B.M.; Langefeld, C.D.; Kerr, B.A.; Agah, M. Comparative study of prostate cancer biophysical and migratory characteristics via iterative mechanoelectrical properties (iMEP) and standard migration assays. Sens. Actuators B Chem. 2020, 321, 128522. [Google Scholar] [CrossRef] [PubMed]
- Hulkower, K.I.; Herber, R.L. Cell migration and invasion assays as tools for drug discovery. Pharmaceutics 2011, 3, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Culley, K.L.; Dragomir, C.L.; Chang, J.; Wondimu, E.B.; Coico, J.; Plumb, D.A.; Otero, M.; Goldring, M.B. Mouse models of osteoarthritis: Surgical model of posttraumatic osteoarthritis induced by destabilization of the medial meniscus. Methods Mol. Biol. 2015, 1226, 143–173. [Google Scholar] [PubMed]
- Glasson, S.S.; Blanchet, T.J.; Morris, E.A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Chambers, M.G.; Van Den Berg, W.B.; Little, C.B. The OARSI histopathology initiative—Recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr. Cartil. 2010, 18 (Suppl. 3), S17–S23. [Google Scholar] [CrossRef] [PubMed]
- Tse, B.W.; Russell, P.J.; Lochner, M.; Förster, I.; Power, C.A. IL-18 inhibits growth of murine orthotopic prostate carcinomas via both adaptive and innate immune mechanisms. PLoS ONE 2011, 6, e24241. [Google Scholar] [CrossRef]
- Ge, H.X.; Zou, F.M.; Li, Y.; Liu, A.M.; Tu, M. JNK pathway in osteoarthritis: Pathological and therapeutic aspects. J. Recept. Signal Transduct. Res. 2017, 37, 431–436. [Google Scholar] [CrossRef]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. Ther. 2017, 19, 248. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosas, S.; Kwok, A.; Moore, J.; Shi, L.; Smith, T.L.; Tallant, E.A.; Kerr, B.A.; Willey, J.S. Osteoarthritis as a Systemic Disease Promoted Prostate Cancer In Vivo and In Vitro. Int. J. Mol. Sci. 2024, 25, 6014. https://doi.org/10.3390/ijms25116014
Rosas S, Kwok A, Moore J, Shi L, Smith TL, Tallant EA, Kerr BA, Willey JS. Osteoarthritis as a Systemic Disease Promoted Prostate Cancer In Vivo and In Vitro. International Journal of Molecular Sciences. 2024; 25(11):6014. https://doi.org/10.3390/ijms25116014
Chicago/Turabian StyleRosas, Samuel, Andy Kwok, Joseph Moore, Lihong Shi, Thomas L. Smith, E. Ann Tallant, Bethany A. Kerr, and Jeffrey S. Willey. 2024. "Osteoarthritis as a Systemic Disease Promoted Prostate Cancer In Vivo and In Vitro" International Journal of Molecular Sciences 25, no. 11: 6014. https://doi.org/10.3390/ijms25116014