
Exploring the Complexity and Promise of Tumor Immunotherapy in Drug Development
Abstract
1. Introduction
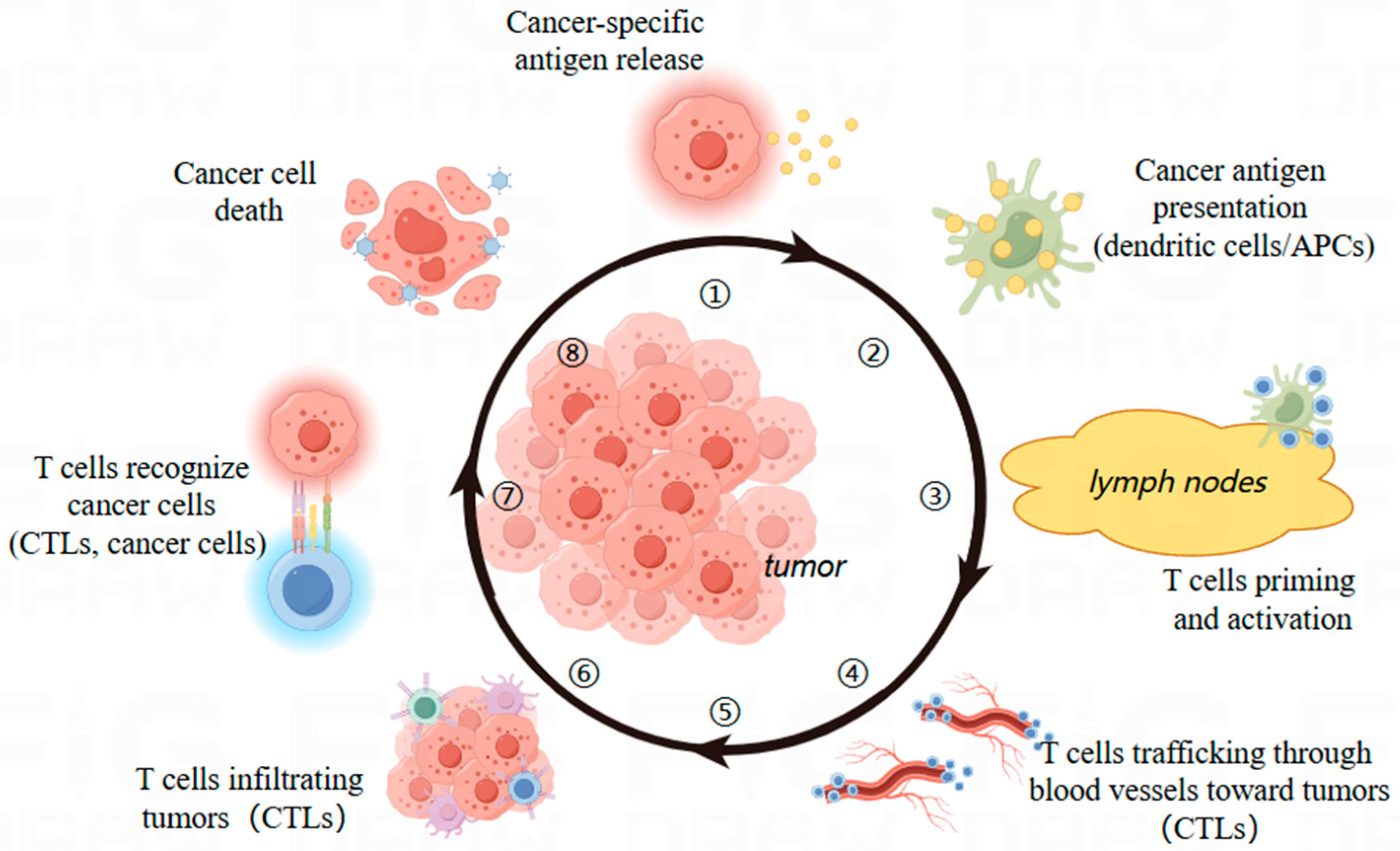
2. Immunotherapy: Booming in Tumor Treatment
2.1. Definition and Classification of Immunotherapy
2.2. Dynamics of T Cells in Immunotherapy
2.3. Advantages of Immunotherapy
3. Immunotherapy via Diverse Mechanisms
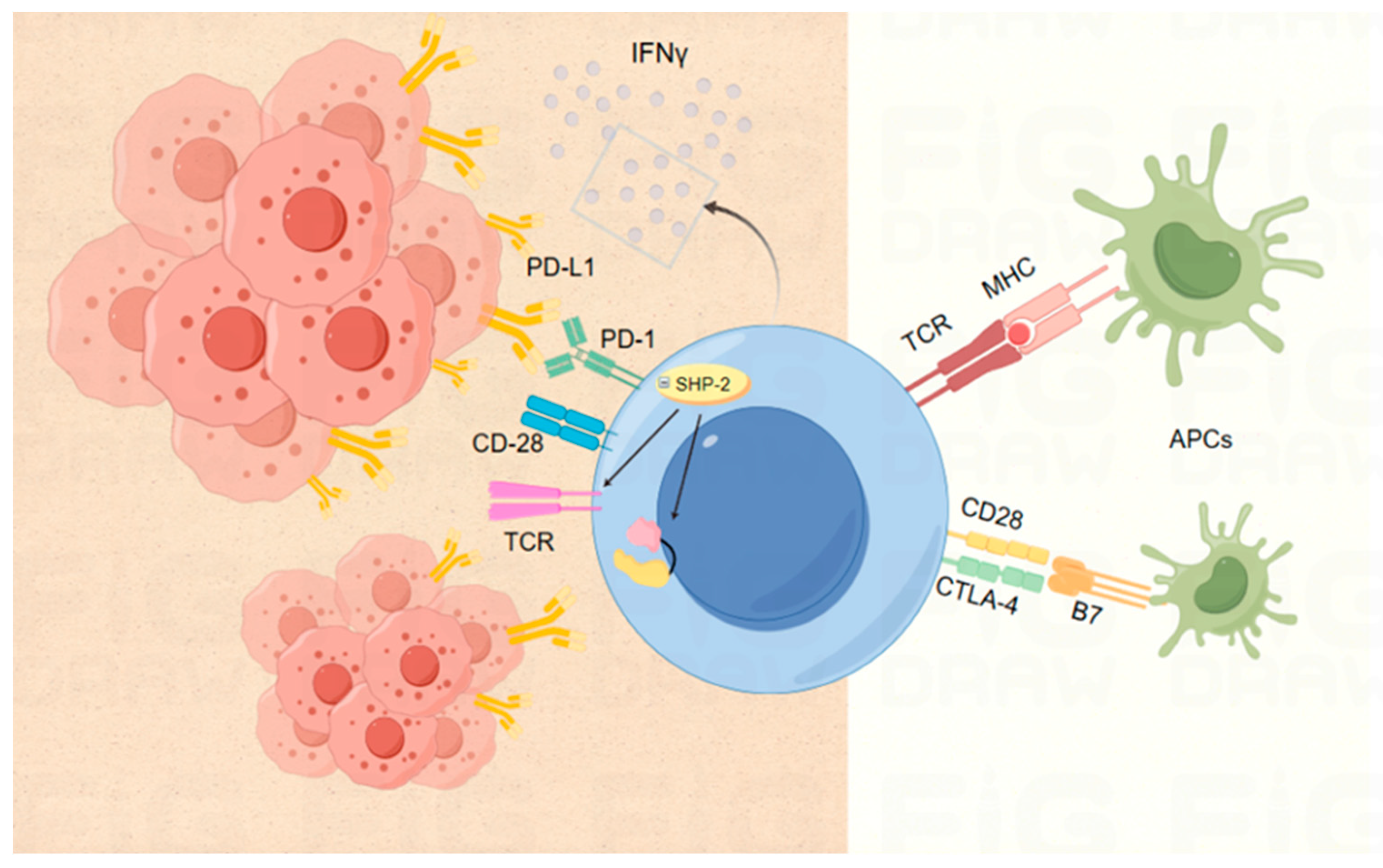
3.1. ICIs Based on Various Targets
3.1.1. Targets PD-1 and PD-L1
3.1.2. Target CTLA-4
3.1.3. Co-stimulation or Co-inhibition of Other Immune Checkpoints
3.1.4. Double Immunotherapy
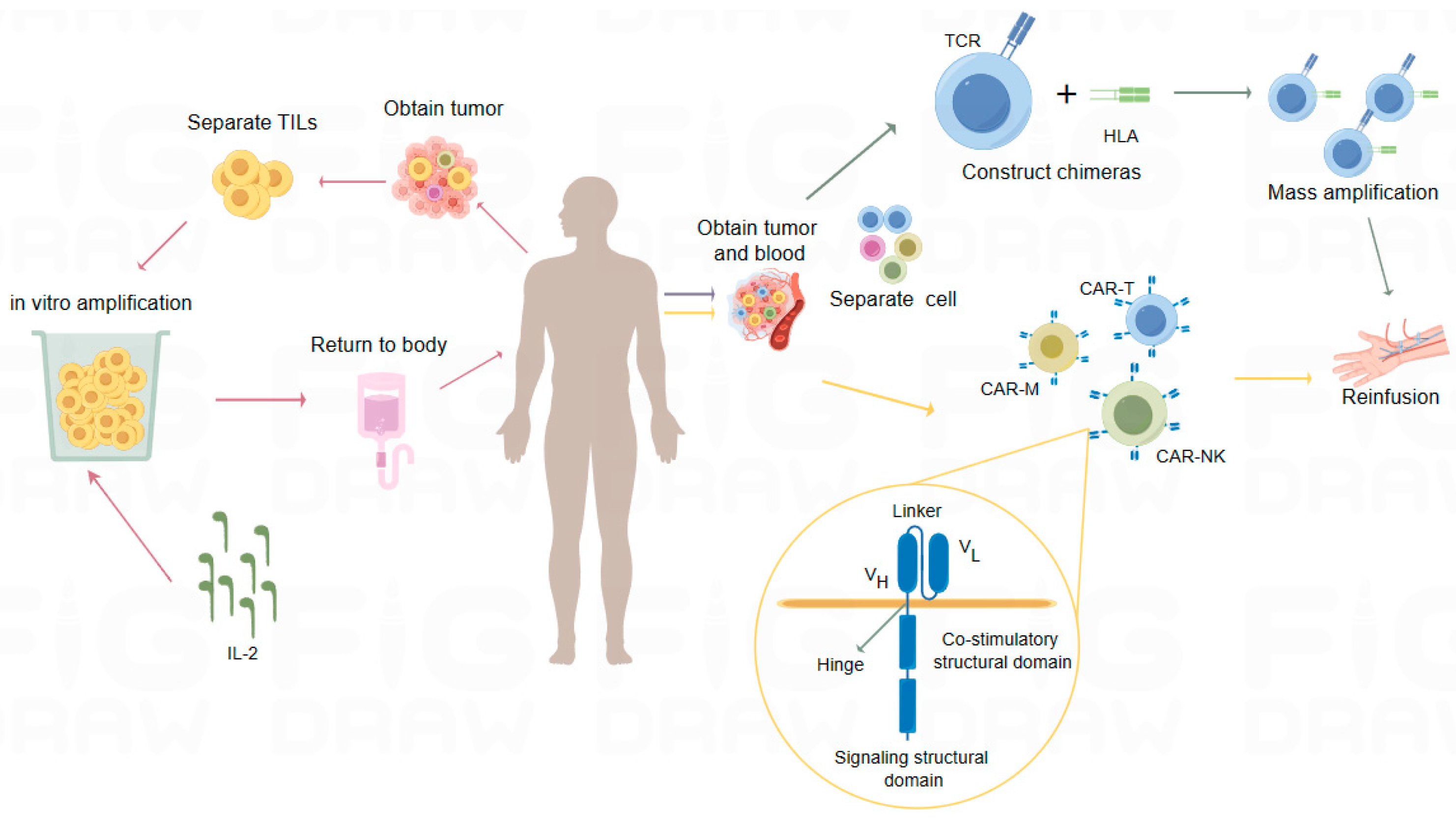
3.2. Adoptive Cell Therapy (ACT)
3.2.1. TILs
3.2.2. T-Cell Receptor Chimeric T-Cell Therapy (TCR-T)
3.2.3. Cell Therapy Based on CAR Technology
3.3. Cytokine Therapy
3.4. Cancer Vaccines
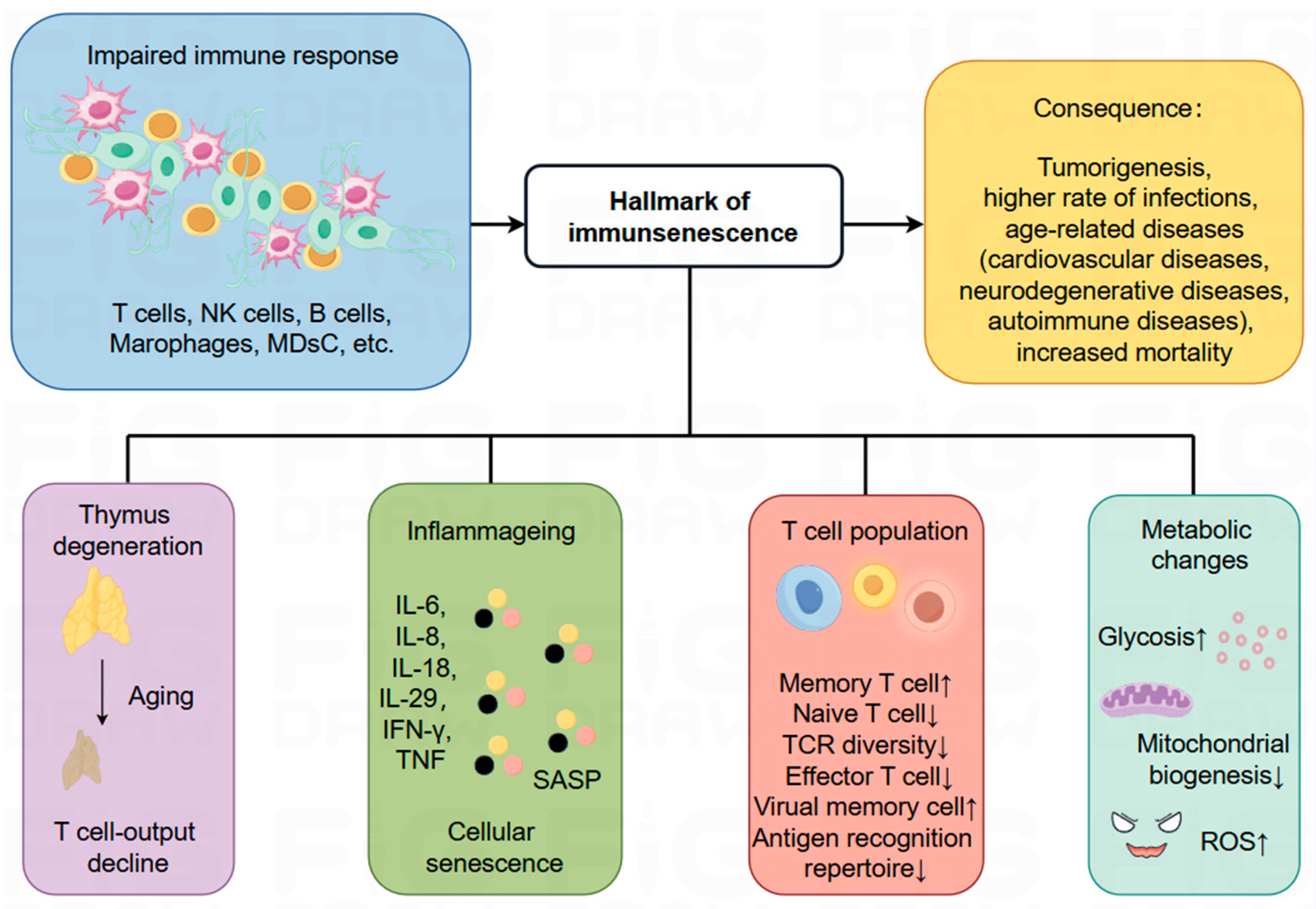
4. A Key Player in Influencing the Development and Treatment of Cancer: Immunosenescence
4.1. The Process of Immunosenescence and Related Markers
4.2. Immunosenescence and Cancer
4.3. Impact of Immune Senescence on Tumor Immunotherapy
4.4. Therapeutic Strategies for Immune Aging
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CD66a | Carcino-Embryonic Antigen-related Cellular Adhesion Molecule1, CEACAM1 |
| Galectin-9 | Lectin, Galactoside-Binding, Soluble 9 |
| HMGB1 | High-Mobility Group Box 1 |
| PS | Phosphatidylserine |
References
- Ibrahim, A.; El Baldi, M.; Mohammed, S.; El Rhazi, K.; Benazzouz, B. Cancer statistics in Yemen: Incidence and mortality, in 2020. BMC Public Health 2024, 24, 962. [Google Scholar] [CrossRef]
- Wilkes, G.M. Targeted Therapy: Attacking Cancer with Molecular and Immunological Targeted Agents. Asia Pac. J. Oncol. Nurs. 2018, 5, 137–155. [Google Scholar] [CrossRef]
- Wujcik, D. Science and mechanism of action of targeted therapies in cancer treatment. Semin. Oncol. Nurs. 2014, 30, 139–146. [Google Scholar] [CrossRef]
- Tavare, A.N.; Perry, N.J.; Benzonana, L.L.; Takata, M.; Ma, D. Cancer recurrence after surgery: Direct and indirect effects of anesthetic agents. Int. J. Cancer 2012, 130, 1237–1250. [Google Scholar] [CrossRef]
- Manna, P.; Dewanjee, S.; Joardar, S.; Chakraborty, P.; Bhattacharya, H.; Bhanja, S.; Bhattacharyya, C.; Bhowmik, M.; Bhowmick, S.; Saha, A.; et al. Carnosic acid attenuates doxorubicin-induced cardiotoxicity by decreasing oxidative stress and its concomitant pathological consequences. Food Chem. Toxicol. 2022, 166, 113205, Erratum in Food Chem. Toxicol. 2023, 181, 114099. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Masoud, V.; Pagès, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef]
- Luo, Y.; Sun, X.; Huang, L.; Yan, J.; Yu, B.Y.; Tian, J. Artemisinin-Based Smart Nanomedicines with Self-Supply of Ferrous Ion to Enhance Oxidative Stress for Specific and Efficient Cancer Treatment. ACS Appl. Mater. Interfaces 2019, 11, 29490–29497. [Google Scholar] [CrossRef]
- Ai, L.J.; Li, G.D.; Chen, G.; Sun, Z.Q.; Zhang, J.N.; Liu, M. Molecular subtyping and the construction of a predictive model of colorectal cancer based on ion channel genes. Eur. J. Med. Res. 2024, 29, 219. [Google Scholar] [CrossRef]
- Shahid, K.; Khalife, M.; Dabney, R.; Phan, A.T. Immunotherapy and targeted therapy-the new roadmap in cancer treatment. Ann. Transl. Med. 2019, 7, 595. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chen, C.; Ke, J.; Zhou, X.E.; Yi, W.; Brunzelle, J.S.; Li, J.; Yong, E.L.; Xu, H.E.; Melcher, K. Structural basis for molecular recognition of folic acid by folate receptors. Nature 2013, 500, 486–489. [Google Scholar] [CrossRef]
- Han, S.; Wu, J. Three-dimensional (3D) scaffolds as powerful weapons for tumor immunotherapy. Bioact. Mater. 2022, 17, 300–319. [Google Scholar] [CrossRef]
- Hamilton, P.T.; Anholt, B.R.; Nelson, B.H. Tumour immunotherapy: Lessons from predator-prey theory. Nat. Rev. Immunol. 2022, 22, 765–775. [Google Scholar] [CrossRef]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. Aaps j 2021, 23, 39. [Google Scholar] [CrossRef]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef]
- Xiang, M.; Li, H.; Zhan, Y.; Ma, D.; Gao, Q.; Fang, Y. Functional CRISPR screens in T cells reveal new opportunities for cancer immunotherapies. Mol. Cancer 2024, 23, 73. [Google Scholar] [CrossRef]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Lu, H.; Li, J.; Yan, X.; Xiao, M.; Hao, J.; Alekseev, A.; Khong, H.; Chen, T.; et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 2019, 567, 525–529. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef]
- Kamdar, M.; Solomon, S.R.; Arnason, J.; Johnston, P.B.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): Results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet 2022, 399, 2294–2308. [Google Scholar] [CrossRef]
- Dang, Q.; Li, B.; Jin, B.; Ye, Z.; Lou, X.; Wang, T.; Wang, Y.; Pan, X.; Hu, Q.; Li, Z.; et al. Cancer immunometabolism: Advent, challenges, and perspective. Mol. Cancer 2024, 23, 72. [Google Scholar] [CrossRef]
- Yang, L.; Ning, Q.; Tang, S.S. Recent Advances and Next Breakthrough in Immunotherapy for Cancer Treatment. J. Immunol. Res. 2022, 2022, 8052212. [Google Scholar] [CrossRef]
- Darvishi, M.; Tosan, F.; Nakhaei, P.; Manjili, D.A.; Kharkouei, S.A.; Alizadeh, A.; Ilkhani, S.; Khalafi, F.; Zadeh, F.A.; Shafagh, S.G. Recent progress in cancer immunotherapy: Overview of current status and challenges. Pathol. Res. Pract. 2023, 241, 154241. [Google Scholar] [CrossRef]
- Tjader, N.P.; Toland, A.E. Immunotherapy for colorectal cancer: Insight from inherited genetics. Trends Cancer 2024, 10, 444–456. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Rivoltini, L.; Carrabba, M.; Huber, V.; Castelli, C.; Novellino, L.; Dalerba, P.; Mortarini, R.; Arancia, G.; Anichini, A.; Fais, S.; et al. Immunity to cancer: Attack and escape in T lymphocyte-tumor cell interaction. Immunol. Rev. 2002, 188, 97–113. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Matteson, E.L.; Moder, K.G.; Flies, D.B.; Zhu, G.; Tamura, H.; Driscoll, C.L.; Chen, L. Costimulating aberrant T cell responses by B7-H1 autoantibodies in rheumatoid arthritis. J. Clin. Investig. 2003, 111, 363–370. [Google Scholar] [CrossRef]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008, 111, 3635–3643. [Google Scholar] [CrossRef]
- Harris, N.L.; Ronchese, F. The role of B7 costimulation in T-cell immunity. Immunol. Cell Biol. 1999, 77, 304–311. [Google Scholar] [CrossRef]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef]
- Riley, J.L.; Mao, M.; Kobayashi, S.; Biery, M.; Burchard, J.; Cavet, G.; Gregson, B.P.; June, C.H.; Linsley, P.S. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 11790–11795. [Google Scholar] [CrossRef]
- Egen, J.G.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 2002, 16, 23–35. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Sansom, D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2022, 185, 576. [Google Scholar] [CrossRef] [PubMed]
- de Mingo Pulido, Á.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74.e66. [Google Scholar] [CrossRef] [PubMed]
- Andreae, S.; Piras, F.; Burdin, N.; Triebel, F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223). J. Immunol. 2002, 168, 3874–3880. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Andrews, L.P.; Workman, C.J.; Vignali, D.A.A. Intratumoral regulatory T cells: Markers, subsets and their impact on anti-tumor immunity. Immunology 2019, 157, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Chocarro, L.; Bocanegra, A.; Blanco, E.; Fernández-Rubio, L.; Arasanz, H.; Echaide, M.; Garnica, M.; Ramos, P.; Piñeiro-Hermida, S.; Vera, R.; et al. Cutting-Edge: Preclinical and Clinical Development of the First Approved Lag-3 Inhibitor. Cells 2022, 11, 2351. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Fu, T.; He, Q.; Sharma, P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011, 71, 5445–5454. [Google Scholar] [CrossRef]
- Gray, C.C.; Armstead, B.E.; Chung, C.S.; Chen, Y.; Ayala, A. VISTA Non-redundantly Regulates Proliferation and CD69low γδ T cell Accumulation in the Intestine in Murine Sepsis. J. Leukoc. Biol. 2023, 115, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Met, Ö.; Jensen, K.M.; Chamberlain, C.A.; Donia, M.; Svane, I.M. Principles of adoptive T cell therapy in cancer. Semin. Immunopathol. 2019, 41, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Ohshita, A.; Kawabuchi, Y.; Ohta, K.; Shimizu, K.; Minami, K.; Hihara, J.; Miyahara, E.; Toge, T. Adoptive immunotherapy of cancer using activated autologous lymphocytes—Current status and new strategies. Hum. Cell 2003, 16, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef] [PubMed]
- Shirakashi, M.; Maruya, M.; Hirota, K.; Tsuruyama, T.; Matsuo, T.; Watanabe, R.; Murata, K.; Tanaka, M.; Ito, H.; Yoshifuji, H.; et al. Effect of Impaired T Cell Receptor Signaling on the Gut Microbiota in a Mouse Model of Systemic Autoimmunity. Arthritis Rheumatol. 2022, 74, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Traver, M.K.; Paul, S.; Schaefer, B.C. T Cell Receptor Activation of NF-κB in Effector T Cells: Visualizing Signaling Events Within and Beyond the Cytoplasmic Domain of the Immunological Synapse. Methods Mol. Biol. 2017, 1584, 101–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, G.; Wan, X. Challenges and new technologies in adoptive cell therapy. J. Hematol. Oncol. 2023, 16, 97. [Google Scholar] [CrossRef]
- Dhillon, S. Tebentafusp: First Approval. Drugs 2022, 82, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S. Cytokine Release Syndrome after Chimeric Antigen Receptor Transduced T-Cell Therapy in Cancers: A Systematic Review. Saudi J. Kidney Dis. Transpl. 2022, 33, 795–823. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Dwivedi, A.; Karulkar, A.; Ghosh, S.; Rafiq, A.; Purwar, R. Corrigendum: Lymphocytes in Cellular Therapy: Functional Regulation of CAR T Cells. Front. Immunol. 2019, 10, 401. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, J.; Zheng, H.; Yang, S.; Hua, Y.; Huang, N.; Kleeff, J.; Liao, Q.; Wu, W. Adoptive cellular immunotherapy for solid neoplasms beyond CAR-T. Mol. Cancer 2023, 22, 28. [Google Scholar] [CrossRef]
- Pan, K.; Farrukh, H.; Chittepu, V.; Xu, H.; Pan, C.X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef]
- Tsai, H.C.; Pietrobon, V.; Peng, M.; Wang, S.; Zhao, L.; Marincola, F.M.; Cai, Q. Current strategies employed in the manipulation of gene expression for clinical purposes. J. Transl. Med. 2022, 20, 535. [Google Scholar] [CrossRef]
- Cassetta, L.; Kitamura, T. Macrophage targeting: Opening new possibilities for cancer immunotherapy. Immunology 2018, 155, 285–293. [Google Scholar] [CrossRef]
- Siegers, G.M.; Dutta, I.; Lai, R.; Postovit, L.M. Functional Plasticity of Gamma Delta T Cells and Breast Tumor Targets in Hypoxia. Front. Immunol. 2018, 9, 1367. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Wakimoto, H.; Martuza, R.L.; Kaufman, H.L.; Rabkin, S.D.; Saha, D. Oncolytic herpes simplex virus expressing IL-2 controls glioblastoma growth and improves survival. J. Immunother. Cancer 2024, 12, e008880. [Google Scholar] [CrossRef]
- Tannir, N.M.; Cho, D.C.; Diab, A.; Sznol, M.; Bilen, M.A.; Balar, A.V.; Grignani, G.; Puente, E.; Tang, L.; Chien, D.; et al. Bempegaldesleukin plus nivolumab in first-line renal cell carcinoma: Results from the PIVOT-02 study. J. Immunother. Cancer 2022, 10, e004419. [Google Scholar] [CrossRef]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef]
- Fallon, J.; Tighe, R.; Kradjian, G.; Guzman, W.; Bernhardt, A.; Neuteboom, B.; Lan, Y.; Sabzevari, H.; Schlom, J.; Greiner, J.W. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 2014, 5, 1869–1884. [Google Scholar] [CrossRef]
- Propper, D.J.; Balkwill, F.R. Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 237–253. [Google Scholar] [CrossRef]
- Perez-Ruiz, E.; Minute, L.; Otano, I.; Alvarez, M.; Ochoa, M.C.; Belsue, V.; de Andrea, C.; Rodriguez-Ruiz, M.E.; Perez-Gracia, J.L.; Marquez-Rodas, I.; et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 2019, 569, 428–432. [Google Scholar] [CrossRef]
- Rastogi, I.; Muralidhar, A.; McNeel, D.G. Vaccines as treatments for prostate cancer. Nat. Rev. Urol. 2023, 20, 544–559. [Google Scholar] [CrossRef]
- Xie, Y.J.; Liu, W.Q.; Li, D.; Hou, J.C.; Coghi, P.S.; Fan, X.X. Overcoming Suppressive Tumor Microenvironment by Vaccines in Solid Tumor. Vaccines 2023, 11, 394. [Google Scholar] [CrossRef]
- Laureano, R.S.; Sprooten, J.; Vanmeerbeerk, I.; Borras, D.M.; Govaerts, J.; Naulaerts, S.; Berneman, Z.N.; Beuselinck, B.; Bol, K.F.; Borst, J.; et al. Trial watch: Dendritic cell (DC)-based immunotherapy for cancer. Oncoimmunology 2022, 11, 2096363. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Schneider, J. Plasmid DNA and viral vector-based vaccines for the treatment of cancer. Vaccine 2007, 25 (Suppl. S2), B24–B34. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; Scaggiante, B.; Morris, R.; Chai, D.; Catalano, M.; Tardiel-Cyril, D.R.; Neeli, P.; Roviello, G.; Mondani, G.; Li, Y. Therapeutic cancer vaccines: From biological mechanisms and engineering to ongoing clinical trials. Cancer Treat. Rev. 2022, 109, 102429. [Google Scholar] [CrossRef]
- Duong, H.T.T.; Thambi, T.; Yin, Y.; Kim, S.H.; Nguyen, T.L.; Phan, V.H.G.; Kim, J.; Jeong, J.H.; Lee, D.S. Degradation-regulated architecture of injectable smart hydrogels enhances humoral immune response and potentiates antitumor activity in human lung carcinoma. Biomaterials 2020, 230, 119599. [Google Scholar] [CrossRef]
- Chen, Z.; Meng, C.; Mai, J.; Liu, Y.; Li, H.; Shen, H. An mRNA vaccine elicits STING-dependent antitumor immune responses. Acta Pharm. Sin. B 2023, 13, 1274–1286. [Google Scholar] [CrossRef]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Del Rivero, J.; Cordes, L.M.; Klubo-Gwiezdzinska, J.; Madan, R.A.; Nieman, L.K.; Gulley, J.L. Endocrine-Related Adverse Events Related to Immune Checkpoint Inhibitors: Proposed Algorithms for Management. Oncologist 2020, 25, 290–300. [Google Scholar] [CrossRef]
- Schaft, N.; Dörrie, J.; Schuler, G.; Schuler-Thurner, B.; Sallam, H.; Klein, S.; Eisenberg, G.; Frankenburg, S.; Lotem, M.; Khatib, A. The future of affordable cancer immunotherapy. Front. Immunol. 2023, 14, 1248867. [Google Scholar] [CrossRef] [PubMed]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef]
- Candeias, S.M.; Gaipl, U.S. The Immune System in Cancer Prevention, Development and Therapy. Anticancer. Agents Med. Chem. 2016, 16, 101–107. [Google Scholar] [CrossRef]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef]
- Finn, O.J. Immuno-oncology: Understanding the function and dysfunction of the immune system in cancer. Ann. Oncol. 2012, 23 (Suppl. S8), viii6–viii9. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Pawelec, G.; Alan, C.; Larbi, A. On the immunological theory of aging. Interdiscip. Top. Gerontol. 2014, 39, 163–176. [Google Scholar] [CrossRef]
- Palmer, S.; Albergante, L.; Blackburn, C.C.; Newman, T.J. Thymic involution and rising disease incidence with age. Proc. Natl. Acad. Sci. USA 2018, 115, 1883–1888. [Google Scholar] [CrossRef]
- Accardi, G.; Caruso, C. Immune-inflammatory responses in the elderly: An update. Immun. Ageing 2018, 15, 11. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8⁺ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef]
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2. [Google Scholar] [CrossRef]
- Palmer, D.B. The effect of age on thymic function. Front. Immunol. 2013, 4, 316. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Understanding immunosenescence to improve responses to vaccines. Nat. Immunol. 2013, 14, 428–436. [Google Scholar] [CrossRef]
- Pawelec, G. Hallmarks of human “immunosenescence”: Adaptation or dysregulation? Immun. Ageing 2012, 9, 15. [Google Scholar] [CrossRef]
- Flores, K.G.; Li, J.; Sempowski, G.D.; Haynes, B.F.; Hale, L.P. Analysis of the human thymic perivascular space during aging. J. Clin. Investig. 1999, 104, 1031–1039. [Google Scholar] [CrossRef]
- Lynch, H.E.; Goldberg, G.L.; Chidgey, A.; Van den Brink, M.R.; Boyd, R.; Sempowski, G.D. Thymic involution and immune reconstitution. Trends Immunol. 2009, 30, 366–373. [Google Scholar] [CrossRef]
- Wertheimer, A.M.; Bennett, M.S.; Park, B.; Uhrlaub, J.L.; Martinez, C.; Pulko, V.; Currier, N.L.; Nikolich-Žugich, D.; Kaye, J.; Nikolich-Žugich, J. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J. Immunol. 2014, 192, 2143–2155. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging as Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Effros, R.B.; Dagarag, M.; Spaulding, C.; Man, J. The role of CD8+ T-cell replicative senescence in human aging. Immunol. Rev. 2005, 205, 147–157. [Google Scholar] [CrossRef]
- Ershler, W.B.; Keller, E.T. Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu. Rev. Med. 2000, 51, 245–270. [Google Scholar] [CrossRef]
- Bruunsgaard, H.; Andersen-Ranberg, K.; Hjelmborg, J.; Pedersen, B.K.; Jeune, B. Elevated levels of tumor necrosis factor alpha and mortality in centenarians. Am. J. Med. 2003, 115, 278–283. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Yi, F.; Frazzette, N.; Cruz, A.C.; Klebanoff, C.A.; Siegel, R.M. Beyond Cell Death: New Functions for TNF Family Cytokines in Autoimmunity and Tumor Immunotherapy. Trends Mol. Med. 2018, 24, 642–653. [Google Scholar] [CrossRef]
- Okoye, I.S.; Xu, L.; Walker, J.; Elahi, S. The glucocorticoids prednisone and dexamethasone differentially modulate T cell function in response to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade. Cancer Immunol. Immunother. 2020, 69, 1423–1436. [Google Scholar] [CrossRef]
- Gong, W.; Hoffmann, J.M.; Stock, S.; Wang, L.; Liu, Y.; Schubert, M.L.; Neuber, B.; Hückelhoven-Krauss, A.; Gern, U.; Schmitt, A.; et al. Comparison of IL-2 vs IL-7/IL-15 for the generation of NY-ESO-1-specific T cells. Cancer Immunol. Immunother. 2019, 68, 1195–1209. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Liu, Y.X.; Xu, B.W.; Chen, Y.J.; Fu, X.Q.; Zhu, P.L.; Bai, J.X.; Chou, J.Y.; Yin, C.L.; Li, J.K.; Wang, Y.P.; et al. Inhibiting the Src/STAT3 signaling pathway contributes to the anti-melanoma mechanisms of dioscin. Oncol. Lett. 2020, 19, 2508–2514. [Google Scholar] [CrossRef]
- So, J.Y.; Skrypek, N.; Yang, H.H.; Merchant, A.S.; Nelson, G.W.; Chen, W.D.; Ishii, H.; Chen, J.M.; Hu, G.; Achyut, B.R.; et al. Induction of DNMT3B by PGE2 and IL6 at Distant Metastatic Sites Promotes Epigenetic Modification and Breast Cancer Colonization. Cancer Res. 2020, 80, 2612–2627. [Google Scholar] [CrossRef]
- Patel, H.J.; Patel, B.M. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2017, 170, 56–63. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Maynard, J.P.; Ertunc, O.; Kulac, I.; Baena-Del Valle, J.A.; De Marzo, A.M.; Sfanos, K.S. IL8 Expression Is Associated with Prostate Cancer Aggressiveness and Androgen Receptor Loss in Primary and Metastatic Prostate Cancer. Mol. Cancer Res. 2020, 18, 153–165. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Doran, M.F.; Pond, G.R.; Crowson, C.S.; O’Fallon, W.M.; Gabriel, S.E. Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum. 2002, 46, 625–631. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. Aging of the T cell compartment in mice and humans: From no naive expectations to foggy memories. J. Immunol. 2014, 193, 2622–2629. [Google Scholar] [CrossRef]
- Sceneay, J.; Goreczny, G.J.; Wilson, K.; Morrow, S.; DeCristo, M.J.; Ubellacker, J.M.; Qin, Y.; Laszewski, T.; Stover, D.G.; Barrera, V.; et al. Interferon Signaling Is Diminished with Age and Is Associated with Immune Checkpoint Blockade Efficacy in Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 1208–1227. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-derived γδ regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Wang, W.; Chen, J.X.; Liao, R.; Deng, Q.; Zhou, J.J.; Huang, S.; Sun, P. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell Biol. 2002, 22, 3389–3403. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Liu, X.; Si, F.; Bagley, D.; Ma, F.; Zhang, Y.; Tao, Y.; Shaw, E.; Peng, G. Blockades of effector T cell senescence and exhaustion synergistically enhance antitumor immunity and immunotherapy. J. Immunother. Cancer 2022, 10, e005020. [Google Scholar] [CrossRef]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. [Google Scholar] [CrossRef]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef]
- Muller, W.A. Localized signals that regulate transendothelial migration. Curr. Opin. Immunol. 2016, 38, 24–29. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef]
- Hughes, P.E.; Caenepeel, S.; Wu, L.C. Targeted Therapy and Checkpoint Immunotherapy Combinations for the Treatment of Cancer. Trends Immunol. 2016, 37, 462–476. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, J.; Liu, M.; Xie, Z.; Lei, X.; Yang, X.; Huang, S.; Deng, X.; Wang, Z.; Tang, G. Small-molecule modulators of tumor immune microenvironment. Bioorg. Chem. 2024, 145, 107251. [Google Scholar] [CrossRef]
- Bueno, L.; de Alwis, D.P.; Pitou, C.; Yingling, J.; Lahn, M.; Glatt, S.; Trocóniz, I.F. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-beta kinase antagonist, in mice. Eur. J. Cancer 2008, 44, 142–150. [Google Scholar] [CrossRef]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Rao, K.S.; Lee, H.J.; Park, S.J.; Park, H.J.; Lee, K.; Sheen, Y.Y.; et al. Discovery of N-((4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline (EW-7197): A highly potent, selective, and orally bioavailable inhibitor of TGF-β type I receptor kinase as cancer immunotherapeutic/antifibrotic agent. J. Med. Chem. 2014, 57, 4213–4238. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, X.L.; Liu, X.; Hu, X.G.; Fu, W.J.; Li, Q.; Wang, Y.; Ping, Y.F.; Zhang, X.; Bian, X.W.; et al. Elevated ASCL2 expression in breast cancer is associated with the poor prognosis of patients. Am. J. Cancer Res. 2017, 7, 955–961. [Google Scholar] [PubMed]
- Ferrara, R.; Mezquita, L.; Auclin, E.; Chaput, N.; Besse, B. Immunosenescence and immunecheckpoint inhibitors in non-small cell lung cancer patients: Does age really matter? Cancer Treat. Rev. 2017, 60, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking aging to chronic disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Kugel, C.H., 3rd; Douglass, S.M.; Webster, M.R.; Kaur, A.; Liu, Q.; Yin, X.; Weiss, S.A.; Darvishian, F.; Al-Rohil, R.N.; Ndoye, A.; et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin. Cancer Res. 2018, 24, 5347–5356. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.E.; Naigeon, M.; Goldschmidt, V.; Roulleaux Dugage, M.; Seknazi, L.; Danlos, F.X.; Champiat, S.; Marabelle, A.; Michot, J.M.; Massard, C.; et al. Immunosenescence, inflammaging, and cancer immunotherapy efficacy. Expert. Rev. Anticancer. Ther. 2022, 22, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Marur, S.; Singh, H.; Mishra-Kalyani, P.; Larkins, E.; Keegan, P.; Sridhara, R.; Blumenthal, G.M.; Pazdur, R. FDA analyses of survival in older adults with metastatic non-small cell lung cancer in controlled trials of PD-1/PD-L1 blocking antibodies. Semin. Oncol. 2018, 45, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Elias, R.; Giobbie-Hurder, A.; McCleary, N.J.; Ott, P.; Hodi, F.S.; Rahma, O. Efficacy of PD-1 & PD-L1 inhibitors in older adults: A meta-analysis. J. Immunother. Cancer 2018, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, T.F.; Muss, H.B.; Shachar, S.S.; Moschos, S.J. Comparison of efficacy of immune checkpoint inhibitors (ICIs) between younger and older patients: A systematic review and meta-analysis. Cancer Treat. Rev. 2016, 45, 30–37. [Google Scholar] [CrossRef]
- Ridolfi, L.; De Rosa, F.; Petracci, E.; Tanda, E.T.; Marra, E.; Pigozzo, J.; Marconcini, R.; Guida, M.; Cappellini, G.C.A.; Gallizzi, G.; et al. Anti-PD1 antibodies in patients aged ≥ 75 years with metastatic melanoma: A retrospective multicentre study. J. Geriatr. Oncol. 2020, 11, 515–522. [Google Scholar] [CrossRef]
- Fu, Z.; Xu, H.; Yue, L.; Zheng, W.; Pan, L.; Gao, F.; Liu, X. Immunosenescence and cancer: Opportunities and challenges. Medicine 2023, 102, e36045. [Google Scholar] [CrossRef]
- Hou, C.; Wang, Z.; Lu, X. Impact of immunosenescence and inflammaging on the effects of immune checkpoint inhibitors. Cancer Pathog. Ther. 2024, 2, 24–30. [Google Scholar] [CrossRef] [PubMed]
- McCay, C.M.; Maynard, L.A.; Sperling, G.; Barnes, L.L. Retarded growth, life span, ultimate body size and age changes in the albino rat after feeding diets restricted in calories. Nutr. Rev. 1975, 33, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, F.; Terrien, J.; Marchal, J.; Dal-Pan, A.; Djelti, F.; Hardy, I.; Chahory, S.; Cordonnier, N.; Desquilbet, L.; Hurion, M.; et al. Caloric restriction increases lifespan but affects brain integrity in grey mouse lemur primates. Commun. Biol. 2018, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Omodei, D.; Fontana, L. Calorie restriction and prevention of age-associated chronic disease. FEBS Lett. 2011, 585, 1537–1542. [Google Scholar] [CrossRef]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Passos, J.F.; Khosla, S.; Tchkonia, T.; Kirkland, J.L. Reducing Senescent Cell Burden in Aging and Disease. Trends Mol. Med. 2020, 26, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Miller, C.M.; Shadrach, J.L.; Wagers, A.J.; Serwold, T. Young, proliferative thymic epithelial cells engraft and function in aging thymuses. J. Immunol. 2015, 194, 4784–4795. [Google Scholar] [CrossRef]
- Sun, L.; Guo, J.; Brown, R.; Amagai, T.; Zhao, Y.; Su, D.M. Declining expression of a single epithelial cell-autonomous gene accelerates age-related thymic involution. Aging Cell 2010, 9, 347–357. [Google Scholar] [CrossRef]
- Henson, S.M.; Snelgrove, R.; Hussell, T.; Wells, D.J.; Aspinall, R. An IL-7 fusion protein that shows increased thymopoietic ability. J. Immunol. 2005, 175, 4112–4118. [Google Scholar] [CrossRef] [PubMed]
- Su, D.M.; Aw, D.; Palmer, D.B. Immunosenescence: A product of the environment? Curr. Opin. Immunol. 2013, 25, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8(+)CD28(-) Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Lorente, M.A.; Cano-Martin, A.C.; Blasco, M.A. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 2019, 10, 4723. [Google Scholar] [CrossRef] [PubMed]
- Lanna, A.; Vaz, B.; D’Ambra, C.; Valvo, S.; Vuotto, C.; Chiurchiù, V.; Devine, O.; Sanchez, M.; Borsellino, G.; Akbar, A.N.; et al. An intercellular transfer of telomeres rescues T cells from senescence and promotes long-term immunological memory. Nat. Cell Biol. 2022, 24, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Fauce, S.R.; Jamieson, B.D.; Chin, A.C.; Mitsuyasu, R.T.; Parish, S.T.; Ng, H.L.; Kitchen, C.M.; Yang, O.O.; Harley, C.B.; Effros, R.B. Telomerase-based pharmacologic enhancement of antiviral function of human CD8+ T lymphocytes. J. Immunol. 2008, 181, 7400–7406. [Google Scholar] [CrossRef]
- Giallongo, C.; Parrinello, N.L.; La Cava, P.; Camiolo, G.; Romano, A.; Scalia, M.; Stagno, F.; Palumbo, G.A.; Avola, R.; Li Volti, G.; et al. Monocytic myeloid-derived suppressor cells as prognostic factor in chronic myeloid leukaemia patients treated with dasatinib. J. Cell Mol. Med. 2018, 22, 1070–1080. [Google Scholar] [CrossRef]
- Li, L.; Liu, X.; Sanders, K.L.; Edwards, J.L.; Ye, J.; Si, F.; Gao, A.; Huang, L.; Hsueh, E.C.; Ford, D.A.; et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab. 2019, 29, 103–123.e105. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Huang, X.; Hsueh, E.C.; Zhang, Q.; Ma, C.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012, 120, 2021–2031. [Google Scholar] [CrossRef]
- Zhan, J.K.; Wang, Y.J.; Li, S.; Wang, Y.; Tan, P.; He, J.Y.; Chen, Y.Y.; Deng, H.Q.; Huang, W.; Lin, X.; et al. AMPK/TSC2/mTOR pathway regulates replicative senescence of human vascular smooth muscle cells. Exp. Ther. Med. 2018, 16, 4853–4858. [Google Scholar] [CrossRef]
- Kusumoto, D.; Seki, T.; Sawada, H.; Kunitomi, A.; Katsuki, T.; Kimura, M.; Ito, S.; Komuro, J.; Hashimoto, H.; Fukuda, K.; et al. Anti-senescent drug screening by deep learning-based morphology senescence scoring. Nat. Commun. 2021, 12, 257. [Google Scholar] [CrossRef]
- Charron, D. Autologous white blood cell transfusion: Toward a younger immunity. Hum. Immunol. 2007, 68, 805–812. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Bannister, C.A.; Holden, S.E.; Jenkins-Jones, S.; Morgan, C.L.; Halcox, J.P.; Schernthaner, G.; Mukherjee, J.; Currie, C.J. Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes. Metab. 2014, 16, 1165–1173. [Google Scholar] [CrossRef]
- Strong, R.; Miller, R.A.; Antebi, A.; Astle, C.M.; Bogue, M.; Denzel, M.S.; Fernandez, E.; Flurkey, K.; Hamilton, K.L.; Lamming, D.W.; et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 2016, 15, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Kapahi, P.; Chen, D.; Rogers, A.N.; Katewa, S.D.; Li, P.W.; Thomas, E.L.; Kockel, L. With TOR, less is more: A key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010, 11, 453–465. [Google Scholar] [CrossRef]
- Ha, C.W.; Huh, W.K. Rapamycin increases rDNA stability by enhancing association of Sir2 with rDNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2011, 39, 1336–1350. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Baur, J.A.; Boyd, A.R.; de Cabo, R.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nelson, J.F.; et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 191–201. [Google Scholar] [CrossRef]
- Popovich, I.G.; Anisimov, V.N.; Zabezhinski, M.A.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Blagosklonny, M.V. Lifespan extension and cancer prevention in HER-2/neu transgenic mice treated with low intermittent doses of rapamycin. Cancer Biol. Ther. 2014, 15, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Pham, C.G.; Albanese, S.K.; Dong, Y.; Oyama, T.; Lee, C.H.; Rodrik-Outmezguine, V.; Yao, Z.; Han, S.; Chen, D.; et al. Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J. Clin. Investig. 2016, 126, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Lee, K.Y.; Kim, J.R.; Choi, H.C. Interaction between mTOR pathway inhibition and autophagy induction attenuates adriamycin-induced vascular smooth muscle cell senescence through decreased expressions of p53/p21/p16. Exp. Gerontol. 2018, 109, 51–58. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Drug | Monotherapy | Combination Therapy | Clinical Treatment | Report |
|---|---|---|---|---|---|
| PD-1 | Nivolumab (Opdivo) | NSCLC, HNSC, GAC and AEG, EAC, STAD, LC, ESCA, BLCA, SCLC, HCC, SKCM, HL, RCC | Fluoropyrimidines + Platinum: ESCC; Cisplatin/Carboplatin: NSCLC; Ipilimumab: NSCLC, CRC, RCC, SKCM, HCC, MPM, cSCC; Relatlimab: SKCM | Front-line treatment: MPM, STAD, AEG, ESCA; Metastatic disease: NSCLC, HNSC, STAD, ESCA; Adjuvant: ESCA, BLCA; Neoadjuvant: NSCLC; Maintenance: NSCLC, HNSC, GAC | NMPA, FDA, EMA |
| Pembrolizumab (Keytruda) | SKCM, ESCC, NSCLC, HNSC, CRC, ESCA, HCC, BC, NSCLC, CHL, PMBL, BLCA, STAD, CCA, NMIBC | Pemetrexed + Carboplatinum: NSCLC; Carboplatin + Paclitaxel: NSCLC; Paclitaxel/Nab-paclitaxel/Gemcitabine + Carboplatin: TNBC; Platinum-based + Platinum-based: AEG, HNSC, ESCA; Gemcitabine + Cisplatin: BTC; Trastuzumab + Fluorouracil/Platinum: STAD, GEJ; Axitinib: RCC; Lenvatinib: UCEC | Front-line treatment: NSCLC, HNSC, CRC, ESCA, BTC; Metastatic disease: SKCM, NSCLC, ESCC, CRC; Neoadjuvant: TNBC; Maintenance: HCC, CRC | NMPA, FDA, EMA | |
| Cemiplimab (Libtayo) | NSCLC, cSCC, BCC | Front-line treatment: NSCLC; Metastatic disease: cSCC; Maintenance: BCC | FDA, EMA | ||
| Dostarlimab (Jemperli) | UCEC | Maintenance: UCEC | FDA, EMA | ||
| Retifanlimab (Zynyz) | EC, MCC, SCAC | Metastatic disease: SCAC, MCC; Maintenance: SCAC, EC | FDA | ||
| HLX10 (Serplulimab) | SKCM, NPC, BLCA | Platinum: ESCC; Pemetrexed + Platinum: NSCLC; Axitinib: RCC | Front-line treatment: NPC, ESCC, NSCLC; Metastatic disease: SKCM, NPC, BLCA, RCC; Adjuvant: NSCLC; Maintenance: NPC, BLCA | NMPA | |
| Sintilimab Injection | HL | Pemetrexed + Platinum: NSCLC; Gemcitabine + Platinum: NSCLC; Bevacizumab: HCC; Cisplatin + Paclitaxel/Cisplatin + Fluoropyrimidine containing: ESCC; Oxaliplatin + Capecitabine: G/GEJ; Bevacizumab + Pemetrexed + Cisplatin: NSCLC | Front-line treatment: NSCLC, HCC, ESCA, G/GEJ; Metastatic disease: HCC, ESCA, NSCLC; Maintenance: HL, NSCLC | NMPA | |
| Camrelizumab (SHR-1210) | HL, HCC, ESCC, NPC | Pemetrexed + Carboplatin: NSCLC; Cisplatin + Gemcitabine: NPC; Paclitaxel + Carboplatin: NSCLC; Paclitaxel + Cisplatin: ESCC; Apatinib: HCC | Front-line treatment: NSCLC, NPC, ESCC, HCC; Metastatic disease: HCC, ESCC, NPC; Maintenance: NPC, ESCC, HCC, HL | NMPA | |
| Tislelizumab | HL, BLCA, HCC, NSCLC, GAC, UCEC, HCCA, PACA, SCC | Paclitaxel/Albumin Paclitaxel + Carboplatin: NSCLC; Pemetrexed + Platinum: NSCLC; Gemcitabine + Cisplatin: NPC; Fluorouracil + Platinum: G/GEJ, ESCC; | Front-line treatment: NSCLC, G/GEJ, ESCC, HCC; Metastatic disease: BLCA, NSCLC, ESCC; Maintenance: NSCLC, HL, BLCA, HCC, ESCC | NMPA | |
| Penpulimab (AK105) | HL | Paclitaxel + Carboplatin: NSCLC | Front-line treatment: HL; Metastatic disease: NSCLC | NMPA | |
| Zimberelimab (GLS-010) | r/r cHL, CCA | Metastatic disease: r/r Chl; Maintenance: CCA | NMPA | ||
| Serplulimab (HLX10) | CRC, GAC, UCEC, G/GEJ, HCCA, PACA, HCC | Carboplatin + Albumin paclitaxel: sqNSCLC; Carboplatin + Etoposide: ES-SCLC; Fluorouracil + Platinum: ESCC | Front-line treatment: sqNSCLC, ES-SCLC, RSCC; Metastatic disease: RSCC | NMPA | |
| Pucotenlimab | CRC, GAC, BC, PCa, HCC, LCA, HL | Metastatic disease: CRC, GAC, BC, PCa, HCC, LCA, HL | NMPA | ||
| PD-L1 | Durvalumab (Imfinzi) | NSCLC, CA, ASTS, TNBC | Etoposide + Platinum/Carboplatin: ES-SCLC; Gemcitabine + Cisplatin: BTC; Tremelimumab: HCC | Front-line treatment: NSCLS, SCLS, BTC | NMPA, FDA, EMA |
| Atezolizumab (Tecentriq) | NSCLC | Carboplatin + Etoposide: SCLC; Bevacizumab: HCC; Pemetrexed + Platinum: NSCLC | Front-line treatment: SCLC, HCC, NSCLC; Adjuvant: NSCLC | NMPA, FDA, EMA | |
| Avelumab (Bavencio) | MCC, BLCA | Axitinib: RCC | Front-line treatment: MCC, BLCA, RCC | FDA, EMA | |
| Envolizumab | CRC, GAC, UCEC, G/GEJ, HCCA, PACA, HCC | Maintenance: CRC, GAC, UCEC, G/GEJ, HCCA, PACA, HCC | NMPA | ||
| Sugilizumab | NSCLC | Paclitaxel + Carboplatin: NSCLC; Fluorouracil + Platinum: ESCC, G/GEJ | Front-line treatment: NSCLC, ESCC, G/GEJ | NMPA | |
| Adebelizumab | Etoposide + Carboplatin: ES-SCLC | Front-line treatment: ES-SCLC | NMPA | ||
| Sokazolizumab | CCA | Metastatic disease: CCA | NMPA | ||
| CTLA-4 | Ipilimumab (Yervoy) | SKCM | Navulizumab: MPM, NSCLC, CRC, RCC, SKCM, HCC | Front-line treatment: MPM, NSCLC, CRC, RCC; Second-line treatment: HCC | NMPA, FDA |
| Tremelimumab | Durvalumab: HCC | Front-line treatment: HCC | FDA | ||
| PD-L1/ CTLA-4 Bispecific Antibody | Cardunolizumab | R/MCC | Front-line treatment: R/MCC | NMPA | |
| LAG-3 | Relatlimab | Nivolumab: SKCM | Neoadjuvant: SKCM | FDA |
| TILs | TCR-T | CAR-T | CAR-NK | |
|---|---|---|---|---|
| Cell Source | Tumor tissue | PBMC, iPSC | PBMC, iPSC, UCB | PBMC, iPSC, hESC, UCB, BM, cell line |
| Theory | Extraction of TILs from tumor samples | Transduction of TCR into T cells | Transduction of CAR into T cells | Transduction of CAR by autologous NK cells or allogeneic NK cells |
| Gene transfer | / | Easy | Easy | Difficult |
| Persistence | Low | Moderate | Moderate | Low |
| Infiltration capacity | High | Low | Low | Low |
| Toxicological | Low OTOT and CRS | CRS | CRS, OTOT, ICANS, GvHD | Low OTOT CRS, ICANS, and GvHD |
| Dominance | Multi-target excitation; high tumor specificity; potential anti-tumor activity | Recognition against multiple tumor antigens; high number of T cells; | High number of T cells; recognition against multiple tumor antigens | Natural anti-tumor activity; no need for MHC molecules to be involved |
| Drawbacks | Time-consuming and costly | Poor tumor-specific binding capacity; limited by MHC | Tumor antigen heterogeneity and tumor antigen loss | Limited efficacy of CAR transduction |
| Naturopathy | Drug | Target | Disease | Favor | Time |
|---|---|---|---|---|---|
| TILs | Lifileucel | CD19 | SKCM | FDA | 2024 |
| TCR-T | Tebentafusp | Gp100 | UM | FDA | 2022 |
| CAR-T | Kymriah | CD19 | ALL, NHL (DLBCL, HGBL) | FDA | 2017 |
| Yescarta | CD19 | NHL (DLBCL, FL, HGBL, PMBL | FDA | 2017 | |
| Tecartus | CD19 | RR/MM, ALL | FDA | 2020 | |
| Breyanzi | CD19 | NHL, (DLBCL, G3BFL, PMBL) | FDA | 2021 | |
| Abecma | BCMA | RR/MM | FDA | 2021 | |
| Carvykti | BCMA | RR/MM | FDA, NMPA | 2022 | |
| Axicabtagene ciloleucel | CD19 | r/r LBCL (DLBCL, PMBL, HGBL, DLBCL) | NMPA | 2021 | |
| Carteyva | CD19 | r/r LBCL | NMPA | 2021 | |
| Equecabtagene autoleucel | BCMA | RR/MM | NMPA | 2023 | |
| Inaticabtagene autoleucel injection | CD19 | r/r B-ALL | NMPA | 2023 | |
| CT053 (Zevorcabtagene autoleucel) | BCMA | RR/MM | NMPA | 2023 |
| NCT– Number | Study Start | Tumor Type | Interventions | Phase | Country |
|---|---|---|---|---|---|
| TILs | |||||
| NCT05475847 | 2022.07 | CCA | ATIL (C-TIL052A) Injection | 1 | China |
| NCT05878028 | 2022.09 | NSCLC | L-TIL + Tislelizumab + Docetaxel | 2 | China |
| NCT05451784 | 2022.07 | TNBC-METS | PD1+ TILs (NUMARZU-001) | 2 | Spain |
| NCT05676749 | 2024.02 | MNSCLC | C-TIL051 | 1 | USA |
| NCT05438797 | 2021.04 | APCAN | Adoptive TIL-TCM transfer therapy | 1 | |
| NCT05681780 | 2023.01 | NSCLC | TIL, Nivolumab, Cyclophosphamide | 2 | USA |
| NCT05869539 | 2023.06 | AM | TIL t+ ANV419 | 1 | Switzerland |
| TCR-T | |||||
| NCT05438667 | 2022.06 | PACA | TCR-T therapy | 1 | China |
| NCT06119256 | 2023.08 | EBV-AIAHSCT | EBV-TCR-T cells | 1 | China |
| NCT06135922 | 2023.08 | EBV-HLH | EBV-TCR-T cells | 1 | China |
| NCT04509726 | 2023.03 | NPC | EBV-specific TCR-T cell | 2 | China |
| NCT05122221 | 2022.07 | CCA, HNC, ANAL-CA | Fludarabine + Cyclophosphamide, Interleukin-2, CRTE7A2-01 TCR-T Cell | 1 | China |
| NCT04520711 | 2022.02 | MEN | TCR-transduced T cells, CDX-1140, Pembrolizumab | 1 | USA |
| CAR-T | |||||
| NCT06132711 | 2023.11 | MM | APRIL-BAFF-Bicephali CAR-T cells | 2 | China |
| NCT05420493 | 2021.09 | RNHL, RNFHL, NHL | CAR-T cells | 1 | China |
| NCT05749133 | 2023.04 | MM | CAR-T Cells Injection | 2 | China |
| NCT06429150 | 2024.05 | MM, PCT | CAR-T cells | 2 | Russian |
| NCT05596266 | 2022.10 | T-ALL | CD5 CAR-T | 1 | China |
| NCT05333302 | 2020.10 | B-ALL, LBCL | CD19 CAR-T-cells, Tocilizumab | 1 | Belarus |
| NCT05535855 | 2024.01 | ALL | CD19 Directed CAR T Cell | 1 | USA |
| CAR-NK | |||||
| NCT05472558 | 2022.09 | BNHL | Anti-CD19 CAR-NK | 1 | China |
| NCT05673447 | 2023.03 | DLBCL | Anti-CD19 CAR NK cells | 1 | China |
| NCT04847466 | 2021.12 | GEJ, HNSCC | N-803, Pembrolizumab | 2 | USA |
| NCT06325748 | 2024.07 | AML, MDS | SENTI-202 | 1 | USA |
| NCT06045091 | 2023.06 | MM, PCL | CAR-NK cells injection | 1 | China |
| NCT06421701 | 2024.06 | SLE | Anti-CD19 CAR-NK cells | 1 | China |
| NCT06307054 | 2024.03 | AML | Anti-CLL-1 CAR NK cells | 1 | China |
| Treatment | Clinical Trial/Drugs | Target | References |
|---|---|---|---|
| IL-7 | IL-7 | Thymic | [162] |
| KGF (keratinocyte growth factor) | KGF | Thymic | [162] |
| IL-22 | IL-22 | Thymic | [162] |
| Ghrelin | Ghrelin | Thymic | [162] |
| Third-gen CAR-T cells containing CD28 + CD137 | NCT02186860 | CD28 | [163] |
| Second-gen CMV-selected CAR-T cells against HER2 containing CD28.zeta signaling domain | NCT01109095 | CD28 | [163] |
| TAB08 | NCT01990157 | CD28 | [163] |
| Rapamycin | Rapamycin | MTOR | [157] |
| Metformin | Metformin | Mitochondrial respiration | [157] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Y.; He, C.; Liu, C.; Shao, B.; Wang, D.; Wu, P. Exploring the Complexity and Promise of Tumor Immunotherapy in Drug Development. Int. J. Mol. Sci. 2024, 25, 6444. https://doi.org/10.3390/ijms25126444
Feng Y, He C, Liu C, Shao B, Wang D, Wu P. Exploring the Complexity and Promise of Tumor Immunotherapy in Drug Development. International Journal of Molecular Sciences. 2024; 25(12):6444. https://doi.org/10.3390/ijms25126444
Chicago/Turabian StyleFeng, Yiyuan, Caiying He, Chang Liu, Bingjie Shao, Dong Wang, and Peijie Wu. 2024. "Exploring the Complexity and Promise of Tumor Immunotherapy in Drug Development" International Journal of Molecular Sciences 25, no. 12: 6444. https://doi.org/10.3390/ijms25126444
APA StyleFeng, Y., He, C., Liu, C., Shao, B., Wang, D., & Wu, P. (2024). Exploring the Complexity and Promise of Tumor Immunotherapy in Drug Development. International Journal of Molecular Sciences, 25(12), 6444. https://doi.org/10.3390/ijms25126444