
Discovering Inflammation in Atherosclerosis: Insights from Pathogenic Pathways to Clinical Practice

Abstract
:1. Introduction
2. Atherosclerosis
2.1. Definition
2.2. Preclinical Atherosclerosis
3. Inflammation in Atherosclerosis
3.1. The Historical View of Inflammation in Atherosclerosis
3.2. The New Perspective of Inflammation in Atherosclerosis
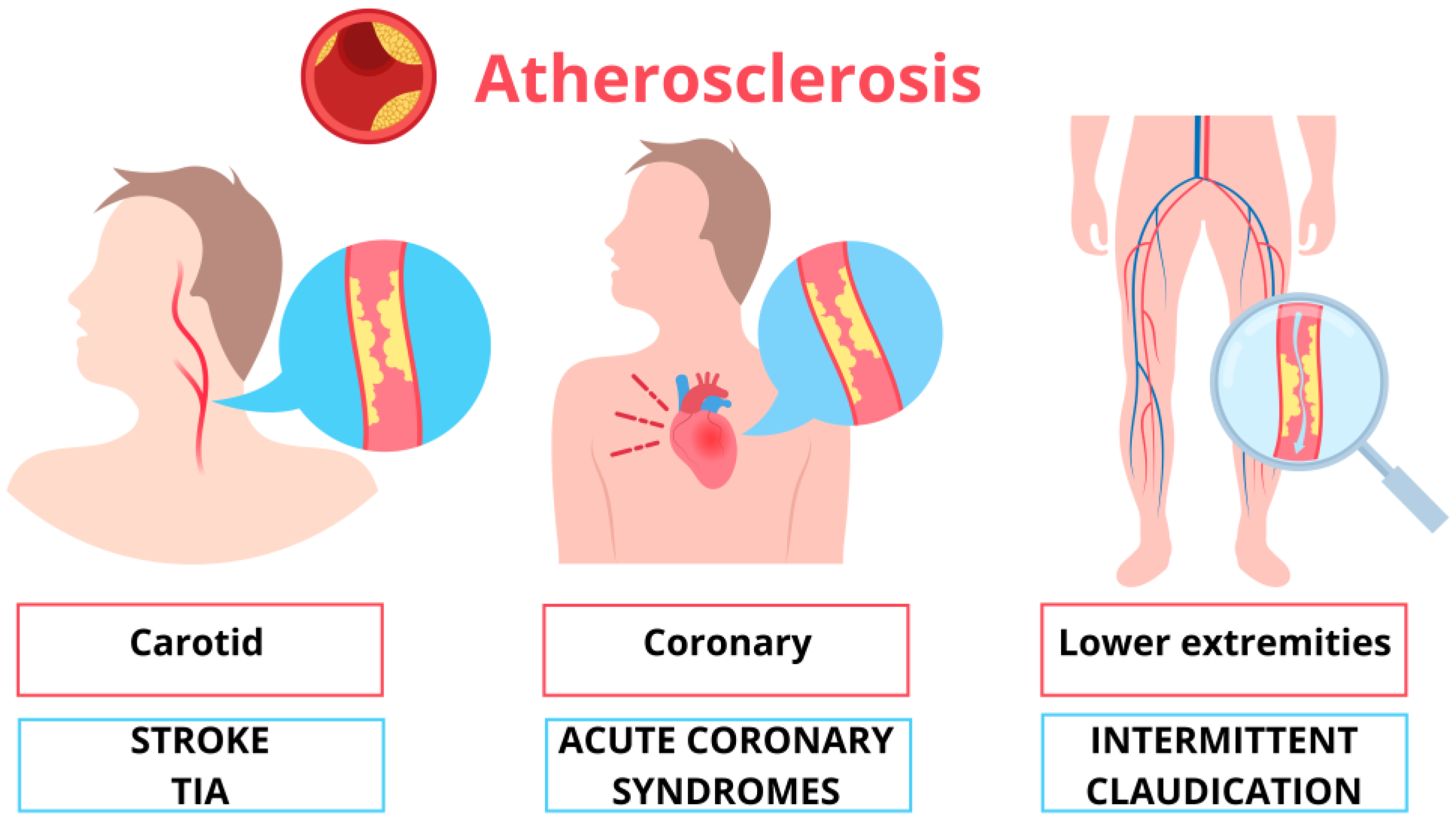
4. Symptoms of Atherosclerosis
5. Carotid Atherosclerosis
5.1. Epidemiology and Clinical Relevance
5.2. Stroke and TIA
5.3. Cognitive Impairment/Dementia
5.4. Carotid Atherosclerosis as a Mirror of Systemic Cardiovascular Disease
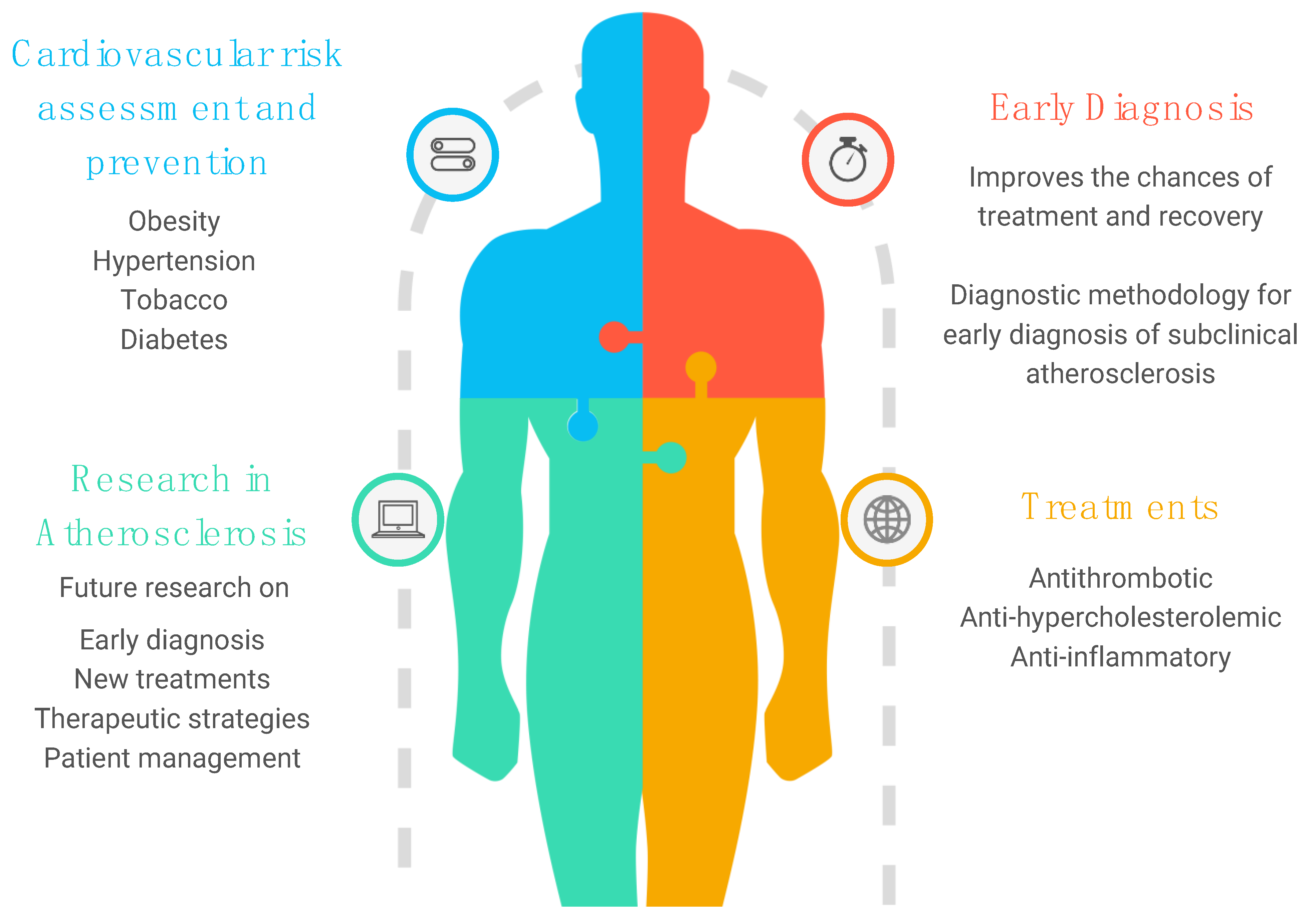
6. Prevention Strategies and Clinical Practice: What Is the Future?
6.1. Cardiovascular Risk Factors
6.2. Anti-Hypercholesterolemic Treatment
6.3. Antithrombotic Treatment in Carotid Artery Atherosclerosis: From Certainties to Dilemma
6.4. Anti-Inflammatory Treatment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Vaduganathan, M.; Mensah, G.A.; Turco, J.V.; Fuster, V.; Roth, G.A. The Global Burden of Cardiovascular Diseases and Risk. J. Am. Coll. Cardiol. 2022, 80, 2361–2371. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 27 April 2024).
- Timmis, A.; Vardas, P.; Townsend, N.; Tobica AKatus, H.; De Smedt, D.; Gale, C.P.; Maggioni, A.P.; Petersen, S.E.; Huculeci, R.; Kazakiewicz, D.; et al. Eiropean Society of Cardiology. European Society of Cardiology: Cardiovascular disease statistics 2021. Eur. Heart J. 2022, 43, 716–799, Erratum in Eur. Heart J. 2022, 43, 799. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.M. Atherosclerotic cardiovascular disease beginning in childhood. Korean Circ. J. 2010, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.P.; Malcom, G.T.; McMahan, C.A.; Tracy, R.E.; Newman, W.P.; Herderick, E.E.; Cornhill, J.F. Prevalence and extent of atherosclerosis in adolescents and young adults: Implications for prevention from the Pathobiological Determinants of Atherosclerosis in Youth Study. JAMA 1999, 281, 727–735. [Google Scholar] [CrossRef]
- Markin, A.M.; Sobenin, I.A.; Grechko, A.V.; Zhang, D.; Orekhov, A.N. Cellular Mechanisms of Human Atherogenesis: Focus on Chronification of Inflammation and Mitochondrial Mutations. Front. Pharmacol. 2020, 11, 642. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, E.; Carpino, G.; Grassi, M.; Musca, A. Morphological aspects of atherosclerosis lesion: Past and present. Clin. Ter. 2006, 157, 135–142. [Google Scholar]
- Hort, W. Arteriosclerosis: Its morphology in the past and today. Basic Res. Cardiol. 1994, 89 (Suppl. S1), 1–15. [Google Scholar] [CrossRef]
- Virchow, R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI—Atheromatous affection of arteries. 1858. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef]
- CLASSIFICATION of atherosclerotic lesions; report of a study group. World Health Organ Tech. Rep. Ser. 1958, 57, 1–20.
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47 (Suppl. S8), C13–C18. [Google Scholar] [CrossRef] [PubMed]
- Pahwa, R.; Jialal, I. Atherosclerosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK507799/ (accessed on 27 April 2024).
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Zanchetti, A.; Hennig, M.; Hollweck, R.; Bond, G.; Tang, R.; Cuspidi, C.; Parati, G.; Facchetti, R.; Mancia, G. Baseline values but not treatment-induced changes in carotid intima-media thickness predict incident cardiovascular events in treated hypertensive patients: Findings in the European Lacidipine Study on Atherosclerosis (ELSA). Circulation 2009, 120, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, D.H.; Polak, J.F.; Kronmal, R.A.; Manolio, T.A.; Burke, G.L.; Wolfson, S.K. Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N. Engl. J. Med. 1999, 340, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation; atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Corrado, E.; Rizzo, M.; Muratori, I.; Coppola, G.; Novo, S. Association of elevated fibrinogen and C-reactive protein levels with carotid lesions in patients with newly diagnosed hypertension or type II diabetes. Arch. Med. Res. 2006, 37, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Corrado, E.; Rizzo, M.; Coppola, G.; Fattouch, K.; Novo, G.; Marturana, I.; Ferrara, F.; Novo, S. An update on the role of markers of inflammation in atherosclerosis. J. Atheroscler. Thromb. 2010, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, M.; Di Noia, D.; Di Michele, L.; Corriero, F.; Di Biase, M.; Biasco, M.G.; Novo, S.; Strano, A.; Rizzon, P. The incidence of asymptomatic extracoronary atherosclerosis in patients with coronary atherosclerosis. Int. Angiol. 1993, 12, 25–28. [Google Scholar]
- Gonçalves, I.; Moses, J.; Dias, N.; Pedro, L.M.; Fernandes e Fernandes, J.; Nilsson, J.; Ares, M.P.S. Changes related to age and cerebrovascular symptoms in the extracellular matrix of human carotid plaques. Stroke 2003, 34, 616–622. [Google Scholar] [CrossRef]
- Mannami, T.; Konishi, M.; Baba, S.; Nishi, N.; Terao, A. Prevalence of asymptomatic carotid atherosclerotic lesions detected by high-resolution ultrasonography and its relation to cardiovascular risk factors in the general population of a Japanese city: The Suita study. Stroke 1997, 28, 518–525. [Google Scholar] [CrossRef]
- Corrado, E.; Rizzo, M.; Tantillo, R.; Muratori, I.; Bonura, F.; Vitale, G.; Novo, S. Markers of inflammation and infection influence the outcome of patients with baseline asymptomatic carotid lesions: A 5-year follow-up study. Stroke 2006, 37, 482–486. [Google Scholar] [CrossRef]
- Coppola, G.; Corrado, E.; Muratori, I.; Tantillo, R.; Vitale, G.; Lo Coco, L.; Novo, S. Increased levels of C-reactive protein and fibrinogen influence the risk of vascular events in patients with NIDDM. Int. J. Cardiol. 2006, 106, 16–20. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.A. Atherosclerosis and the Arterial Smooth Muscle Cell. Science 1973, 180, 1332–1339. [Google Scholar] [CrossRef]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.A.; Barrett, E.J.; Lindner, J.R.; Clark, M.G.; Rattigan, S. Inhibiting NOS blocks microvascular recruitment and blunts muscle glucose uptake in response to insulin. Am. J. Physiol.-Endocrinol. Metab. 2003, 285, E123–E129. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Natural History and Histological Classification of Atherosclerotic Lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef]
- Newby, A.C. Metalloproteinases and Vulnerable Atherosclerotic Plaques. Trends Cardiovasc. Med. 2007, 17, 253–258. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Zernecke, A.; Winkels, H.; Cochain, C.; Williams, J.W.; Wolf, D.; Soehnlein, O.; Robbins, C.S.; Monaco, C.; Park, I.; McNamara, C.A.; et al. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ. Res. 2020, 127, 402–426. [Google Scholar] [CrossRef]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.-E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698.e14. [Google Scholar] [CrossRef]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated Accumulation of Desmosterol Integrates Macrophage Lipid Metabolism and Inflammatory Responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef]
- Depuydt, M.A.C.; Schaftenaar, F.H.; Prange, K.H.M.; Boltjes, A.; Hemme, E.; Delfos, L.; de Mol, J.; de Jong, M.J.M.; Bernabé Kleijn, M.N.A.; Peeters, J.A.H.M.; et al. Single-cell T cell receptor sequencing of paired human atherosclerotic plaques and blood reveals autoimmune-like features of expanded effector T cells. Nat. Cardiovasc. Res. 2023, 2, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Lu, S.; Zhang, C.; Ma, Z.; Su, R.; Li, Y.; Sun, T.; Li, Y.; Hong, M.; et al. Pairing of single-cell RNA analysis and T cell antigen receptor profiling indicates breakdown of T cell tolerance checkpoints in atherosclerosis. Nat. Cardiovasc. Res. 2023, 2, 290–306. [Google Scholar] [CrossRef]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xiang, X.; Nie, L.; Guo, X.; Zhang, F.; Wen, C.; Xia, Y.; Mao, L. The emerging role of Th1 cells in atherosclerosis and its implications for therapy. Front. Immunol. 2023, 13, 1079668. [Google Scholar] [CrossRef]
- Buono, C.; Binder, C.J.; Stavrakis, G.; Witztum, J.L.; Glimcher, L.H.; Lichtman, A.H. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc. Natl. Acad. Sci. USA 2005, 102, 1596–1601. [Google Scholar] [CrossRef]
- Davenport, P.; Tipping, P.G. The Role of Interleukin-4 and Interleukin-12 in the Progression of Atherosclerosis in Apolipoprotein E-Deficient Mice. Am. J. Pathol. 2003, 163, 1117–1125. [Google Scholar] [CrossRef]
- Cardilo-Reis, L.; Gruber, S.; Schreier, S.M.; Drechsler, M.; Papac-Milicevic, N.; Weber, C.; Wagner, O.; Stangl, H.; Soehnlein, O.; Binder, C.J. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol. Med. 2012, 4, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Dale, B.L.; Pandey, A.K.; Chen, Y.; Smart, C.D.; Laroumanie, F.; Ao, M.; Xiao, L.; Dikalova, A.E.; Dikalov, S.I.; Elijovich, F.; et al. Critical role of IL-21 and T follicular helper cells in hypertension and vascular dysfunction. JCI Insight 2019, 4, e129278. [Google Scholar] [CrossRef] [PubMed]
- Foks, A.C.; Lichtman, A.H.; Kuiper, J. Treating Atherosclerosis With Regulatory T Cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.-K.L.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Gjurich, B.N.; Phillips, T.; Galkina, E.V. The IL-17A/IL-17RA Axis Plays a Proatherogenic Role via the Regulation of Aortic Myeloid Cell Recruitment. Circ. Res. 2012, 110, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Winship, A.; Tay, C.; Kanellakis, P.; Hosseini, H.; Cao, A.; Li, P.; Tipping, P.; Bobik, A. Toh B-H Cytotoxic and Proinflammatory CD8+ T Lymphocytes Promote Development of Vulnerable Atherosclerotic Plaques in ApoE-Deficient Mice. Circulation 2013, 127, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- van Duijn, J.; Kritikou, E.; Benne, N.; van der Heijden, T.; van Puijvelde, G.H.; Kröner, M.J.; Schaftenaar, F.H.; Foks, A.C.; Wezel, A.; Smeets, H.; et al. CD8+ T-cells contribute to lesion stabilization in advanced atherosclerosis by limiting macrophage content and CD4+ T-cell responses. Cardiovasc. Res. 2019, 115, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Sage, A.P.; Mallat, Z.; Binder, C.J. Targeting B Cells in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.; Newland, S.A.; Jiang, W.; Giakomidi, D.; Zhao, X.; Clement, M.; Masters, L.; Corovic, A.; Zhang, X.; Drago, F.; et al. Marginal zone B cells produce “natural” atheroprotective IgM antibodies in a T cell–dependent manner. Cardiovasc. Res. 2024, 120, 318–328. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Theurl, I.; Gerhardt, L.M.S.; Robbins, C.S.; Weber, G.F.; Gonen, A.; Iwamoto, Y.; Degousee, N.; Holderried, T.A.W.; Winter, C.; et al. Innate Response Activator B Cells Aggravate Atherosclerosis by Stimulating T Helper-1 Adaptive Immunity. Circulation 2014, 129, 1677–1687. [Google Scholar] [CrossRef]
- Drechsler, M.; Duchene, J.; Soehnlein, O.; Mobilization, C.C. Recruitment, and Fate of Monocytes in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1050–1055. [Google Scholar] [CrossRef]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef]
- Aiello, R.J.; Bourassa, P.-A.K.; Lindsey, S.; Weng, W.; Natoli, E.; Rollins, B.J.; Milos, P.M. Monocyte Chemoattractant Protein-1 Accelerates Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1518–1525. [Google Scholar] [CrossRef]
- Živković, L.; Asare, Y.; Bernhagen, J.; Dichgans, M.; Georgakis, M.K. CCL2/CCR2 inhibition in atherosclerosis: A meta-analysis of preclinical studies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Georgakis, M.K.; van der Laan, S.W.; Asare, Y.; Mekke, J.M.; Haitjema, S.; Schoneveld, A.H.; de Jager, S.C.A.; Nurmohamed, N.S.; Kroon, J.; Stroes, E.S.G.; et al. Monocyte-Chemoattractant Protein-1 Levels in Human Atherosclerotic Lesions Associate With Plaque Vulnerability. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2038–2048. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Gill, D.; Rannikmäe, K.; Traylor, M.; Anderson, C.D.; Lee, J.-M.; Kamatani, Y.; Hopewell, J.C.; Worrall, B.B.; Bernhagen, J.; et al. Genetically Determined Levels of Circulating Cytokines and Risk of Stroke. Circulation 2019, 139, 256–268. [Google Scholar] [CrossRef]
- Döring, Y.; Noels, H.; van der Vorst, E.P.C.; Neideck, C.; Egea, V.; Drechsler, M.; Mandl, M.; Pawig, L.; Jansen, Y.; Schröder, K.; et al. Vascular CXCR4 Limits Atherosclerosis by Maintaining Arterial Integrity. Circulation 2017, 136, 388–403. [Google Scholar] [CrossRef]
- Zernecke, A.; Bot, I.; Djalali-Talab, Y.; Shagdarsuren, E.; Bidzhekov, K.; Meiler, S.; Krohn, R.; Schober, A.; Sperandio, M.; Soehnlein, O.; et al. Protective Role of CXC Receptor 4/CXC Ligand 12 Unveils the Importance of Neutrophils in Atherosclerosis. Circ. Res. 2008, 102, 209–217. [Google Scholar] [CrossRef]
- Döring, Y.; Jansen, Y.; Cimen, I.; Aslani, M.; Gencer, S.; Peters, L.J.F.; Duchene, J.; Weber, C.; van der Vorst, E.P.C. B-Cell-Specific CXCR4 Protects Against Atherosclerosis Development and Increases Plasma IgM Levels. Circ. Res. 2020, 126, 787–788. [Google Scholar] [CrossRef]
- Cimen, I.; Natarelli, L.; Abedi Kichi, Z.; Henderson, J.M.; Farina, F.M.; Briem, E.; Aslani, M.; Megens, R.T.A.; Jansen, Y.; Mann-Fallenbuchel, E.; et al. Targeting a cell-specific microRNA repressor of CXCR4 ameliorates atherosclerosis in mice. Sci. Transl. Med. 2023, 15, eadf3357. [Google Scholar] [CrossRef]
- Heller, E.A.; Liu, E.; Tager, A.M.; Yuan, Q.; Lin, A.Y.; Ahluwalia, N.; Jones, K.; Koehn, S.L.; Lok, V.M.; Aikawa, E.; et al. Chemokine CXCL10 Promotes Atherogenesis by Modulating the Local Balance of Effector and Regulatory T Cells. Circulation 2006, 113, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, S.K.; Peng, L.; Li, Y.; Lu, S.; Sun, T.; Carnevale, L.; Perrotta, M.; Ma, Z.; Förstera, B.; Stanic, K.; et al. Neuroimmune cardiovascular interfaces control atherosclerosis. Nature 2022, 605, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, S.K.; Weber, C.; Yin, C.; Habenicht, A.J.R. The dawn has come for new therapeutics to treat atherosclerosis: Targeting neuroimmune cardiovascular interfaces in artery brain circuits. Clin. Transl. Med. 2022, 12, e1040. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Moos, M.P.W.; Gräbner, R.; Pédrono, F.; Fan, J.; Kaiser, B.; John, N.; Schmidt, S.; Spanbroek, R.; Lötzer, K.; et al. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat. Med. 2004, 10, 966–973. [Google Scholar] [CrossRef]
- Moos, M.P.W.; John, N.; Gräbner, R.; Noßmann, S.; Günther, B.; Vollandt, R.; Funk, C.D.; Kaiser, B.; Habenicht, A.J.R. The Lamina Adventitia Is the Major Site of Immune Cell Accumulation in Standard Chow-Fed Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2386–2391. [Google Scholar] [CrossRef] [PubMed]
- Hinterdobler, J.; Simin, S.; Jin, H.; Meesmann, A.; Steinsiek, A.-L.; Zimmermann, A.-S.; Wobst, J.; Müller, P.; Mauersberger, C.; Vilne, B.; et al. Acute mental stress drives vascular inflammation and promotes plaque destabilization in mouse atherosclerosis. Eur. Heart J. 2021, 42, 4077–4088. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cao, Y.; Xu, X.; Wang, C.; Ni, Q.; Lv, X.; Yang, C.; Zhang, Z.; Qi, X.; Song, G. Sleep Deprivation Promotes Endothelial Inflammation and Atherogenesis by Reducing Exosomal miR-182-5p. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 995–1014. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, M.P.; Pober, J.S.; Wheeler, M.E.; Cotran, R.S.; Gimbrone, M.A. Interleukin-1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am. J. Pathol. 1985, 121, 394–403. [Google Scholar]
- Libby, P.; Warner, S.J.; Friedman, G.B. Interleukin 1: A mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J. Clin. Investig. 1988, 81, 487–498. [Google Scholar] [CrossRef]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; Liu, W.; Thomas, D.G.; Hajebrahimi, M.A.; Pircher, J.; et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021, 592, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The low-density lipoprotein pathway and its relation to atherosclerosis. Annu. Rev. Biochem. 1977, 46, 897–930. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Wiviott, S.D.; Blazing, M.A.; De Ferrari, G.M.; Park, J.-G.; Murphy, S.A.; White, J.A.; Tershakovec, A.M.; Cannon, C.P.; Braunwald, E. Long-term Safety and Efficacy of Achieving Very Low Levels of Low-Density Lipoprotein Cholesterol: A Prespecified Analysis of the IMPROVE-IT Trial. JAMA Cardiol. 2017, 2, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Ananthramaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Ansell, B.J.; Fonarow, G.C.; Vahabzadeh, K.; Hama, S.; Hough, G.; Kamranpour, N.; et al. The oxidation hypothesis of atherogenesis: The role of oxidized phospholipids and HDL. J. Lipid Res. 2004, 45, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.I.; Choi, S.-H.; Wiesner, P.; Fang, L.; Harkewicz, R.; Hartvigsen, K.; Boullier, A.; Gonen, A.; Diehl, C.J.; Que, X.; et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 2011, 108, 235–248. [Google Scholar] [CrossRef]
- Okafor, O.N.; Gorog, D.A. Endogenous Fibrinolysis: An Important Mediator of Thrombus Formation and Cardiovascular Risk. J. Am. Coll. Cardiol. 2015, 65, 1683–1699. [Google Scholar] [CrossRef]
- Berenson, G.S.; Srinivasan, S.R.; Bao, W.; Newman, W.P.; Tracy, R.E.; Wattigney, W.A. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N. Engl. J. Med. 1998, 338, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Almohtasib, Y.; Fancher, A.J.; Sawalha, K. Emerging Trends in Atherosclerosis: Time to Address Atherosclerosis From a Younger Age. Cureus 2024, 16, e56635. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Friera, L.; Peñalvo, J.L.; Fernández-Ortiz, A.; Ibañez, B.; López-Melgar, B.; Laclaustra, M.; Oliva, B.; Mocoroa, A.; Mendiguren, J.; Martínez de Vega, V.; et al. Prevalence, Vascular Distribution, and Multiterritorial Extent of Subclinical Atherosclerosis in a Middle-Aged Cohort: The PESA (Progression of Early Subclinical Atherosclerosis) Study. Circulation 2015, 131, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Sucato, V.; Madaudo, C.; Galassi, A.R. Classification, Diagnosis, and Treatment of Coronary Microvascular Dysfunction. J. Clin. Med. 2022, 11, 4610. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.R.; Beiser, A.; Seshadri, S.; Benjamin, E.J.; Polak, J.F.; Vasan, R.S.; Au, R.; DeCarli, C.; Wolf, P.A. Carotid artery atherosclerosis, MRI indices of brain ischemia, aging, and cognitive impairment: The Framingham study. Stroke 2009, 40, 1590–1596. [Google Scholar] [CrossRef]
- Yew, B.; Nation, D.A.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrovascular resistance: Effects on cognitive decline, cortical atrophy, and progression to dementia. Brain 2017, 140, 1987–2001. [Google Scholar] [CrossRef] [PubMed]
- Sander, K.; Bickel, H.; Förstl, H.; Etgen, T.; Briesenick, C.; Poppert, H.; Sander, D. Carotid- intima media thickness is independently associated with cognitive decline. The INVADE study. Int. J. Geriatr. Psychiatry 2010, 25, 389–394. [Google Scholar] [CrossRef]
- Moresoli, P.; Habib, B.; Reynier, P.; Secrest, M.H.; Eisenberg, M.J.; Filion, K.B. Carotid Stenting Versus Endarterectomy for Asymptomatic Carotid Artery Stenosis: A Systematic Review and Meta-Analysis. Stroke 2017, 48, 2150–2157. [Google Scholar] [CrossRef]
- Hollander, M.; Bots, M.L.; Del Sol, A.I.; Koudstaal, P.J.; Witteman, J.C.M.; Grobbee, D.E.; Hofman, A.; Breteler, M.M.B. Carotid plaques increase the risk of stroke and subtypes of cerebral infarction in asymptomatic elderly: The Rotterdam study. Circulation 2002, 105, 2872–2877. [Google Scholar] [CrossRef]
- Störk, S.; van den Beld, A.W.; von Schacky, C.; Angermann, C.E.; Lamberts, S.W.J.; Grobbee, D.E.; Bots, M.L. Carotid artery plaque burden, stiffness, and mortality risk in elderly men: A prospective, population-based cohort study. Circulation 2004, 110, 344–348. [Google Scholar] [CrossRef]
- Song, P.; Fang, Z.; Wang, H.; Cai, Y.; Rahimi, K.; Zhu, Y.; Fowkes, F.G.R.; Fowkes, F.J.I.; Rudan, I. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: A systematic review, meta-analysis, and modelling study. Lancet Glob. Health 2020, 8, e721–e729. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2197–2223. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- de Weerd, M.; Greving, J.P.; Hedblad, B.; Lorenz, M.W.; Mathiesen, E.B.; O’Leary, D.H.; Rosvall, M.; Sitzer, M.; Buskens, E.; Bots, M.L. Prevalence of asymptomatic carotid artery stenosis in the general population: An individual participant data meta-analysis. Stroke 2010, 41, 1294–1297. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Avolio, A.P.; Mergner, W.J.; Robinowitz, M.; Herderick, E.E.; Cornhill, J.F.; Guo, S.Y.; Liu, T.H.; Ou, D.Y.; O’Rourke, M. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am. J. Pathol. 1991, 139, 1119–1129. [Google Scholar] [PubMed]
- Gasbarrino, K.; Gorgui, J.; Nauche, B.; Côté, R.; Daskalopoulou, S.S. Circulating adiponectin and carotid intima-media thickness: A systematic review and meta-analysis. Metabolism 2016, 65, 968–986. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.W.; Markus, H.S.; Bots, M.L.; Rosvall, M.; Sitzer, M. Prediction of clinical cardiovascular events with carotid intima-media thickness: A systematic review and meta-analysis. Circulation 2007, 115, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Darabian, S.; Hormuz, M.; Latif, M.A.; Pahlevan, S.; Budoff, M.J. The role of carotid intimal thickness testing and risk prediction in the development of coronary atherosclerosis. Curr. Atheroscler. Rep. 2013, 15, 306. [Google Scholar] [CrossRef]
- Gupta, A.; Baradaran, H.; Schweitzer, A.D.; Kamel, H.; Pandya, A.; Delgado, D.; Dunning, A.; Mushlin, A.I.; Sanelli, P.C. Carotid plaque MRI and stroke risk: A systematic review and meta-analysis. Stroke 2013, 44, 3071–3077. [Google Scholar] [CrossRef]
- Reinhard, M.; Schwarzer, G.; Briel, M.; Altamura, C.; Palazzo, P.; King, A.; Bornstein, N.M.; Petersen, N.; Motschall, E.; Hetzel, A.; et al. Cerebrovascular reactivity predicts stroke in high-grade carotid artery disease. Neurology 2014, 83, 1424–1431. [Google Scholar] [CrossRef]
- Furlan, A.J.; Whisnant, J.P.; Baker, H.L. Long-term prognosis after carotid artery occlusion. Neurology 1980, 30, 986–988. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, H.C.; Weiner, M.; Hynan, L.S.; Cullum, C.M.; Khera, A.; Lacritz, L.H. Subclinical atherosclerosis and subsequent cognitive function. Atherosclerosis 2015, 241, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Martinić-Popović, I.; Lovrencić-Huzjan, A.; Demarin, V. Assessment of subtle cognitive impairment in stroke-free patients with carotid disease. Acta Clin. Croat. 2009, 48, 231–240. [Google Scholar] [PubMed]
- Rocque, B.G.; Jackson, D.; Varghese, T.; Hermann, B.; McCormick, M.; Kliewer, M.; Mitchell, C.; Dempsey, R.J. Impaired cognitive function in patients with atherosclerotic carotid stenosis and correlation with ultrasound strain measurements. J. Neurol. Sci. 2012, 322, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.E.; Wang, X.; Mitchell, C.C.; Kundu, B.; Jackson, D.C.; Wilbrand, S.M.; Varghese, T.; Hermann, B.P.; Rowley, H.A.; Johnson, S.C.; et al. The relationship between carotid artery plaque stability and white matter ischemic injury. Neuroimage Clin. 2015, 9, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Poredos, P.; Golob, M.; Jensterle, M. Interrelationship between peripheral arterial occlusive disease, carotid atherosclerosis and flow mediated dilation of the brachial artery. Int. Angiol. 2003, 22, 83–87. [Google Scholar]
- Amato, M.; Montorsi, P.; Ravani, A.; Oldani, E.; Galli, S.; Ravagnani, P.M.; Tremoli, E.; Baldassarre, D. Carotid intima-media thickness by B-mode ultrasound as surrogate of coronary atherosclerosis: Correlation with quantitative coronary angiography and coronary intravascular ultrasound findings. Eur. Heart J. 2007, 28, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, N.; Kogame, N.; Iijima, R.; Nakamura, M.; Sugi, K. Carotid artery intima-media thickness and plaque score can predict the SYNTAX score. Eur. Heart J. 2012, 33, 113–119. [Google Scholar] [CrossRef]
- Mattace-Raso, F.; van Popele, N.M.; Schalekamp, M.A.D.H.; van der Cammen, T.J.M. Intima-media thickness of the common carotid arteries is related to coronary atherosclerosis and left ventricular hypertrophy in older adults. Angiology 2002, 53, 569–574. [Google Scholar] [CrossRef]
- Nakamura, S.; Iihara, K.; Matayoshi, T.; Yasuda, H.; Yoshihara, F.; Kamide, K.; Horio, T.; Miyamoto, S.; Kawano, Y. The incidence and risk factors of renal artery stenosis in patients with severe carotid artery stenosis. Hypertens. Res. 2007, 30, 839–844. [Google Scholar] [CrossRef]
- Martínez-González, M.A.; Salas-Salvadó, J.; Estruch, R.; Corella, D.; Fitó, M.; Ros, E.; PREDIMED INVESTIGATORS. Benefits of the Mediterranean Diet: Insights From the PREDIMED Study. Prog. Cardiovasc. Dis. 2015, 58, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, D.M.; Leip, E.P.; Larson, M.G.; D’Agostino, R.B.; Beiser, A.; Wilson, P.W.F.; Wolf, P.A.; Levy, D. Prediction of lifetime risk for cardiovascular disease by risk factor burden at 50 years of age. Circulation 2006, 113, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Juhola, J.; Magnussen, C.G.; Berenson, G.S.; Venn, A.; Burns, T.L.; Sabin, M.A.; Srinivasan, S.R.; Daniels, S.R.; Davis, P.H.; Chen, W.; et al. Combined effects of child and adult elevated blood pressure on subclinical atherosclerosis: The International Childhood Cardiovascular Cohort Consortium. Circulation 2013, 128, 217–224. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Shipley, M.; Markus, H.; ACES Investigators. The effect of medical treatments on stroke risk in asymptomatic carotid stenosis. Stroke 2013, 44, 542–546. [Google Scholar] [CrossRef]
- Mostaza, J.M.; Lahoz, C.; Salinero-Fort, M.A.; de Burgos-Lunar, C.; Laguna, F.; Estirado, E.; García-Iglesias, F.; González-Alegre, T.; Cornejo-Del-Río, V.; Sabín, C.; et al. Carotid atherosclerosis severity in relation to glycemic status: A cross-sectional population study. Atherosclerosis 2015, 242, 377–382. [Google Scholar] [CrossRef] [PubMed]
- AbuRahma, A.F.; Srivastava, M.; Stone, P.A.; Richmond, B.K.; AbuRahma, Z.; Jackson, W.; Dean, L.S.; Mousa, A.Y. Effect of statins on early and late clinical outcomes of carotid endarterectomy and the rate of post-carotid endarterectomy restenosis. J. Am. Coll. Surg. 2015, 220, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, A.; Kakkos, S.; Abbott, A.; Naylor, A.R.; Richards, T.; Mikhailidis, D.P.; Geroulakos, G.; Nicolaides, A.N. Long-term Mortality in Patients with Asymptomatic Carotid Stenosis: Implications for Statin Therapy. Eur. J. Vasc. Endovasc. Surg. 2015, 50, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Merwick, Á.; Albers, G.W.; Arsava, E.M.; Ay, H.; Calvet, D.; Coutts, S.B.; Cucchiara, B.L.; Demchuk, A.M.; Giles, M.F.; Mas, J.-L.; et al. Reduction in early stroke risk in carotid stenosis with transient ischemic attack associated with statin treatment. Stroke 2013, 44, 2814–2820. [Google Scholar] [CrossRef]
- Avgerinos, E.D.; Kakisis, J.D.; Moulakakis, K.G.; Giannakopoulos, T.G.; Sfyroeras, G.; Antonopoulos, C.N.; Kadoglou, N.P.; Liapi, C.D. Statins influence long term restenosis and cardiovascular events following carotid endarterectomy. Curr. Vasc. Pharmacol. 2015, 13, 239–247. [Google Scholar] [CrossRef]
- Makris, G.C.; Lavida, A.; Nicolaides, A.N.; Geroulakos, G. The effect of statins on carotid plaque morphology: A LDL-associated action or one more pleiotropic effect of statins? Atherosclerosis 2010, 213, 8–20. [Google Scholar] [CrossRef]
- Kim, B.-K.; Hong, S.-J.; Lee, Y.-J.; Hong, S.J.; Yun, K.H.; Hong, B.-K.; Heo, J.H.; Rha, S.-W.; Cho, Y.-H.; Lee, S.-J.; et al. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): A randomised, open-label, non-inferiority trial. Lancet 2022, 400, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; Kearney, P.; et al. Aspirin in the primary and secondary prevention of vascular disease collaborative meta analysis of individual participant data from randomized trials. Lancet 2009, 373, 1849–1860. [Google Scholar] [PubMed]
- Abdelaziz, H.K.; Saad, M.; Pothineni, N.V.K.; Megaly, M.; Potluri, R.; Saleh, M.; Kon, D.L.C.; Roberts, D.H.; Bhatt, D.L.; Aronow, H.D.; et al. Aspirin for Primary Prevention of Cardiovascular Events. J. Am. Coll. Cardiol. 2019, 73, 2915–2929. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.L.; Roddick, A.J. Association of Aspirin Use for Primary Prevention With Cardiovascular Events and Bleeding Events: A Systematic Review and Meta-analysis. JAMA 2019, 321, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Sprynger, M.; Rigo, F.; Moonen, M.; Aboyans, V.; Edvardsen, T.; de Alcantara, M.L.; Brodmann, M.; Naka, K.K.; Kownator, S.; Simova, I.; et al. Focus on echovascular imaging assessment of arterial disease: Complement to the ESC guidelines (PARTIM 1) in collaboration with the Working Group on Aorta and Peripheral Vascular Diseases. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 1195–1221. [Google Scholar] [CrossRef] [PubMed]
- Nambi, V.; Chambless, L.; Folsom, A.R.; He, M.; Hu, Y.; Mosley, T.; Volcik, K.; Boerwinkle, E.; Ballantyne, C.M. Carotid intima-media thickness and presence or absence of plaque improves prediction of coronary heart disease risk: The ARIC (Atherosclerosis Risk in Communities) study. J. Am. Coll. Cardiol. 2010, 55, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Côté, R.; Battista, R.N.; Abrahamowicz, M.; Langlois, Y.; Bourque, F.; Mackey, A. Lack of effect of aspirin in asymptomatic patients with carotid bruits and substantial carotid narrowing. The Asymptomatic Cervical Bruit Study Group. Ann. Intern. Med. 1995, 123, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Mills, K.; Romero, J.; Li, Y.; Hu, Z.; Cao, Y.; Huang, H.; Xu, Y.; Jiang, L. Comparative effects of lipid lowering, hypoglycemic, antihypertensive and antiplatelet medications on carotid artery intima-media thickness progression: A network meta-analysis. Cardiovasc. Diabetol. 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett Brendan, M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. Mass. Med. Soc. 2017, 377, 1119–1131. [Google Scholar]
- Nidorf, S.M.; Fiolet, A.T.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.; Salem, H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. New Engl. J. Med. Mass. Med. Soc. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Tardif, J.; Kouz, S.; Waters, D.; Bertrand, O.; Diaz, R.; Maggioni, A.; Pinto, F.; Ibrahim, R.; Gamra, H.; Kiwan, G.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. Mass. Med. Soc. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.C.; Rothman, A.M.K.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.C.; Quinn, S.; Nasis, A.; Hiew, C.; Roberts-Thomson, P.; Adams, H.; Sriamareswaran, R.; Htun, N.M.; Wilson, W.; Stub, D.; et al. Colchicine in Patients with Acute Coronary Syndrome. Circulation 2020, 142, 1890–1900. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Patient Category | Prevention Goals (STEP 1) | Intensified/Additional Prevention Goals (STEP 2) |
|---|---|---|
| Apparently healthy persons | Stop smoking and lifestyle SBP < 140 to 130 mmHg LDL-C < 2.6 mmol/L (100 mg/dL) | SBP < 130 mmHg LDL-C < 1.8 mmol/L (70 mg/dL) and >50% reduction in high-risk patients LDL-C < 1.4 mmol/L (55 mg/dL) and >50% reduction in very-high-risk patients |
| Patients with CKD | Stop smoking and lifestyle optimization SBP < 140 down to 130 mmHg LDL-C < 2.6 mmol/L (100 mg/dL) and >50% LDL-C reduction Otherwise according to ASCVD and DM history | LDL-C < 1.8 mmol/L (70 mg/dL) in high-risk patients and <1.4 mmol/L (55 mg/dL) in very-high risk patients |
| Patients with FH | Stop smoking and lifestyle optimization SBP < 140 down to 130 mmHg LDL-C < 2.6 mmol/L (100 mg/dL) and >50% LDL-C Reduction. Otherwise according to ASCVD and DM history | LDL-C < 1.8 mmol/L (70 mg/dL) in high-risk patients and <1.4 mmol/L (55 mg/dL) in very-high risk patients |
| Patients with type 2 DM without established ASCVD or severe TOD | Stop smoking and lifestyle optimization SBP < 140 down to 130 mmHg LDL-C < 2.6 mmol/L (100 mg/dL) HbA1c < 53 mmol/mol (7.0%) | SBP < 130 mmHg LDL-C < 1.8 mmol/L (70 mg/dL) and >50% reduction SGLT2 inhibitor or GLP-1RA |
| Patients with type 2 DM with established ASCVD or severe TOD | Stop smoking and lifestyle optimization SBP < 140 down to 130 mmHg LDL-C < 1.8 mmol/L (70 mg/dL) HbA1c < 64 mmol/mol (8.0%) SGLT2 inhibitor or GLP1-RA CVD: antiplatelet therapy | SBP < 130 mmHg LDL-C < 1.4 mmol/L (55 mg/dL) and >50% reduction SGLT2 inhibitor or GLP-1RA if not already on May additionally consider novel upcoming treatments: DAPT, dual pathway inhibition, colchicine, icosapent ethyl, etc. |
| Patients with established ASCVD | Stop smoking and lifestyle optimization SBP < 140 down to 130 mmHg Intensive oral lipid-lowering therapy aiming at >50% LDL-C reduction and LDL-C < 1.8 mmol/L (70 mg/dL) Antiplatelet therapy | SBP < 130 mmHg LDL-C < 1.4 mmol/L (55 mg/dL) May additionally consider novel upcoming treatments: DAPT, dual pathway inhibition, colchicine, icosapent ethyl, etc. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madaudo, C.; Coppola, G.; Parlati, A.L.M.; Corrado, E. Discovering Inflammation in Atherosclerosis: Insights from Pathogenic Pathways to Clinical Practice. Int. J. Mol. Sci. 2024, 25, 6016. https://doi.org/10.3390/ijms25116016
Madaudo C, Coppola G, Parlati ALM, Corrado E. Discovering Inflammation in Atherosclerosis: Insights from Pathogenic Pathways to Clinical Practice. International Journal of Molecular Sciences. 2024; 25(11):6016. https://doi.org/10.3390/ijms25116016
Chicago/Turabian StyleMadaudo, Cristina, Giuseppe Coppola, Antonio Luca Maria Parlati, and Egle Corrado. 2024. "Discovering Inflammation in Atherosclerosis: Insights from Pathogenic Pathways to Clinical Practice" International Journal of Molecular Sciences 25, no. 11: 6016. https://doi.org/10.3390/ijms25116016