
Phylogeny and Metabolic Potential of New Giant Sulfur Bacteria of the Family Beggiatoaceae from Coastal-Marine Sulfur Mats of the White Sea †
 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Biotop Features
2.2. Morphology of Filamentous Colorless Sulfur Bacteria from Sulfur Mats
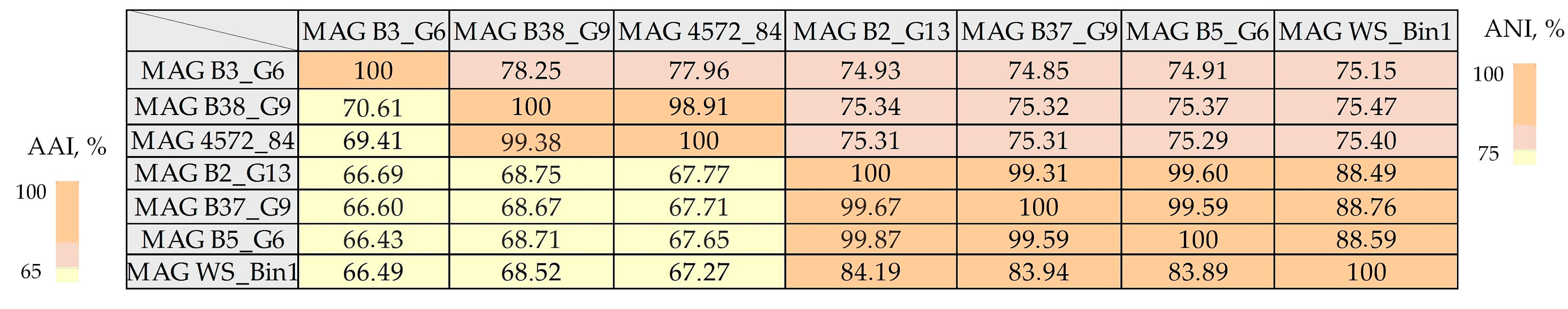
2.3. Assembly of Beggiatoaceae MAGs
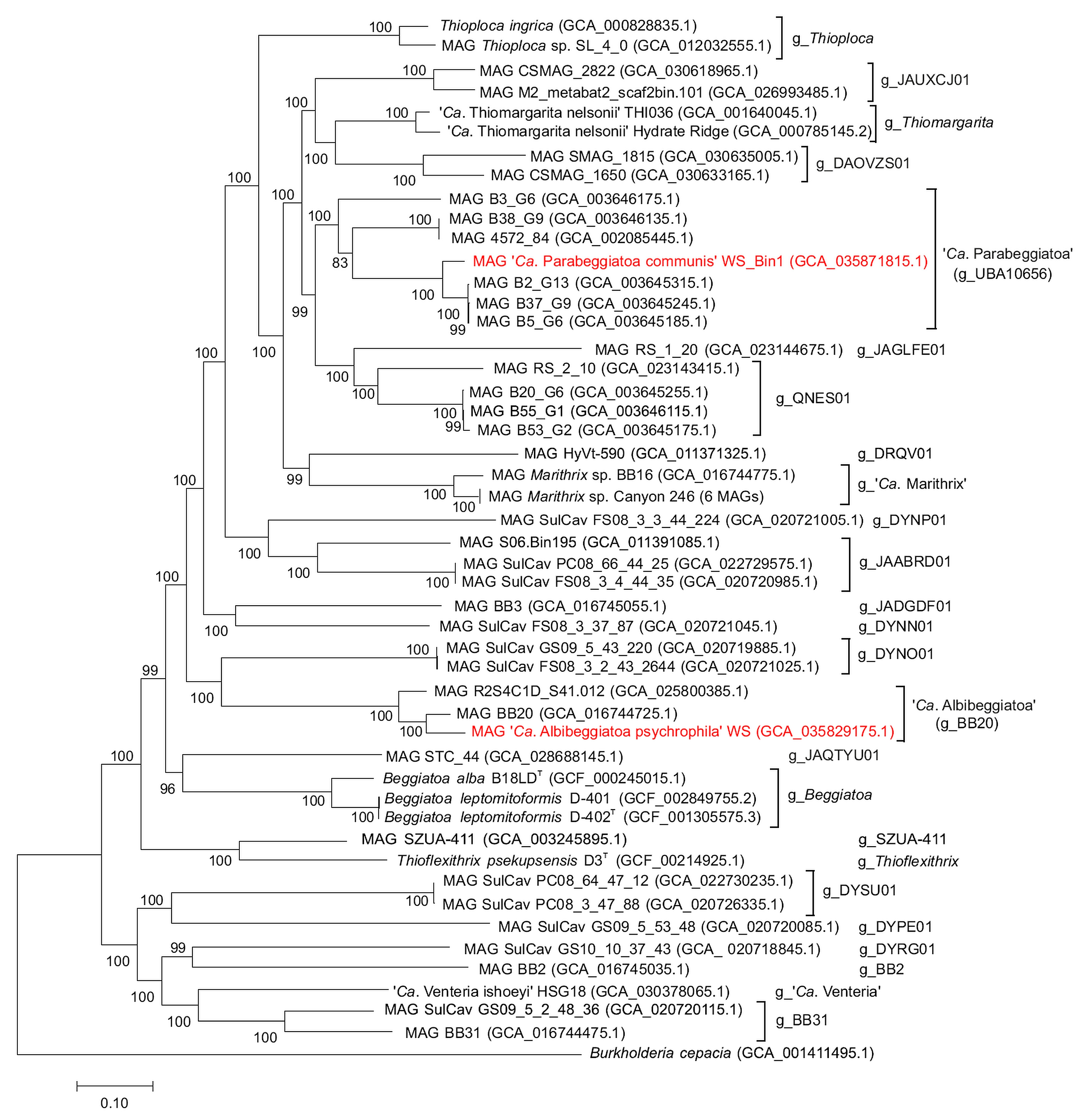
2.4. Phylogenetic Analysis of the MAG Beggiatoaceae sp. WS_Bin1
2.5. Phylogenetic Analysis of the MAG Beggiatoaceae sp. WS_Bin3
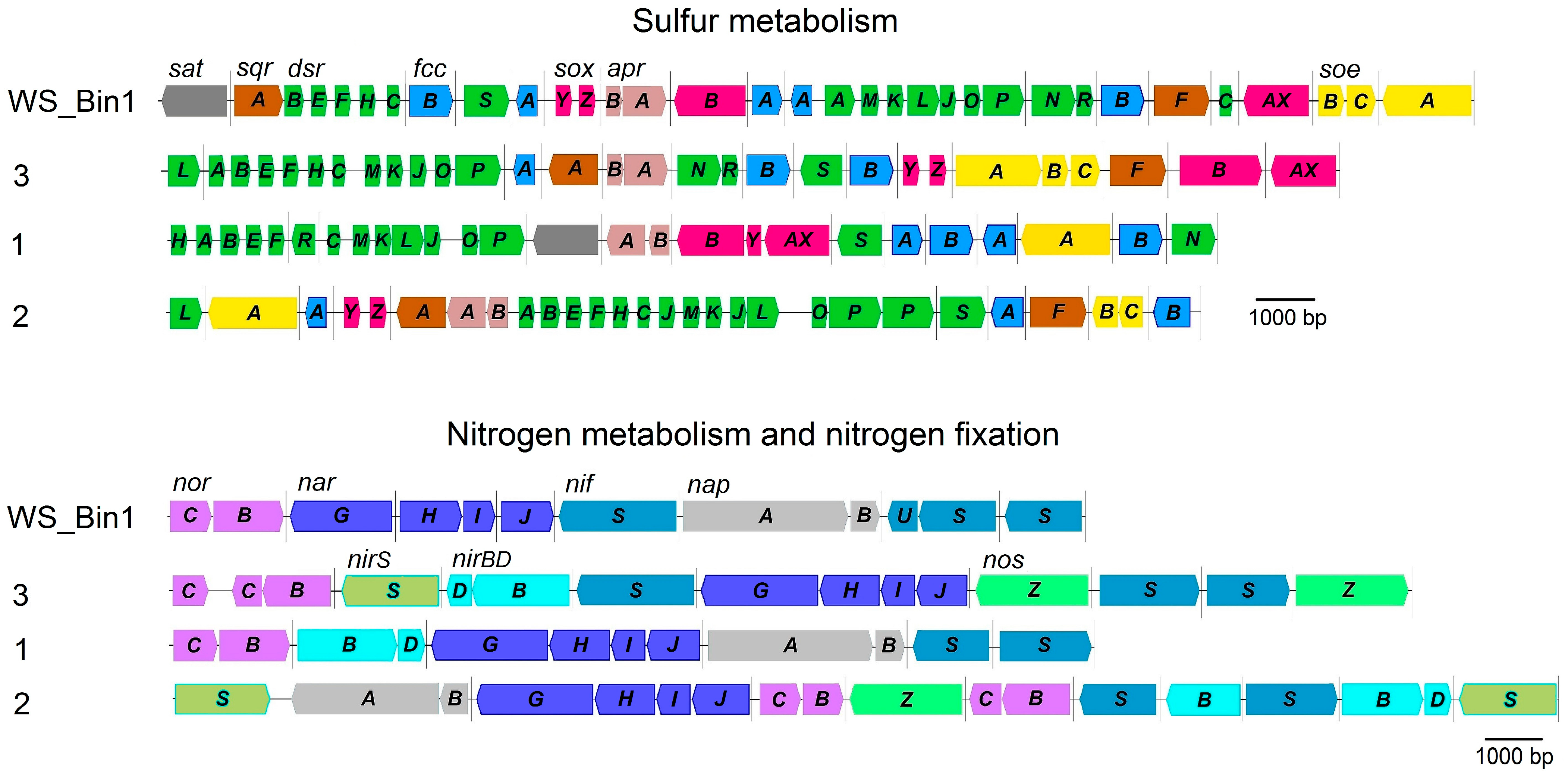
2.6. Genome Analysis of the Main Metabolic Pathways of Representatives of ‘Candidatus Parabeggiatoa’
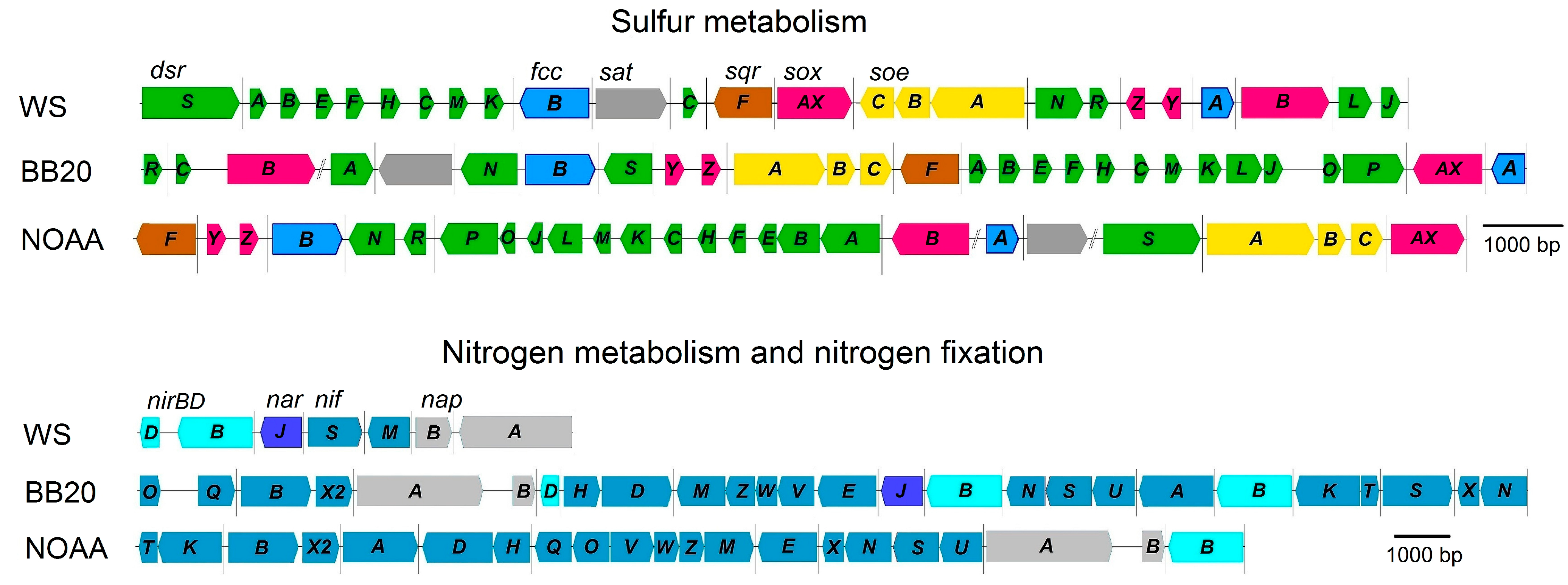
2.7. Genome Analysis of the Main Metabolic Pathways of Representatives of ‘Candidatus Albibeggiatoa’ gen. nov.
2.8. Description of New Genus and Species
2.8.1. Description of ‘Candidatus Albibeggiatoa’ gen. nov.
2.8.2. Description of ‘Candidatus Albibeggiatoa psychrophila’ sp. nov.
2.8.3. Emended Description of ‘Candidatus Parabeggiatoa’ Salman et al., 2011 (Lists of names of Prokaryotic Candidatus Taxa 2020) [34]
3. Materials and Methods
3.1. Geography and Physicochemical Characteristics of Environmental Sampling Sites for Metagenomic Characterization of MAG Beggiatoaceae sp. WS_Bin1 and MAG Beggiatoaceae sp. WS_Bin3
3.2. Underwater Photography
3.3. Microscopy and Microphotography
3.4. Genome DNA Isolation and Purification
3.5. Metagenome Sequencing and Assembly of MAGs
3.6. Genome Analysis and Annotation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mussmann, M.; Schulz, H.N.; Strotmann, B.; Kjaer, T.; Nielsen, L.P.; Rosselló-Mora, R.A.; Amann, R.I.; Jørgensen, B.B. Phylogeny and distribution of nitrate-storing Beggiatoa spp. in coastal marine sediments. Environ. Microbiol. 2003, 5, 523–533. [Google Scholar] [CrossRef]
- Salman, V.; Amann, R.; Girnth, A.C.; Polerecky, L.; Bailey, J.V.; Høgslund, S.; Jessen, G.; Pantoja, S.; Schulz-Vogt, H.N. A single-cell sequencing approach to the classification of large, vacuolated sulfur bacteria. Syst. Appl. Microbiol. 2011, 34, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Hinck, S.; Mussmann, M.; Salman, V.; Neu, T.R.; Lenk, S.; de Beer, D.; Jonkers, H.M. Vacuolated Beggiatoa-like filaments from different hypersaline environments form a novel genus. Environ. Microbiol. 2011, 13, 3194–3205. [Google Scholar] [CrossRef]
- Grünke, S.; Lichtschlag, A.; de Beer, D.; Felden, J.; Salman, V.; Ramette, A.; Schulz-Vogt, H.N.; Boetius, A. Mats of psychrophilic thiotrophic bacteria associated with cold seeps of the Barents Sea. Biogeosciences 2012, 9, 2947–2960. [Google Scholar] [CrossRef]
- Dubinina, G.; Savvichev, A.; Orlova, M.; Gavrish, E.; Verbarg, S.; Grabovich, M. Beggiatoa leptomitoformis sp. nov., the first freshwater member of the genus capable of chemolithoautotrophic growth. Int. J. Syst. Evol. Microbiol. 2017, 67, 197–204. [Google Scholar] [CrossRef]
- Gureeva, M.V.; Belousova, E.V.; Dubinina, G.A.; Novikov, A.A.; Kopitsyn, D.S.; Grabovich, M.Y. Thioflexithrix psekupsensis gen. nov., sp. nov., a filamentous gliding sulfur bacterium from the family Beggiatoaceae. Int. J. Syst. Evol. Microbiol. 2019, 69, 798–804. [Google Scholar] [CrossRef]
- Strohl, W.R.; Larkin, J.M. Enumeration, isolation, and characterization of Beggiatoa from freshwater sediments. Appl. Environ. Microbiol. 1978, 36, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.C.; Castenholz, R.W. Use of reduced sulfur compounds by Beggiatoa sp. J. Bacteriol. 1981, 147, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.M.; Henk, M.C. Filamentous sulfide-oxidizing bacteria at hydrocarbon seeps of the Gulf of Mexico. Microsc. Res. Tech. 1996, 33, 23–31. [Google Scholar] [CrossRef]
- Berg, J.S.; Schwedt, A.; Kreutzmann, A.C.; Kuypers, M.M.; Milucka, J. Polysulfides as intermediates in the oxidation of sulfide to sulfate by Beggiatoa spp. Appl. Environ. Microbiol. 2014, 80, 629–636. [Google Scholar] [CrossRef]
- Rudenko, T.S.; Tarlachkov, S.V.; Shatskiy, N.D.; Grabovich, M.Y. Comparative genomics of Beggiatoa leptomitoformis strains D-401 and D-402T with contrasting physiology but extremely high level of genomic identity. Microorganisms 2020, 8, 928. [Google Scholar] [CrossRef] [PubMed]
- Breed, R.S.; Murray, E.G.D.; Hitchens, A.P. (Eds.) Bergey’s Manual of Determinative Bacteriology, 6th ed.; The Williams and Wilkins Co.: Baltimore, MD, USA, 1948. [Google Scholar]
- Garrity, G.M.; Bell, J.A.; Lilburn, T.; Family, I. Thiotrichaceae fam. nov. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Garrity, B.M., Brenner, D.J., Krieg, N.R., Staley, J.T., Eds.; Springer: New York, NY, USA, 2005; Volume 2, p. 131. [Google Scholar]
- Danovaro, R.; Canals, M.; Tangherlini, M.; Dell’Anno, A.; Gambi, C.; Lastras, G.; Amblas, D.; Sanchez-Vidal, A.; Frigola, J.; Calafat, A.M.; et al. A submarine volcanic eruption leads to a novel microbial habitat. Nat. Ecol. Evol. 2017, 1, 144. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.; Ishoey, T.; Espinoza, C.; Pérez-Pantoja, D.; Manghisi, A.; Morabito, M.; Salas-Burgos, A.; Gallardo, V.A. Genomic features of “Candidatus Venteria ishoeyi”, a new sulfur-oxidizing macrobacterium from the Humboldt Sulfuretum off Chile. PLoS ONE 2017, 12, e0188371. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Dombrowski, N.; Seitz, K.W.; Teske, A.P.; Baker, B.J. Genomic insights into potential interdependencies in microbial hydrocarbon and nutrient cycling in hydrothermal sediments. Microbiome 2017, 5, 106. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, N.; Teske, A.P.; Baker, B.J. Expansive microbial metabolic versatility and biodiversity in dynamic Guaymas Basin hydrothermal sediments. Nat. Commun. 2018, 9, 4999. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Rosselló-Móra, R.; Amann, R. Uncultivated microbes in need of their own taxonomy. ISME J. 2017, 11, 2399–2406. [Google Scholar] [CrossRef]
- Rosales, S.M.; Huebner, L.K.; Clark, A.S.; McMinds, R.; Ruzicka, R.R.; Muller, E.M. Bacterial metabolic potential and micro-eukaryotes enriched in stony coral tissue loss disease lesions. Front. Mar. Sci. 2022, 8, 776859. [Google Scholar] [CrossRef]
- Ravin, N.V.; Smolyakov, D.D.; Markov, N.D.; Beletsky, A.V.; Mardanov, A.V.; Rudenko, T.S.; Grabovich, M.Y. tilS and rpoB: New molecular markers for phylogenetic and biodiversity studies of the genus Thiothrix. Microorganisms 2023, 11, 2521. [Google Scholar] [CrossRef] [PubMed]
- Saravanakumar, C.; Dineshkumar, N.; Alavandi, S.V.; Salman, V.; Poornima, M.; Kalaimani, N. Enrichment and identification of large filamentous sulfur bacteria related to Beggiatoa species from brackishwater ecosystems of Tamil Nadu along the southeast coast of India. Syst. Appl. Microbiol. 2012, 35, 396–403. [Google Scholar] [CrossRef]
- Kappler, U.; Maher, M.J. The bacterial SoxAX cytochromes. Cell. Mol. Life Sci. 2013, 70, 977–992. [Google Scholar] [CrossRef]
- Sauvé, V.; Roversi, P.; Leath, K.J.; Garman, E.F.; Antrobus, R.; Lea, S.M.; Berks, B.C. Mechanism for the hydrolysis of a sulfur-sulfur bond based on the crystal structure of the thiosulfohydrolase SoxB. J. Biol. Chem. 2009, 284, 21707–21718. [Google Scholar] [CrossRef] [PubMed]
- Otte, S.; Kuenen, J.G.; Nielsen, L.P.; Paerl, H.W.; Zopfi, J.; Schulz, H.N.; Teske, A.; Strotmann, B.; Gallardo, V.A.; Jorgensen, B.B. Nitrogen, carbon, and sulfur metabolism in natural thioploca samples. Appl. Environ. Microbiol. 1999, 65, 3148–3157. [Google Scholar] [CrossRef]
- Sayama, M. Presence of nitrate-accumulating sulfur bacteria and their influence on nitrogen cycling in a shallow coastal marine sediment. Appl. Environ. Microbiol. 2001, 67, 3481–3487. [Google Scholar] [CrossRef]
- Zopfi, J.; Kjaer, T.; Nielsen, L.P.; Jørgensen, B.B. Ecology of Thioploca spp.: Nitrate and sulfur storage in relation to chemical microgradients and influence of Thioploca spp. on the sedimentary nitrogen cycle. Appl. Environ. Microbiol. 2001, 67, 5530–5537. [Google Scholar] [CrossRef] [PubMed]
- McHatton, S.C.; Barry, J.P.; Jannasch, H.W.; Nelson, D.C. High nitrate concentrations in vacuolate, autotrophic marine Beggiatoa spp. Appl. Environ. Microbiol. 1996, 62, 954–958. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Dunker, R.; Grünke, S.; Røy, H. Filamentous sulfur bacteria, Beggiatoa spp., in arctic marine sediments (Svalbard, 79 degrees N). FEMS Microbiol. Ecol. 2010, 73, 500–513. [Google Scholar] [CrossRef]
- Jewell, T.; Huston, S.L.; Nelson, D.C. Methylotrophy in freshwater Beggiatoa alba strains. Appl. Environ. Microbiol. 2008, 74, 5575–5578. [Google Scholar] [CrossRef]
- Orlova, M.V.; Tarlachkov, S.V.; Kulinchenko, E.I.; Dubinina, G.A.; Tutukina, M.N.; Grabovich, M.Y. Genomics and biochemistry of metabolic pathways for the C1 compounds utilization in colorless sulfur bacterium Beggiatoa leptomitoformis D-402. Indian J. Microbiol. 2018, 58, 415–422. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M.; Parker, C.T.; Chuvochina, M.; Trujillo, M.E. Lists of names of prokaryotic Candidatus taxa. Int. J. Syst. Evol. Microbiol. 2020, 70, 3956–4042. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: A rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 2023, 20, 1203–1212. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome (MAG) | Genome Assembly | Genome Size (MB) | Contigs | GC-Content (%) | Genes | ||
|---|---|---|---|---|---|---|---|
| Protein-Coding | 16S rRNA | tRNA | |||||
| Beggiatoaceae sp. WS_Bin1 | GCA_035871815.1 | 8.64 | 1575 | 41.0 | 6942 | 1 | 45 |
| Beggiatoaceae sp. WS_Bin3 | GCA_035829175.1 | 3.64 | 708 | 38.0 | 2800 | 1 | 33 |
| MAG B3_G6 | GCA_003646175.1 | 8.8 | 1343 | 41.8 | 6453 | 0 | 56 |
| MAG B38_G9 | GCA_003646135.1 | 9.4 | 888 | 42.9 | 6502 | 0 | 64 |
| MAG 4572_84 | GCA_002085445.1 | 8.5 | 406 | 42.8 | 6041 | 0 | 53 |
| MAG B2_G13 | GCA_003645315.1 | 8.3 | 1040 | 41.0 | 6541 | 0 | 43 |
| MAG B37_G9 | GCA_003645245.1 | 8.2 | 977 | 40.9 | 6555 | 0 | 47 |
| MAG B5_G6 | GCA_003645185.1 | 8.4 | 853 | 40.9 | 6645 | 0 | 40 |
| MAG BB20 | GCA_016744725.1 | 4.5 | 296 | 37.9 | 3939 | 0 | 31 |
| Thiotrichaceae sp. R2S4C1D_S41.012 | GCA_025800385.1 | 4.8 | 257 | 38.5 | 3584 | 1 | 38 |
| Metabolic Pathways | ‘Ca. Parabeggiatoa’ | ‘Ca. Albibeggiatoa’ | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WS_Bin1 | Species 3 | Species 1 | Species 2 | WS | BB20 | NOAA | |||||||
| B5_G6 | B2_G13 | B37_G9 | B3_G6 | B38_G9 | 4572_84 | ||||||||
| Nitrogen metabolism | Denitrification, nitrate reductase from the Nar family | NO3− → NO−2 | narGHIJ | narGHIJ | narGHIJ | narGHIJ | − | ||||||
| NO2− → NO | − | nirS | − | nirS | |||||||||
| NO → N2O | cnorBC | cnorBC | cnorBC | cnorBC | |||||||||
| N2O → N2 | − | nosZ | − | nosZ | |||||||||
| Dissimilatory reduction of NO3− to NH4+, nitrate reductase from the Nap family | napAB | nirBD | napAB, nirBD | napAB, nirBD | |||||||||
| Assimilatory reduction of NO3− to NH4+, nitrate reductase from the Nas family | nasA, nasD | nasA, nasD | |||||||||||
| Molecular nitrogen fixation | - | - | nifASUBXX2ENQVWMHDKZTO | ||||||||||
| Dissimilatory sulfur metabolism | Hydrogen sulfide oxidation systems | sqrAF, fccAB | sqrAF, fccAB | sqrAF, fccAB | sqrA, fccAB | fccAB | sqrAF, fccAB | sqrAF, fccAB | sqrF, fccAB | ||||
| Thiosulfate oxidation, Sox-system | soxAXBYZ | soxAXBYZ | soxAXYZ | soxAXBYZ | soxAXBY | soxAXBYZ | soxBYZ | soxAXBYZ | |||||
| Elemental sulfur oxidation system, rDSR | dsrABEFHCMKLJOPNRS | ||||||||||||
| Sulfite oxidation systems | Direct way | soeABC | soeABC | soeAB | soeA | soeA | soeABC | soeABC | soeABC | ||||
| Indirect way | aprAB, sat | aprAB | aprAB | aprAB, sat | aprAB, sat | aprAB, sat | aprAB | sat | |||||
| Carbon metabolism | Krebs cycle | + | + | ||||||||||
| Glyoxylate pathway | − | + | |||||||||||
| Pentose-phosphate pathway | + | + | |||||||||||
| Type of RuBisCO | II | IAq | II | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravin, N.V.; Rudenko, T.S.; Beletsky, A.V.; Smolyakov, D.D.; Mardanov, A.V.; Grabovich, M.Y.; Muntyan, M.S. Phylogeny and Metabolic Potential of New Giant Sulfur Bacteria of the Family Beggiatoaceae from Coastal-Marine Sulfur Mats of the White Sea. Int. J. Mol. Sci. 2024, 25, 6028. https://doi.org/10.3390/ijms25116028
Ravin NV, Rudenko TS, Beletsky AV, Smolyakov DD, Mardanov AV, Grabovich MY, Muntyan MS. Phylogeny and Metabolic Potential of New Giant Sulfur Bacteria of the Family Beggiatoaceae from Coastal-Marine Sulfur Mats of the White Sea. International Journal of Molecular Sciences. 2024; 25(11):6028. https://doi.org/10.3390/ijms25116028
Chicago/Turabian StyleRavin, Nikolai V., Tatyana S. Rudenko, Alexey V. Beletsky, Dmitry D. Smolyakov, Andrey V. Mardanov, Margarita Yu. Grabovich, and Maria S. Muntyan. 2024. "Phylogeny and Metabolic Potential of New Giant Sulfur Bacteria of the Family Beggiatoaceae from Coastal-Marine Sulfur Mats of the White Sea" International Journal of Molecular Sciences 25, no. 11: 6028. https://doi.org/10.3390/ijms25116028