
Distinct Responses to Menin Inhibition and Synergy with DOT1L Inhibition in KMT2A-Rearranged Acute Lymphoblastic and Myeloid Leukemia
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
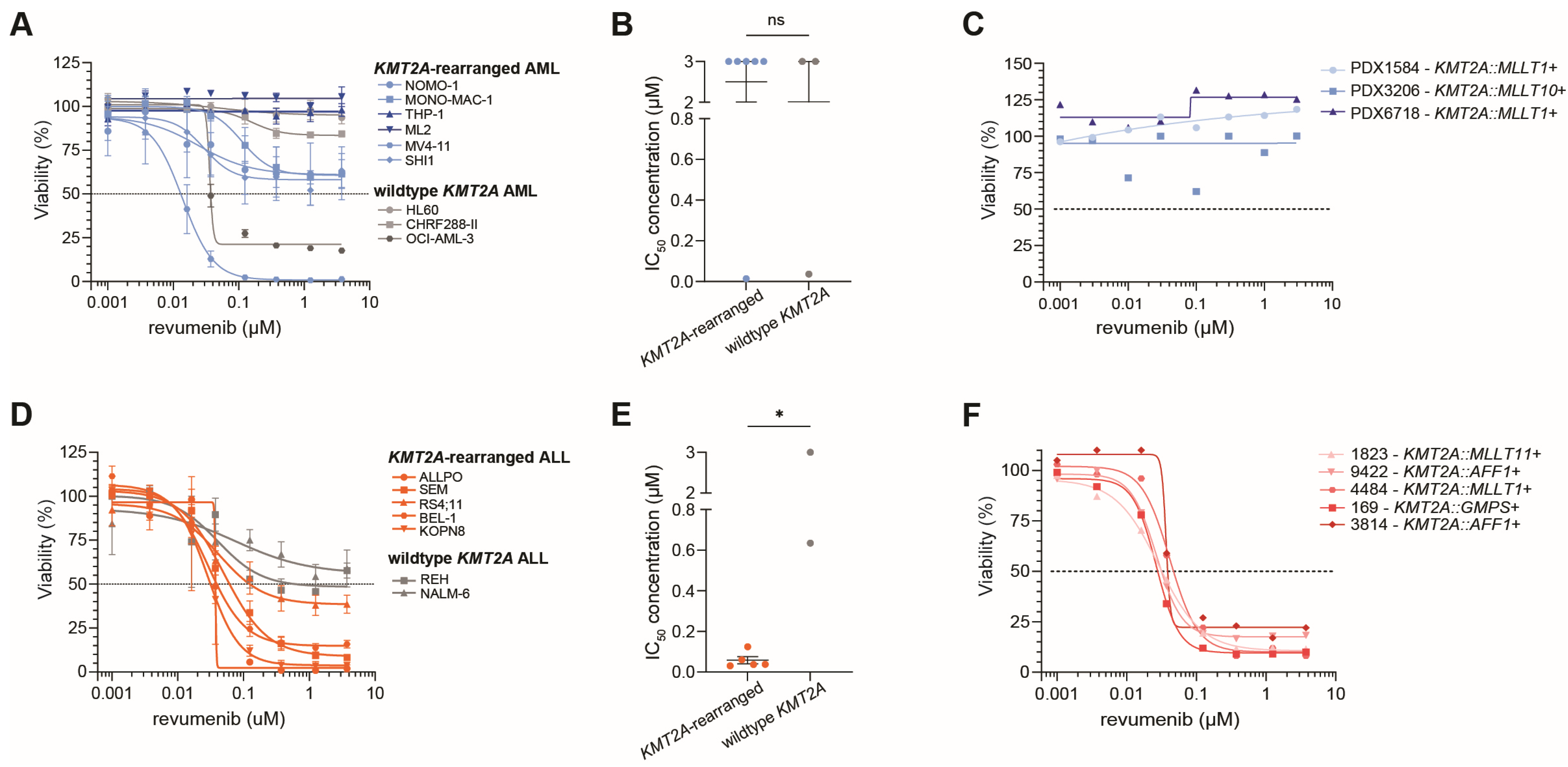
2.1. Diverse Responses to Revumenib in KMT2A-Rearranged ALL and AML Samples
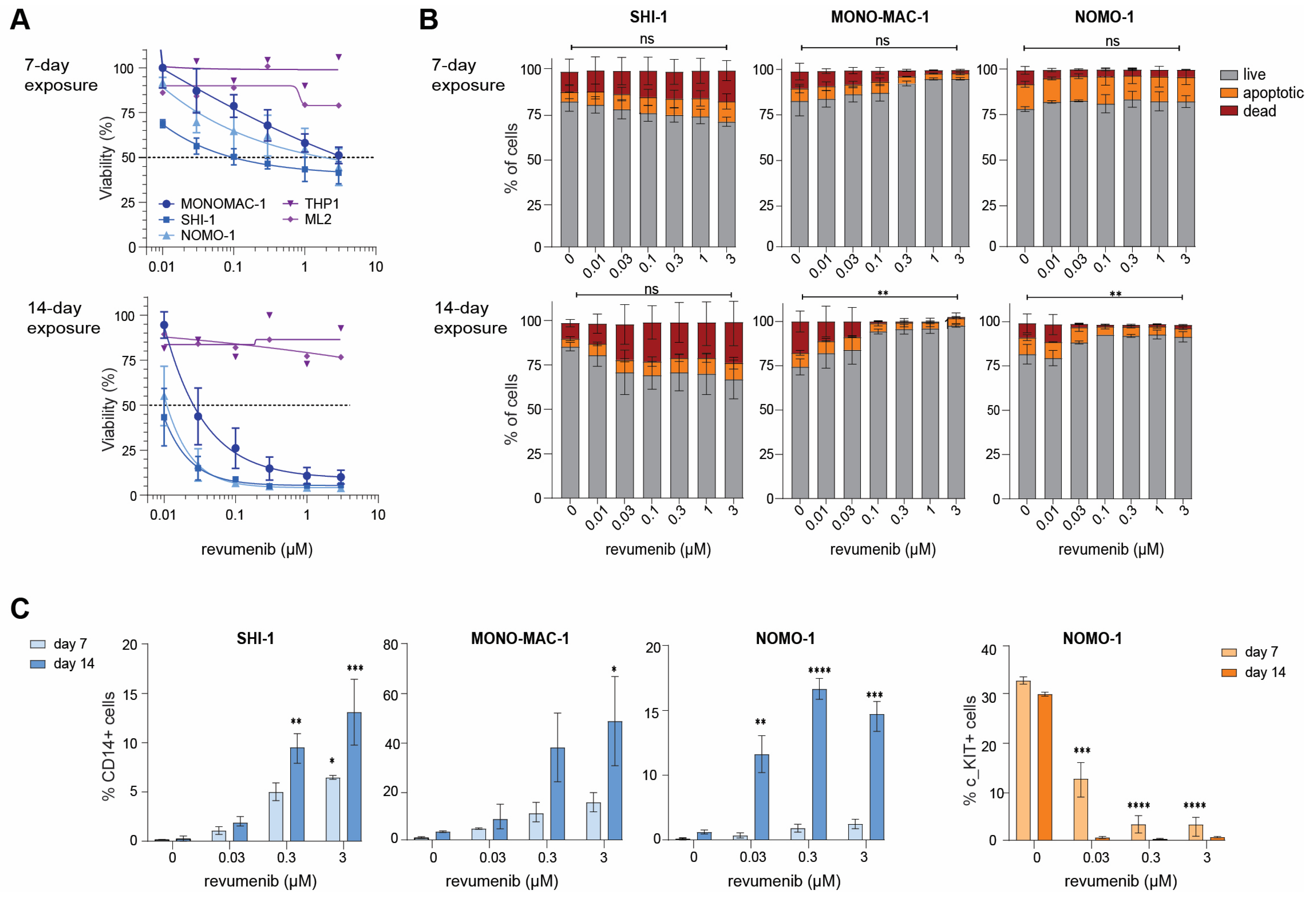
2.2. Delayed Revumenib Responses and Differentiation in KMT2A-Rearranged AML Cells
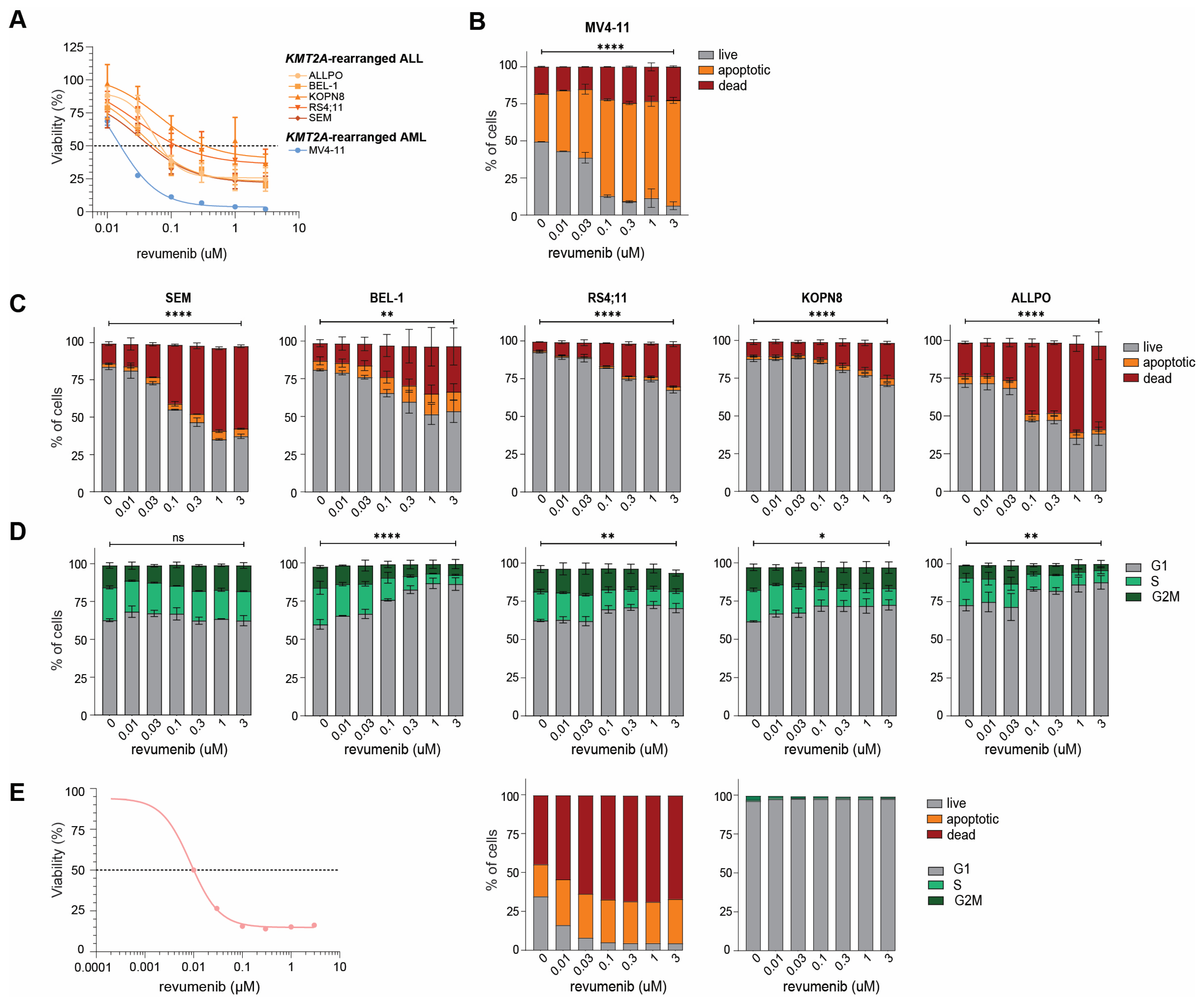
2.3. Rapid Apoptosis Induction in Response to Revumenib in KMT2A-Rearranged ALL Cells
2.4. Acquired Resistance to Revumenib in KMT2A-Rearranged ALL Cells
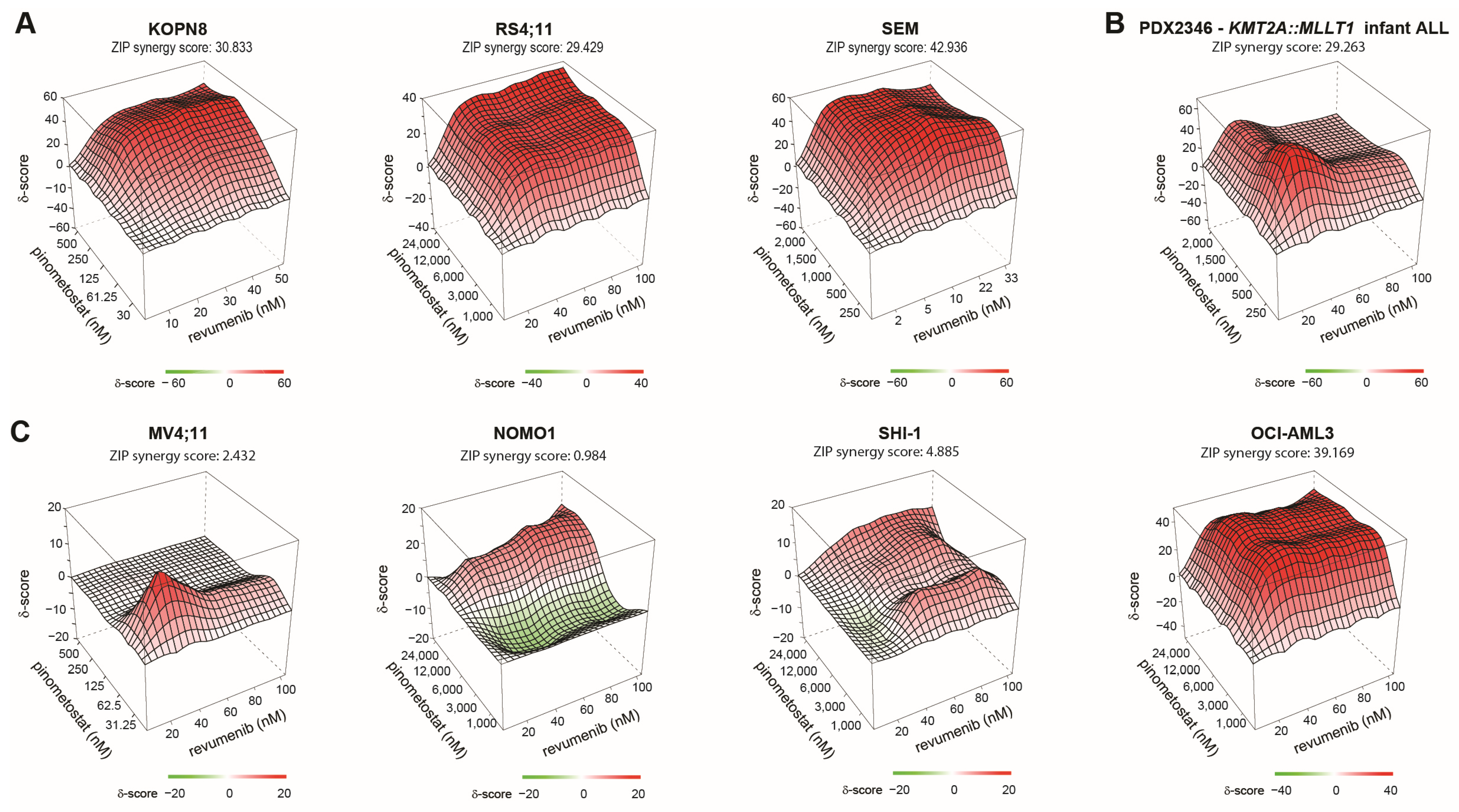
2.5. Synergy between Revumenib and Pinometostat in KMT2A-Rearranged ALL
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Cell Line Models
4.3. Therapeutic Agents and Viability Assays
4.4. In Vitro Exposure to Revumenib and Trypan Blue Exclusion
4.5. Establishment of Acquired Revumenib Resistance in SEM Cells
4.6. Flow Cytometry (FACS) Analyses
4.7. Drug Synergy Experiments
4.8. Menin Mutation Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pieters, R.; Mullighan, C.G.; Hunger, S.P. Advancing Diagnostics and Therapy to Reach Universal Cure in Childhood ALL. J. Clin. Oncol. 2023, 41, 5579–5591. [Google Scholar] [CrossRef] [PubMed]
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- van Weelderen, R.E.; Klein, K.; Harrison, C.J.; Jiang, Y.; Abrahamsson, J.; Arad-Cohen, N.; Bart-Delabesse, E.; Buldini, B.; De Moerloose, B.; Dworzak, M.N.; et al. Measurable Residual Disease and Fusion Partner Independently Predict Survival and Relapse Risk in Childhood KMT2A-Rearranged Acute Myeloid Leukemia: A Study by the International Berlin-Frankfurt-Münster Study Group. J. Clin. Oncol. 2023, 41, 2963–2974. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; De Lorenzo, P.; Ancliffe, P.; Aversa, L.A.; Brethon, B.; Biondi, A.; Campbell, M.; Escherich, G.; Ferster, A.; Gardner, R.A.; et al. Outcome of Infants Younger Than 1 Year With Acute Lymphoblastic Leukemia Treated With the Interfant-06 Protocol: Results From an International Phase III Randomized Study. J. Clin. Oncol. 2019, 37, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- van der Sluis, I.M.; de Lorenzo, P.; Kotecha, R.S.; Attarbaschi, A.; Escherich, G.; Nysom, K.; Stary, J.; Ferster, A.; Brethon, B.; Locatelli, F.; et al. Blinatumomab Added to Chemotherapy in Infant Lymphoblastic Leukemia. N. Engl. J. Med. 2023, 388, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Murai, M.J.; Chruszcz, M.; Reddy, G.; Grembecka, J.; Cierpicki, T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem. 2011, 286, 31742–31748. [Google Scholar] [CrossRef]
- Bernt, K.M.; Armstrong, S.A. A role for DOT1L in MLL-rearranged leukemias. Epigenomics 2011, 3, 667–670. [Google Scholar] [CrossRef]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Feng, Z.; Lemieux, M.E.; Faber, J.; Vempati, S.; Sinha, A.U.; Xia, X.; Jesneck, J.; Bracken, A.P.; Silverman, L.B.; et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14, 355–368. [Google Scholar] [CrossRef]
- Yokoyama, A.; Somervaille, T.C.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef]
- Xu, Y.; Yue, L.; Wang, Y.; Xing, J.; Chen, Z.; Shi, Z.; Liu, R.; Liu, Y.C.; Luo, X.; Jiang, H.; et al. Discovery of Novel Inhibitors Targeting the Menin-Mixed Lineage Leukemia Interface Using Pharmacophore- and Docking-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1847–1855. [Google Scholar] [CrossRef]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Aguilar, A.; Huang, L.; Xu, T.; Zheng, K.; McEachern, D.; Przybranowski, S.; Foster, C.; Zawacki, K.; Liu, Z.; et al. Discovery of M-808 as a Highly Potent, Covalent, Small-Molecule Inhibitor of the Menin-MLL Interaction with Strong In Vivo Antitumor Activity. J. Med. Chem. 2020, 63, 4997–5010. [Google Scholar] [CrossRef]
- Zhang, M.; Aguilar, A.; Xu, S.; Huang, L.; Chinnaswamy, K.; Sleger, T.; Wang, B.; Gross, S.; Nicolay, B.N.; Ronseaux, S.; et al. Discovery of M-1121 as an Orally Active Covalent Inhibitor of Menin-MLL Interaction Capable of Achieving Complete and Long-Lasting Tumor Regression. J. Med. Chem. 2021, 64, 10333–10349. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Zheng, K.; Xu, T.; Xu, S.; Huang, L.; Fernandez-Salas, E.; Liu, L.; Bernard, D.; Harvey, K.P.; Foster, C.; et al. Structure-Based Discovery of M-89 as a Highly Potent Inhibitor of the Menin-Mixed Lineage Leukemia (Menin-MLL) Protein-Protein Interaction. J. Med. Chem. 2019, 62, 6015–6034. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673.e611. [Google Scholar] [CrossRef] [PubMed]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Boettcher, S.; Daver, N.; Mill, C.P.; Sasaki, K.; Birdwell, C.E.; Davis, J.A.; Takahashi, K.; Kadia, T.M.; DiNardo, C.D.; et al. Effective Menin inhibitor-based combinations against AML with MLL rearrangement or NPM1 mutation (NPM1c). Blood Cancer J. 2022, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Tao, W.; Mak, P.Y.; Ostermann, L.B.; Mak, D.; McGeehan, G.; Ordentlich, P.; Andreeff, M. Menin inhibition decreases Bcl-2 and synergizes with venetoclax in NPM1/FLT3-mutated AML. Blood 2021, 138, 1637–1641. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Daver, N.; Boettcher, S.; Mill, C.P.; Sasaki, K.; Birdwell, C.E.; Davis, J.A.; Das, K.; Takahashi, K.; Kadia, T.M.; et al. Activity of menin inhibitor ziftomenib (KO-539) as monotherapy or in combinations against AML cells with MLL1 rearrangement or mutant NPM1. Leukemia 2022, 36, 2729–2733. [Google Scholar] [CrossRef]
- Dzama, M.M.; Steiner, M.; Rausch, J.; Sasca, D.; Schönfeld, J.; Kunz, K.; Taubert, M.C.; McGeehan, G.M.; Chen, C.W.; Mupo, A.; et al. Synergistic targeting of FLT3 mutations in AML via combined menin-MLL and FLT3 inhibition. Blood 2020, 136, 2442–2456. [Google Scholar] [CrossRef]
- Tubío-Santamaría, N.; Jayavelu, A.K.; Schnoeder, T.M.; Eifert, T.; Hsu, C.J.; Perner, F.; Zhang, Q.; Wenge, D.V.; Hansen, F.M.; Kirkpatrick, J.M.; et al. Immunoproteasome function maintains oncogenic gene expression in KMT2A-complex driven leukemia. Mol. Cancer 2023, 22, 196. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature 2023, 615, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; Issa, G.C.; Thirman, M.; DiPersio, J.; Arellano, M.; Blachly, J.S.; Mannis, G.N.; Perl, A.; Dickens, D.S.; McMahon, C.M.; et al. Revumenib Monotherapy in Patients with Relapsed/Refractory KMT2Ar Acute Leukemia: Topline Efficacy and Safety Results from the Pivotal Augment-101 Phase 2 Study. Blood 2023, 142, LBA-5. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature 2023, 615, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Kühn, M.W.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer Discov. 2016, 6, 1166–1181. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Tallman, M.S. Mixed lineage rearranged leukaemia: Pathogenesis and targeting DOT1L. Curr. Opin. Hematol. 2015, 22, 92–96. [Google Scholar] [CrossRef]
- Perner, F.; Armstrong, S.A. Targeting Chromatin Complexes in Myeloid Malignancies and Beyond: From Basic Mechanisms to Clinical Innovation. Cells 2020, 9, 2721. [Google Scholar] [CrossRef]
- Dafflon, C.; Craig, V.J.; Méreau, H.; Gräsel, J.; Schacher Engstler, B.; Hoffman, G.; Nigsch, F.; Gaulis, S.; Barys, L.; Ito, M.; et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 2017, 31, 1269–1277. [Google Scholar] [CrossRef]
- Perner, F.; Gadrey, J.Y.; Xiong, Y.; Hatton, C.; Eschle, B.K.; Weiss, A.; Stauffer, F.; Gaul, C.; Tiedt, R.; Perry, J.A.; et al. Novel inhibitors of the histone methyltransferase DOT1L show potent antileukemic activity in patient-derived xenografts. Blood 2020, 136, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, H.; Stauffer, F.; Caravatti, G.; Vollmer, S.; Machauer, R.; Holzer, P.; Möbitz, H.; Scheufler, C.; Klumpp, M.; et al. Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Med. Chem. Lett. 2016, 7, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Numata, M.; Haginoya, N.; Shiroishi, M.; Hirata, T.; Sato-Otsubo, A.; Yoshikawa, K.; Takata, Y.; Nagase, R.; Kashimoto, Y.; Suzuki, M.; et al. A novel Menin-MLL1 inhibitor, DS-1594a, prevents the progression of acute leukemia with rearranged MLL1 or mutated NPM1. Cancer Cell Int. 2023, 23, 36. [Google Scholar] [CrossRef] [PubMed]
- Bardini, M.; Trentin, L.; Rizzo, F.; Vieri, M.; Savino, A.M.; Garrido Castro, P.; Fazio, G.; Van Roon, E.H.J.; Kerstjens, M.; Smithers, N.; et al. Antileukemic Efficacy of BET Inhibitor in a Preclinical Mouse Model of MLL-AF4(+) Infant ALL. Mol. Cancer Ther. 2018, 17, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Gilan, O.; Lam, E.Y.; Becher, I.; Lugo, D.; Cannizzaro, E.; Joberty, G.; Ward, A.; Wiese, M.; Fong, C.Y.; Ftouni, S.; et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat. Struct. Mol. Biol. 2016, 23, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; Schrappe, M.; De Lorenzo, P.; Hann, I.; De Rossi, G.; Felice, M.; Hovi, L.; LeBlanc, T.; Szczepanski, T.; Ferster, A.; et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): An observational study and a multicentre randomised trial. Lancet 2007, 370, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Kaspers, G.J.; Veerman, A.J.; Pieters, R.; Broekema, G.J.; Huismans, D.R.; Kazemier, K.M.; Loonen, A.H.; Rottier, M.A.; van Zantwijk, C.H.; Hählen, K.; et al. Mononuclear cells contaminating acute lymphoblastic leukaemic samples tested for cellular drug resistance using the methyl-thiazol-tetrazolium assay. Br. J. Cancer 1994, 70, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, A.; Di Berardino, C.; Scanziani, E.; Garofalo, A.; Rivolta, A.; Fontana, G.; Rambaldi, A.; Giavazzi, R.; Biondi, A. A human acute lymphoblastic leukemia line with the T(4;11) translocation as a model of minimal residual disease in SCID mice. Leuk. Res. 1997, 21, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; Loonen, A.H.; Huismans, D.R.; Broekema, G.J.; Dirven, M.W.; Heyenbrok, M.W.; Hählen, K.; Veerman, A.J. In vitro drug sensitivity of cells from children with leukemia using the MTT assay with improved culture conditions. Blood 1990, 76, 2327–2336. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adriaanse, F.R.S.; Schneider, P.; Arentsen-Peters, S.T.C.J.M.; Fonseca, A.M.N.d.; Stutterheim, J.; Pieters, R.; Zwaan, C.M.; Stam, R.W. Distinct Responses to Menin Inhibition and Synergy with DOT1L Inhibition in KMT2A-Rearranged Acute Lymphoblastic and Myeloid Leukemia. Int. J. Mol. Sci. 2024, 25, 6020. https://doi.org/10.3390/ijms25116020
Adriaanse FRS, Schneider P, Arentsen-Peters STCJM, Fonseca AMNd, Stutterheim J, Pieters R, Zwaan CM, Stam RW. Distinct Responses to Menin Inhibition and Synergy with DOT1L Inhibition in KMT2A-Rearranged Acute Lymphoblastic and Myeloid Leukemia. International Journal of Molecular Sciences. 2024; 25(11):6020. https://doi.org/10.3390/ijms25116020
Chicago/Turabian StyleAdriaanse, Fabienne R. S., Pauline Schneider, Susan T. C. J. M. Arentsen-Peters, Ana M. Neves da Fonseca, Janine Stutterheim, Rob Pieters, C. Michel Zwaan, and Ronald W. Stam. 2024. "Distinct Responses to Menin Inhibition and Synergy with DOT1L Inhibition in KMT2A-Rearranged Acute Lymphoblastic and Myeloid Leukemia" International Journal of Molecular Sciences 25, no. 11: 6020. https://doi.org/10.3390/ijms25116020
APA StyleAdriaanse, F. R. S., Schneider, P., Arentsen-Peters, S. T. C. J. M., Fonseca, A. M. N. d., Stutterheim, J., Pieters, R., Zwaan, C. M., & Stam, R. W. (2024). Distinct Responses to Menin Inhibition and Synergy with DOT1L Inhibition in KMT2A-Rearranged Acute Lymphoblastic and Myeloid Leukemia. International Journal of Molecular Sciences, 25(11), 6020. https://doi.org/10.3390/ijms25116020