
Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Dorsi Muscle of Gannan Yak and Jeryak
 , ,
, ,
Abstract
1. Introduction
2. Results
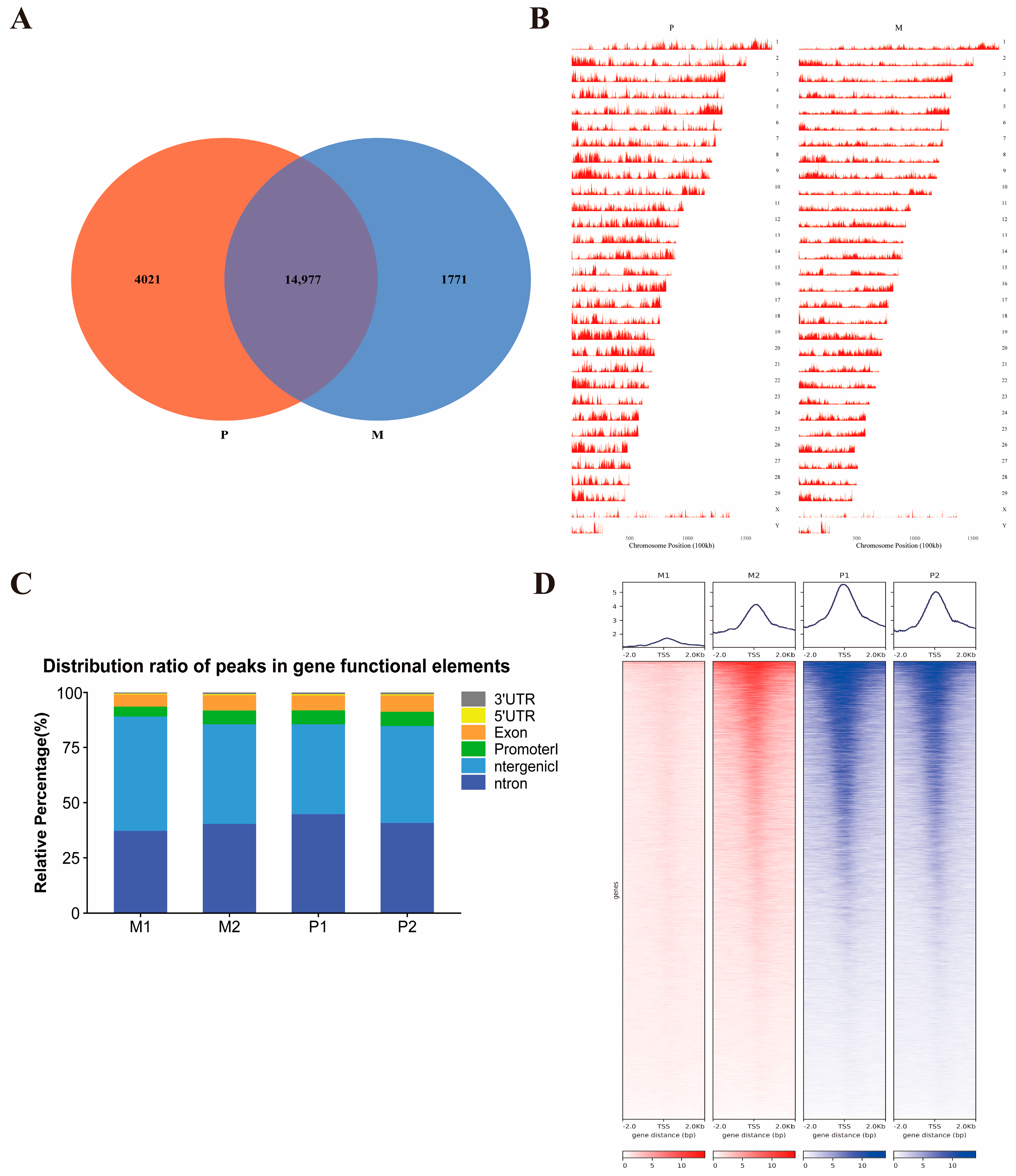
2.1. ATAC-seq Quality Control of the Gannan Yak and Jeryak Muscle Tissues
2.2. Accessibility of Chromatin in the Longissimus Dorsi Muscle of Gannan Yaks and Jeryaks
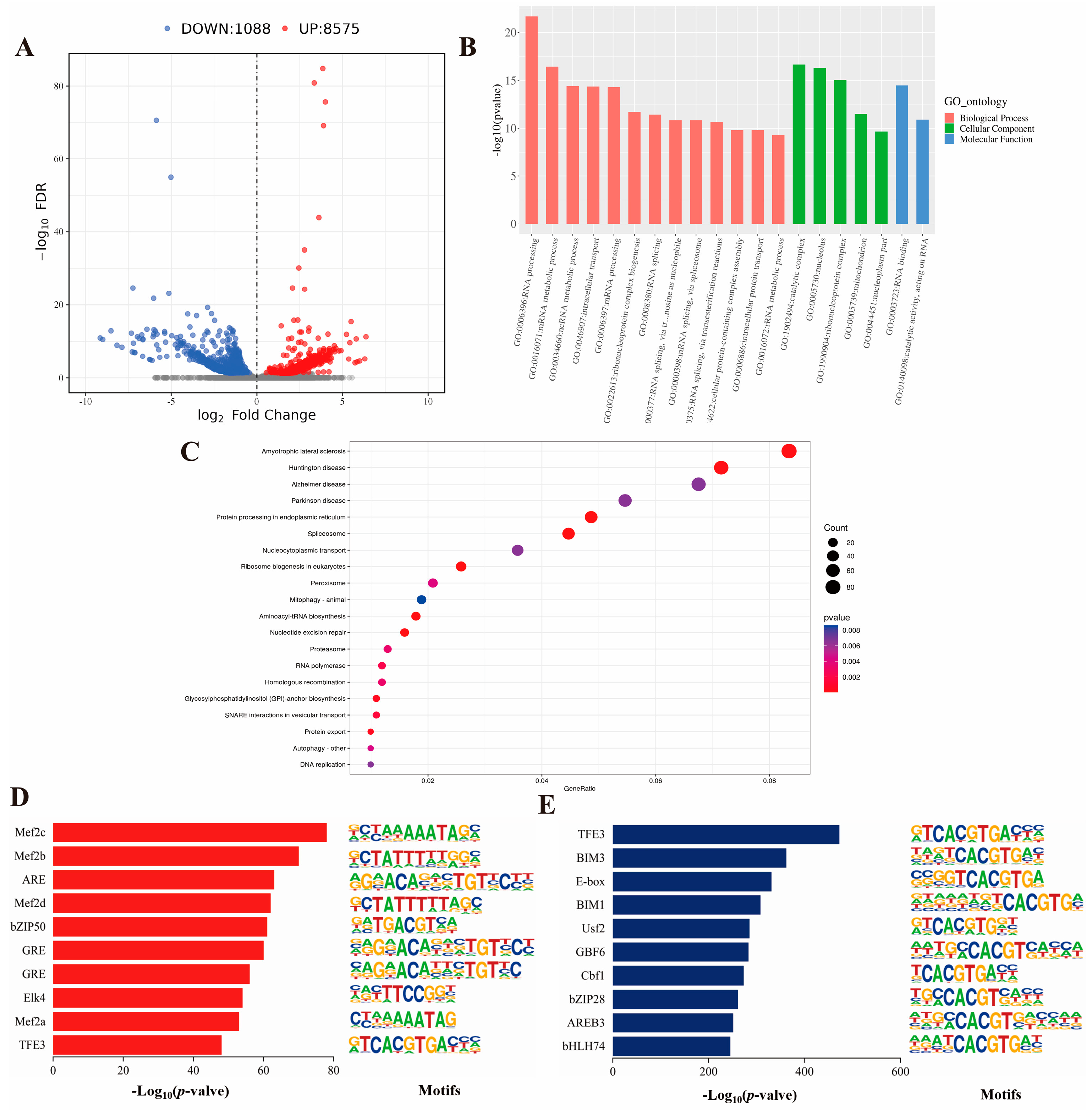
2.3. GO and KEGG Analysis of Genes Corresponding to Differential Peaks and Motif Analysis
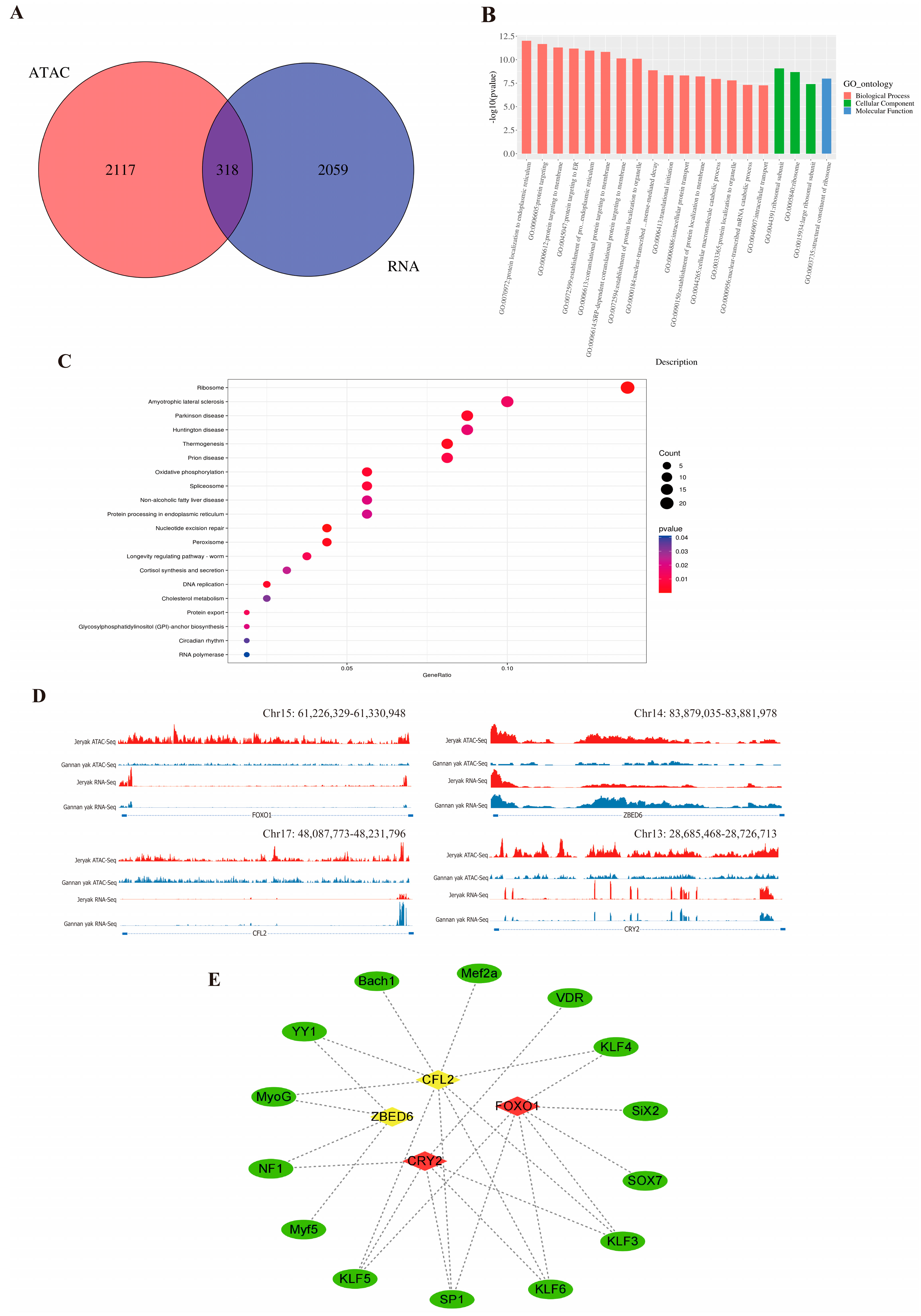
2.4. Integration Analysis of ATAC-seq and RNA-seq
2.5. Integration Validation of the Results by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Sample Collection
4.2. ATAC-seq Library Preparation, Sequencing and Analysis
4.3. GO and KEGG Enrichment Analysis of Differential Peak-Associated Genes
4.4. Integration Analysis of ATAC-seq and RNA-seq
4.5. Identification of Differentially Expressed Genes by qRT-PCR
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, S.Z.; Ma, D.L.; Li, B.M.; Baozhaxi, J.C.; Zhang, T.Y.; Xu, G.Q.; Zhang, H.X.; Wang, L.B.; Lamao, J.B.; Wang, W.B.; et al. Determination of growth and development indexes of Jeryak in Gannan alpine pasture area. Chin. Herbiv. Sci. 2019, 39, 73–75. [Google Scholar]
- Guo, S.Z.; Ma, D.L.; Yu, S.J.; Li, B.M.; Baozhaxi, J.C.; Wang, L.B.; Ma, Z.T.; Zhang, Y.Z.; Niu, X.Y.; Zhou, J.; et al. Observations on the effect of crossbreeding between Jersey cattle and Gannan yaks in alpine pastures. Chin. Bov. Sci. 2018, 44, 32–35. [Google Scholar]
- Song, C.; Huang, Y.; Yang, Z.; Ma, Y.; Chaogetu, B.; Zhuoma, Z.; Chen, H. RNA-Seq Analysis Identifies Differentially Expressed Genes Insubcutaneous Adipose Tissuein Qaidamford Cattle, Cattle-Yak, and Angus Cattle. Animals 2019, 9, 1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Hu, R.; Peng, Q.; Xue, B.; Wang, L. Comparison of carcass characteristics and meat quality between Simmental crossbred cattle, cattle-yaks and Xuanhan yellow cattle. J. Sci. Food Agr. 2021, 101, 3927–3932. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Shang, P.; Zhang, B.; Tian, X.; Nie, R.; Zhang, R.; Zhang, H. Function of the Porcine TRPC1 Gene in Myogenesis and Muscle Growth. Cells 2021, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef] [PubMed]
- Aase-Remedios, M.E.; Coll-Llado, C.; Ferrier, D. More Than One-to-Four via 2 R: Evidence of an Independent Amphioxus Expansion and Two-Gene Ancestral Vertebrate State for MyoD-Related Myogenic Regulatory Factors (MRFs). Mol. Biol. Evol. 2020, 37, 2966–2982. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.V.; Hughes, S.M. Mef2 and the skeletal muscle differentiation program. Semin. Cell Dev. Biol. 2017, 72, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Shang, Y.; Peng, J.; Jiang, S. Histone H3 Methyltransferase Suv39 h1 Prevents Myogenic Terminal Differentiation by Repressing MEF2 Activity in Muscle Cells. Int. J. Mol. Sci. 2016, 17, 1908. [Google Scholar] [CrossRef] [PubMed]
- Buas, M.F.; Kadesch, T. Regulation of skeletal myogenesis by Notch. Exp. Cell Res. 2010, 316, 3028–3033. [Google Scholar] [CrossRef]
- Girardi, F.; Le Grand, F. Wnt Signaling in Skeletal Muscle Development and Regeneration. Prog. Mol. Biol. Transl. 2018, 153, 157–179. [Google Scholar]
- Sakai, H.; Murakami, C.; Usuki, T.; Lu, Q.; Matsumoto, K.I.; Urano, T.; Sakane, F. Diacylglycerol kinase eta regulates C2 C12 myoblast proliferation through the mTOR signaling pathway. Biochimie 2020, 177, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; La, Y.; Bao, P.; Chu, M.; Guo, X.; Wu, X.; Pei, J.; Ding, X.; Liang, C.; Yan, P. Regulatory Role of N6-Methyladenosine in Longissimus Dorsi Development in Yak. Front. Vet. Sci. 2022, 9, 757115. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Dai, R.; Meng, G.; Dingkao, R.; Wang, X.; Ren, W.; Ma, X.; Wu, X.; Chu, M.; La, Y.; et al. Transcriptome-Wide Study of mRNAs and lncRNAs Modified by m(6)A RNA Methylation in the Longissimus Dorsi Muscle Development of Cattle-Yak. Cells 2022, 11, 3654. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Y.; Feng, D.; Yu, Y.; Long, H.; Hu, Z.; Lu, Q.; Zhao, M. Integrated analysis of ATAC-seq and RNA-seq reveals the transcriptional regulation network in SLE. Int. Immunopharmacol. 2023, 116, 109803. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Hou, X.; Liu, X.; Wang, L.; Gao, H.; Zhao, F.; Shi, L.; Shi, L.; Yan, H.; Deng, T.; et al. The landscape of chromatin accessibility in skeletal muscle during embryonic development in pigs. J. Anim. Sci. Biotechnol. 2021, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Vrljicak, P.; Lucas, E.S.; Lansdowne, L.; Lucciola, R.; Muter, J.; Dyer, N.P.; Brosens, J.J.; Ott, S. Analysis of chromatin accessibility in decidualizing human endometrial stromal cells. FASEB J. 2018, 32, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Kissane, S.; Dhandapani, V.; Orsini, L. Protocol for assay of transposase accessible chromatin sequencing in non-model species. STAR Protoc. 2021, 2, 100341. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Si, J.; Yue, J.; Zhao, M.; Qi, W.; Zhu, S.; Mo, J.; Wang, L.; Lan, G.; Liang, J. The Landscape of Accessible Chromatin and Developmental Transcriptome Maps Reveal a Genetic Mechanism of Skeletal Muscle Development in Pigs. Int. J. Mol. Sci. 2023, 24, 6413. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Hu, X.; Zhang, Y.; Li, H.; Zhang, Q.; Cai, W.; Wang, Z.; Zhu, B.; Xu, L.; et al. Transcriptional states and chromatin accessibility during bovine myoblasts proliferation and myogenic differentiation. Cell Prolif. 2022, 55, e13219. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Ma, Z.; Tang, Z.; Yu, L.; Liu, S.; Huang, T.; Wang, P.; Wu, T.; Song, Z.; Zhang, H.; et al. Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Muscle of Luchuan and Duroc Pigs. Front. Nutr. 2021, 8, 742672. [Google Scholar] [CrossRef]
- Shashikant, T.; Ettensohn, C.A. Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol. 2019, 151, 219–235. [Google Scholar] [PubMed]
- Wu, X.; Yang, Y.; Zhong, C.; Guo, Y.; Wei, T.; Li, S.; Lin, H.; Liu, X. Integration of ATAC-seq and RNA-seq Unravels Chromatin Accessibility during Sex Reversal in Orange-Spotted Grouper (Epinephelus coioides). Int. J. Mol. Sci. 2020, 21, 2800. [Google Scholar] [CrossRef]
- Yin, H.; He, H.; Cao, X.; Shen, X.; Han, S.; Cui, C.; Zhao, J.; Wei, Y.; Chen, Y.; Xia, L.; et al. MiR-148 a-3 p Regulates Skeletal Muscle Satellite Cell Differentiation and Apoptosis via the PI3 K/AKT Signaling Pathway by Targeting Meox2. Front. Genet. 2020, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.Z.; Bao, Y.Q.; Ma, D.L.; Li, B.M.; Wang, W.B.; Xu, G.Q.; Hu, J.; Baozhaxi, J.C.; Wang, L.B.; Zhang, H.X.; et al. Determination of slaughter performance and meat quality of Jeryak in alpine pasturing area. Chin. Herbiv. Sci. 2019, 39, 72–74. [Google Scholar]
- Huang, C.; Ge, F.; Ma, X.; Dai, R.; Dingkao, R.; Zhaxi, Z.; Burenchao, G.; Bao, P.; Wu, X.; Guo, X.; et al. Comprehensive Analysis of mRNA, lncRNA, circRNA, and miRNA Expression Profiles and Their ceRNA Networks in the Longissimus Dorsi Muscle of Cattle-Yak and Yak. Front. Genet. 2021, 12, 772557. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Huang, Y. Accessible chromatin reveals regulatory mechanisms underlying cell fate decisions during early embryogenesis. Sci. Rep. 2021, 11, 7896. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Pei, J.; Xiong, L.; Guo, S.; Wang, X.; Kang, Y.; Guo, X. Analysis of Chromatin Openness in Testicle Tissue of Yak and Cattle-Yak. Int. J. Mol. Sci. 2022, 23, 15810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, W.; Liu, X.; Du, X.; Zhang, K.; Zhang, Y.; Song, Y.; Zi, Y.; Qiu, Q.; Lenstra, J.A.; et al. Structural Variants Selected during Yak Domestication Inferred from Long-Read Whole-Genome Sequencing. Mol. Biol. Evol. 2021, 38, 3676–3680. [Google Scholar] [CrossRef] [PubMed]
- Nezhad, F.Y.; Riermeier, A.; Schonfelder, M.; Becker, L.; de Angelis, M.H.; Wackerhage, H. Skeletal muscle phenotyping of Hippo gene-mutated mice reveals that Lats1 deletion increases the percentage of type I muscle fibers. Transgenic Res. 2022, 31, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Copur, A.; Marchiano, F.; Hein, M.Y.; Alpern, D.; Russeil, J.; Luis, N.M.; Mann, M.; Deplancke, B.; Habermann, B.H.; Schnorrer, F. The Hippo pathway controls myofibril assembly and muscle fiber growth by regulating sarcomeric gene expression. eLife 2021, 10, e63726. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, I.; Sanjaya, A.; Lesmana, R.; Yen, P.M.; Goenawan, H. Hippo pathway effectors YAP and TAZ and their association with skeletal muscle ageing. J. Physiol. Biochem. 2021, 77, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Naka, A.; Iida, K.T.; Nakagawa, Y.; Iwasaki, H.; Takeuchi, Y.; Satoh, A.; Matsuzaka, T.; Ishii, K.A.; Kobayashi, K.; Yatoh, S.; et al. TFE3 inhibits myoblast differentiation in C2 C12 cells via down-regulating gene expression of myogenin. Biochem. Biophys. Res. Commun. 2013, 430, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.J.; Cole, C.J.; Josselyn, S.A. Emerging roles for MEF2 transcription factors in memory. Genes Brain Behav. 2014, 13, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Pon, J.R.; Wong, J.; Saberi, S.; Alder, O.; Moksa, M.; Grace, C.S.; Morin, G.B.; Hoodless, P.A.; Hirst, M.; Marra, M.A. MEF2 B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2 B target gene activation. Nat. Commun. 2015, 6, 7953. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Hancock, W.W.; Brancolini, C. MEF2 and the tumorigenic process, hic sunt leones. BBA Rev. Cancer 2018, 1870, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, A.; Sekrecki, M.; Szczesniak, M.W.; Sobczak, K. MEF2 C shapes the microtranscriptome during differentiation of skeletal muscles. Sci. Rep. 2021, 11, 3476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Raza, S.H.; Wei, D.; Yaping, S.; Chao, J.; Jin, W.; Almohaimeed, H.M.; Batarfi, M.A.; Assiri, R.; Aggad, W.S.; et al. Roles of MEF2 A and MyoG in the transcriptional regulation of bovine LATS2 gene. Res. Vet. Sci. 2022, 152, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Nelson, B.R.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of MEF2 A, C, and D for skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 4109–4114. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Zhang, J.; Song, Y.; Song, X.; Wu, H.; Jiao, R.; Li, L.; Zhang, G.; Wei, D. FOXO1 regulates bovine skeletal muscle cells differentiation by targeting MYH3. Int. J. Biol. Macromol. 2024, 260, 129643. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Xue, T.; Liu, S.B.; Geng, S.; Shi, X.; Qian, P.; He, W.; Zheng, J.; Li, Y.; Lou, J.; et al. Loss of CRY2 promotes regenerative myogenesis by enhancing PAX7 expression and satellite cell proliferation. Medcomm 2023, 4, e202. [Google Scholar] [CrossRef]
- Liu, L.; Wang, S.; Tian, W.; Xu, C.; Wei, C.; Cui, K.; Jiang, L.; Wang, D. Effect of Zbed6 Single-Allele Knockout on the Growth and Development of Skeletal Muscle in Mice. Biology 2023, 12, 325. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, T.; Ma, Y.; Wu, X.; Mao, Y.; Yang, Z.; Chen, H. New Insight into Muscle-Type Cofilin (CFL2) as an Essential Mediator in Promoting Myogenic Differentiation in Cattle. Bioengineering 2022, 9, 729. [Google Scholar] [CrossRef] [PubMed]
- Ming, H.; Sun, J.; Pasquariello, R.; Gatenby, L.; Herrick, J.R.; Yuan, Y.; Pinto, C.R.; Bondioli, K.R.; Krisher, R.L.; Jiang, Z. The landscape of accessible chromatin in bovine oocytes and early embryos. Epigenetics 2021, 16, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Akopyan, G.; Garban, H.; Bonavida, B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 2006, 25, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhou, J.; Li, Y.; Zhao, Y.; Yuan, J.; Cao, Y.; Wang, L.; Zhang, Z.; Zhang, B.; Wang, C.C.; et al. YY1 regulates skeletal muscle regeneration through controlling metabolic reprogramming of satellite cells. EMBO J. 2019, 38, e99727. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.H.; Zhong, Y.; Chen, Y.H.; Zhang, Z.W. Research progress on the roles of Kruppel-like factors in muscle tissues. Yi Chuan 2018, 40, 733–748. [Google Scholar] [PubMed]
- Cai, S.; Wang, X.; Xu, R.; Liang, Z.; Zhu, Q.; Chen, M.; Lin, Z.; Li, C.; Duo, T.; Tong, X.; et al. KLF4 regulates skeletal muscle development and regeneration by directly targeting P57 and Myomixer. Cell Death Dis. 2023, 14, 612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.H.; Yin, H.D.; Li, J.J.; Wang, Y.; Yang, C.W.; Jiang, X.S.; Du, H.R.; Liu, Y.P. KLF5 regulates chicken skeletal muscle atrophy via the canonical Wnt/beta-catenin signaling pathway. Exp. Anim. 2020, 69, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Matsumoto, M.; Katoh, Y.; Liu, L.; Ochiai, K.; Aizawa, Y.; Nagatomi, R.; Okuno, H.; Itoi, E.; Igarashi, K. Bach1 promotes muscle regeneration through repressing Smad-mediated inhibition of myoblast differentiation. PLoS ONE 2020, 15, e0236781. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Zhou, M.; Ruan, J.; He, A.; Ma, C.; Wu, W.; Lai, D.; Fan, Y.; Gao, A.; Weng, W.; et al. Genome-Wide Identification, Evolution, and Expression Pattern Analysis of the GATA Gene Family in Tartary Buckwheat (Fagopyrum tataricum). Int. J. Mol. Sci. 2022, 23, 12434. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, B.; Chen, H.; Yin, Q.; Liu, Y.; Xiang, Y.; Zhang, B.; Liu, B.; Wang, Q.; Xia, W.; et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 2016, 534, 652–657. [Google Scholar] [CrossRef]
- Lu, Z.; Hofmeister, B.T.; Vollmers, C.; DuBois, R.M.; Schmitz, R.J. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 2017, 45, e41. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Strbenac, D.; Stirzaker, C.; Statham, A.L.; Song, J.; Speed, T.P.; Clark, S.J. Copy-number-aware differential analysis of quantitative DNA sequencing data. Genome Res. 2012, 22, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Clean Ratio | Q20 | Q30 | GC |
|---|---|---|---|---|---|---|---|---|
| M1 | 91,332,714 | 13,699,907,100 | 90,778,160 | 9,844,432,268 | 99.39% | 98.65% | 95.57% | 49.01% |
| M2 | 78,231,904 | 11,734,785,600 | 77,589,728 | 9,045,505,914 | 99.18% | 98.33% | 94.78% | 50.65% |
| P1 | 116,244,208 | 17,436,631,200 | 115,487,052 | 13,089,463,824 | 99.35% | 98.40% | 94.88% | 50.53% |
| P2 | 99,271,020 | 14,890,653,000 | 98,734,712 | 10,698,377,169 | 99.46% | 98.38% | 94.76% | 49.35% |
| Sample | Total Reads | Mapped Reads | Map Rate | Uniq | Paired |
|---|---|---|---|---|---|
| M1 | 90,778,160 | 77,361,144 | 85.22% | 62,555,109 | 60,227,766 |
| M2 | 77,589,728 | 66,895,805 | 86.22% | 56,009,360 | 53,197,036 |
| P1 | 115,487,052 | 98,334,567 | 85.15% | 83,789,052 | 79,604,294 |
| P2 | 98,734,712 | 82,898,529 | 83.96% | 69,904,662 | 66,443,372 |
| mRNA | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| FOXO1 | ACCCCACAAGGTTTCCGATG | AGTGTCCCCTCTCTTTCCAAC |
| CFL2 | ATTCTGGGCTCCTGAAAGTGC | TCTCTCCAAGTGTGGAACGG |
| ZBED6 | ACTGGACAAGGGCCAACAAA | AAGCTTCCACTGCTTCCAGC |
| PARK7 | TAAGGTCACCGTTGCAGGTC | CCTCCTGGAAGAACCACCAC |
| DDX23 | AACGGCATCGTTCAAGGGAT | TCCCTGTCTCGGTCCTTCTT |
| DDX47 | TCTGCCCATTCTCAACGCAT | CAATGACAGCACACTGCACC |
| ADARB1 | AGCTGAACGAGATCAAGCCC | CTCGAACACCTGTCCGTTGA |
| NPC1 | TGCCACAGGATGGCTATGAC | GGACAGAAACTGCAGAGGCA |
| CRY2 | GATTCCCCACTGAAGGGCTC | GCGGTAGTAGAAGAGACGGC |
| GAPDH | CCACGAGAAGTATAACAACACC | GTCATAAGTCCCTCCACGAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Guo, D.; Wei, Y.; Li, J.; Jia, X.; Niu, Y.; Liu, Z.; Bai, Y.; Chen, Z.; Shi, B.; et al. Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Dorsi Muscle of Gannan Yak and Jeryak. Int. J. Mol. Sci. 2024, 25, 6029. https://doi.org/10.3390/ijms25116029
Zhao Z, Guo D, Wei Y, Li J, Jia X, Niu Y, Liu Z, Bai Y, Chen Z, Shi B, et al. Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Dorsi Muscle of Gannan Yak and Jeryak. International Journal of Molecular Sciences. 2024; 25(11):6029. https://doi.org/10.3390/ijms25116029
Chicago/Turabian StyleZhao, Zhidong, Dashan Guo, Yali Wei, Jingsheng Li, Xue Jia, Yanmei Niu, Zhanxin Liu, Yanbin Bai, Zongchang Chen, Bingang Shi, and et al. 2024. "Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Dorsi Muscle of Gannan Yak and Jeryak" International Journal of Molecular Sciences 25, no. 11: 6029. https://doi.org/10.3390/ijms25116029
APA StyleZhao, Z., Guo, D., Wei, Y., Li, J., Jia, X., Niu, Y., Liu, Z., Bai, Y., Chen, Z., Shi, B., Zhang, X., Hu, J., Wang, J., Liu, X., & Li, S. (2024). Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Dorsi Muscle of Gannan Yak and Jeryak. International Journal of Molecular Sciences, 25(11), 6029. https://doi.org/10.3390/ijms25116029