
Michael Acceptors as Anti-Cancer Compounds: Coincidence or Causality?

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Phosphorylation regulates numerous cellular processes such as the cell cycle, and growth and signal transduction pathways [2].
- Acetylation primarily affects protein stability and interactions and often occurs at the N-terminus [3].
- Ubiquitination signals protein degradation [4].
- Methylation alters the interactions of proteins with DNA, RNA or other proteins [5].
- Glycosylation affects the folding, stability and cell adhesion of proteins [6].
- Oxidation occurs when reactive oxygen species (ROS) donate electrons to proteins. It can extensively modify the primary structure of proteins and peptides and often leads to modifications of higher-order structures. It is implicated in human disease, carcinogenesis and aging [7].
2. Michael Reaction and Covalent Binding to Proteins
2.1. Michael Reaction
2.2. The Kinetics of Inhibition via Michael Reaction
2.3. Reversible and Irreversible Covalent Binding to Proteins
- Drug discovery and development, by covalently binding to specific target proteins, thereby inhibiting their function or altering their activity.
- Inhibition of enzymes by binding to and inactivating them, thereby interfering with the regulation of cellular processes.
- Protein engineering, inducing covalent modifications in proteins modifies their stability, function or other properties.
3. Transcription Factors That May Be Potential Targets of Michael Acceptor Compounds in Neoplastic Metabolism
- They can interact with transcription factors, altering their ability to activate or repress the transcription of certain genes [49].
- They can affect the stability of mRNA and thus influence the production of certain proteins. Changes in mRNA stability have a significant impact on protein production in cells [50].
3.1. Transcription Factor NF-κB
3.2. Transcription Factor PPAR-γ
- PPAR-γ agonists can suppress inflammatory responses in psoriatic skin lesions and attenuate associated comorbidities [63].
- Neurodegenerative diseases, signaling networks, insulin sensitivity, glucose homeostasis, fatty acid oxidation, immune responses, redox balance, cardiovascular integrity and cell fate depend on the PPAR-γ factor [64]. PPAR-γ agonists can reduce amyloid and tau pathologies, and neuroinflammation and improve memory impairment in models of Alzheimer’s disease [65].
- Suppression of PPAR-γ under disease conditions exacerbates inflammatory and fibrogenic factors, contributing to kidney damage [66].
- PPAR-γ is associated with susceptibility to neuronal damage in models of brain disease [67].
3.3. Transcription Factor STAT3
- STAT3 contributes to metastasis and drug resistance; its hyperactivation is associated with a poor clinical prognosis in most human cancers [74].
- STAT3 is observed in both cancer and non-cancer cells in the tumor microenvironment, inhibits the expression of immune activation and promotes immunosuppressive factors, thus contributing to tumor progression [75].
- The interplay between STAT3 and non-coding RNAs has attracted attention and highlighted its regulatory role in gene expression networks [76].
3.4. The Nuclear Export Receptor Exportin-1
3.5. Oncoprotein c-Myc
4. Michael Acceptors in Current Chemotherapy Treatments
4.1. Tyrosine Kinase Inhibitors (TKIs)
4.2. Cyclin-Dependent Kinases (CDKs) Inhibitors
4.3. Aurora Kinase (AURK) Inhibitors
4.4. Bruton’s Tyrosine Kinase (BTK) Inhibitors
4.5. Nitro Fatty Acids (NO2-FAs)
4.6. Anthracycline Family of Chemotherapy Drugs
- Doxorubicin, which is used in the treatment of breast cancer, lung cancer, ovarian cancer, liver cancer, thyroid cancer, leukemias and lymphomas [135].
- Daunorubicin, which is used in the treatment of acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), chronic myeloid leukemia (CML) and Kaposi’s sarcoma.
- Epirubicin, which is used for breast cancer, ovarian cancer, stomach cancer, lung cancer and lymphoma.
- Idarubicin, which is prescribed for acute myeloid leukemia.
4.7. Family of Selective Inhibitors of Nuclear Export (SINEs)
5. Natural Michael Acceptors in Cancer Prevention and Treatment
5.1. Alkaloids
5.2. Terpenes and Terpenoids
5.2.1. Sesquiterpenes
5.2.2. Diterpenoids
5.2.3. Triterpenoids or Steroids
5.2.4. Tetraterpenoids
5.2.5. Hop-Derived Bitter Acids
5.3. Polyketides
- They show anti-cancer activity against various types of cancer, including breast, lung, colon and prostate cancer [217].
- They have anti-inflammatory properties that could be useful in the treatment of inflammatory diseases such as rheumatoid arthritis and inflammatory bowel disease [218].
- They have antiparasitic activity against parasites, including the parasite that causes malaria [223].
5.4. Polyphenols
5.4.1. Esters Derived from Caffeic Acid
- Caffeic acid phenethyl ester (CAPE), a phenolic compound recognized as one of the major active components in propolis, with significant biological activities [237].
- Dactylifric acid (DA), also known as date acid or 5-O-caffeoylshikimic acid, an ester derived from caffeic acid and shikimic acid, found mainly in dates (Phoenix dactylifera fruits) [238].
- Chlorogenic acid (CGA), the ester formed from caffeic acid and quinic acid. The collective term “chlorogenic acids” refers to a family of related polyphenols that include hydroxycinnamic acids bound to quinic acid (such as caffeic acid, ferulic acid and p-coumaric acid). Chlorogenic acids are widely used in foods such as coffee beans, coffee drinks, mate and tea and include numerous isomers, each with different sensory properties [239].
- Rosmarinic acid (RA), isolated from Rosmarinus officinalis L., and commonly known as rosemary.
5.4.2. Chalcones
5.4.3. Curcuminoids
- Protecting cells from free radical damage that contributes to aging and disease [268].
- They show neuroprotective effects by protecting brain cells from oxidative stress and inflammation, both of which are associated with neurodegenerative diseases such as Alzheimer’s and Parkinson’s [269].
- They have anti-inflammatory properties that are crucial in combating chronic inflammation such as arthritis, cancer and cardiovascular disease [270].
- They have shown anti-tumor properties in laboratory studies, which has led to ongoing clinical trials to investigate their effectiveness in cancer treatment [271].
- They have an anti-acidifying effect, which may be beneficial for people with heartburn or acid reflux as it reduces gastric acid secretion.
- They act as a radioprotective agent by protecting cells from radiation-induced damage, which is particularly beneficial for people undergoing radiotherapy for cancer [272].
- They relieve arthritis symptoms by reducing inflammation and associated pain [273].
5.4.4. Flavonoids
5.4.5. Coumarins
- Difficulty in large-scale isolation: they are often difficult to isolate in large quantities, which can hinder their development into drugs.
- Understanding the action mechanism can be challenging, which can slow down their pharmaceutical development.
- Pharmaceutical development challenges: even when they show promising anti-cancer properties, it can be difficult to develop them into a drug that can be used in clinical settings.
- Bioavailability and solubility issues of MA, such as curcumin or resveratrol, have shown potent anti-cancer activity but have poor solubility and bioavailability when administered alone.
6. Antibody and Peptide-Mediated Delivery of Michael Acceptors
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Su, M.-G.; Weng, J.T.-Y.; Hsu, J.B.-K.; Huang, K.-Y.; Chi, Y.-H.; Lee, T.-Y. Investigation and identification of functional post-translational modification sites associated with drug binding and protein-protein interactions. BMC Syst. Biol. 2017, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, T.; Poncet, S.; Derouiche, A.; Shi, L.; Mijakovic, I.; Noirot-Gros, M.-F. Role of Protein Phosphorylation in the Regulation of Cell Cycle and DNA-Related Processes in Bacteria. Front. Microbiol. 2016, 7, 184. [Google Scholar] [CrossRef]
- Ree, R.; Varland, S.; Arnesen, T. Spotlight on protein N-terminal acetylation. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin–Proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Solá, R.J.; Griebenow, K. Effects of glycosylation on the stability of protein pharmaceuticals. J. Pharm. Sci. 2009, 98, 1223–1245. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Impact of Reactive Species on Amino Acids—Biological Relevance in Proteins and Induced Pathologies. Int. J. Mol. Sci. 2022, 23, 14049. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Bhopatkar, A.A.; Uversky, V.N.; Rangachari, V. Disorder and cysteines in proteins: A design for orchestration of conformational see-saw and modulatory functions. Prog. Mol. Biol. Transl. Sci. 2020, 174, 331–373. [Google Scholar] [CrossRef]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Munguira, E.B.; Juan, C.A.; Plou, F.J.; Lebeña, E.P. Electrophilic Compounds in the Human Diet and Their Role in the Induction of the Transcription Factor NRF2. Int. J. Mol. Sci. 2024, 25, 3521. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. From reactive species to disease development: Effect of oxidants and antioxidants on the cellular biomarkers. J. Biochem. Mol. Toxicol. 2023, 37, e23455. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.-T.; Chen, C.; Chen, R.-X.; Li, R.; Chen, W.-L.; Jiang, G.-H.; Du, L.-L. Michael acceptor molecules in natural products and their mechanism of action. Front. Pharmacol. 2022, 13, 1033003. [Google Scholar] [CrossRef] [PubMed]
- Kühn, B.; Brat, C.; Fettel, J.; Hellmuth, N.; Maucher, I.V.; Bulut, U.; Hock, K.J.; Grimmer, J.; Manolikakes, G.; Rühl, M.; et al. Anti-inflammatory nitro-fatty acids suppress tumor growth by triggering mitochondrial dysfunction and activation of the intrinsic apoptotic pathway in colorectal cancer cells. Biochem. Pharmacol. 2018, 155, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Tokoroyama, T. Discovery of the Michael reaction. Eur. J. Org. Chem. 2010, 2010, 2009–2016. [Google Scholar] [CrossRef]
- Michael, A. Ueber die Addition von Natriumacetessig-und Natriummalonsäureäthern zu den Aethern ungesättigter Säuren. J. Für Prakt. Chem. 1887, 35, 349–356. [Google Scholar] [CrossRef]
- Asthalter, T.; Rajagopalan, S.; Kauf, T.; Rabe, V.; Christoffers, J. Monitoring reaction intermediates in the FeCl3-catalyzed michael reaction by nuclear inelastic scattering. J. Phys. Chem. A 2008, 112, 11514–11518. [Google Scholar] [CrossRef] [PubMed]
- Rosiak, A.; Hoenke, C.; Christoffers, J. Synthesis of 3-Phenyl-4-piperidones from Acetophenone by Shapiro and Aza-Michael Reactions and Their Further Derivatization. Eur. J. Org. Chem. 2007, 2007, 4376–4382. [Google Scholar] [CrossRef]
- Christoffers, J.; Koripelly, G.; Rosiak, A.; Roessle, M. Recent advances in metal-catalyzed asymmetric conjugate additions. Synthesis 2007, 2007, 1279–1300. [Google Scholar] [CrossRef]
- Christoffers, J.; Scharl, H. Copper-Catalyzed Asymmetric Michael Reactions with α-Amino Acid Amides: Synthesis of an Optically Active Piperidine Derivative. Eur. J. Org. Chem. 2002, 2002, 1505–1508. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Vicario, J.L.; Badia, D.; Carrillo, L. Organocatalytic enantioselective Michael and hetero-Michael reactions. Synthesis 2007, 2007, 2065–2092. [Google Scholar] [CrossRef]
- Mather, B.D.; Viswanathan, K.; Miller, K.M.; Long, T.E. Michael addition reactions in macromolecular design for emerging technologies. Prog. Polym. Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Enders, D.; Haertwig, A.; Raabe, G.; Runsink, J. Enantioselective Synthesis of Vicinal Amino Alcohols by Oxa-Michael Addition of(−)-N-Formylnorephedrine to Nitroalkenes. Angew. Chem. Int. Ed. 1996, 35, 2388–2390. [Google Scholar] [CrossRef]
- Stewart, I.C.; Bergman, R.G.; Toste, F.D. Phosphine-catalyzed hydration and hydroalkoxylation of activated olefins: Use of a strong nucleophile to generate a strong base. J. Am. Chem. Soc. 2003, 125, 8696–8697. [Google Scholar] [CrossRef] [PubMed]
- Nising, C.F.; Bräse, S. Recent developments in the field of oxa-Michael reactions. Chem. Soc. Rev. 2012, 41, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Nising, C.F.; Bräse, S. The oxa-Michael reaction: From recent developments to applications in natural product synthesis. Chem. Soc. Rev. 2008, 37, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Roselló, M.; Aceña, J.L.; Simón-Fuentes, A.; del Pozo, C. A general overview of the organocatalytic intramolecular aza-Michael reaction. Chem. Soc. Rev. 2014, 43, 7430–7453. [Google Scholar] [CrossRef] [PubMed]
- Wabnitz, T.C.; Yu, J.; Spencer, J.B. Evidence that protons can be the active catalysts in lewis acid mediated hetero-michael addition reactions. Chem.—A Eur. J. 2004, 10, 484–493. [Google Scholar] [CrossRef]
- Krenske, E.H.; Petter, R.C.; Houk, K.N. Kinetics and Thermodynamics of reversible thiol additions to mono- and diactivated michael acceptors: Implications for the design of drugs that bind covalently to cysteines. J. Org. Chem. 2016, 81, 11726–11733. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The thiol-michael addition click reaction: A Powerful and widely used tool in materials chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Allen, C.F.H.; Fournier, J.O.; Humphlett, W.J. the thermal reversibility of the michael reaction: Iv. Thiol adducts. Can. J. Chem. 1964, 42, 2616–2620. [Google Scholar] [CrossRef]
- Avonto, C.; Taglialatela Scafati, O.; Pollastro, F.; Minassi, A.; Di Marzo, V.; De Petrocellis, L.; Appendino, G.B. An NMR spectroscopic method to identify and classify thiol-trapping agents: Revival of Michael acceptors for drug discovery? Angew. Chem. Int. Ed. 2011, 50, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Krenske, E.H.; Petter, R.C.; Zhu, Z.; Houk, K. Transition states and energetics of nucleophilic additions of thiols to substituted α, β-unsaturated ketones: Substituent effects involve enone stabilization, product branching, and solvation. J. Org. Chem. 2011, 76, 5074–5081. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Saint-Dizier, A.; Lannou, M.; Lenzen, A. The Phospha-Michael Addition in Organic Synthesis. Eur. J. Org. Chem. 2006, 2006, 29–49. [Google Scholar] [CrossRef]
- de Freitas Silva, M.; Pruccoli, L.; Morroni, F.; Sita, G.; Seghetti, F.; Viegas, C.; Tarozzi, A. The Keap1/Nrf2-ARE Pathway as a Pharmacological Target for Chalcones. Molecules 2018, 23, 1803. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef]
- Chu, H.-W.; Sethy, B.; Hsieh, P.-W.; Horng, J.-T. Identification of Potential Drug Targets of Broad-Spectrum Inhibitors with a Michael Acceptor Moiety Using Shotgun Proteomics. Viruses 2021, 13, 1756. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Han, X.; Xiao, X.; Zhou, J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules 2022, 27, 7728. [Google Scholar] [CrossRef] [PubMed]
- Serafimova, I.M.; A Pufall, M.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef]
- Pritchard, R.B.; Lough, C.E.; Currie, D.J.; Holmes, H.L. Equilibrium reactions of n-butanethiol with some conjugated heteroenoid compounds. Can. J. Chem. 1968, 46, 775–781. [Google Scholar] [CrossRef]
- Lough, C.E.; Currie, D.J.; Holmes, H.L. Rates of reaction of n-butanethiol with some conjugated heteroenoid compounds. Can. J. Chem. 1968, 46, 771–774. [Google Scholar] [CrossRef]
- Miller, R.M.; Paavilainen, V.O.; Krishnan, S.; Serafimova, I.M.; Taunton, J. Electrophilic Fragment-Based Design of Reversible Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2013, 135, 5298–5301. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Miller, R.M.; Tian, B.; Mullins, R.D.; Jacobson, M.P.; Taunton, J. Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc. 2014, 136, 12624–12630. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Lemus, E.; Hägglund, P.; López-Alarcón, C.; Davies, M.J. Oxidative Crosslinking of Peptides and Proteins: Mechanisms of Formation, Detection, Characterization and Quantification. Molecules 2021, 27, 15. [Google Scholar] [CrossRef] [PubMed]
- Faridoon; Ng, R.; Zhang, G.; Li, J.J. An update on the discovery and development of reversible covalent inhibitors. Med. Chem. Res. 2023, 32, 1039–1062. [Google Scholar] [CrossRef] [PubMed]
- Kiely-Collins, H.; Winter, G.E.; Bernardes, G.J. The role of reversible and irreversible covalent chemistry in targeted protein degradation. Cell Chem. Biol. 2021, 28, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; E Huma, Z.; Duncan, D. Reversible Covalent Inhibition─Desired Covalent Adduct Formation by Mass Action. ACS Chem. Biol. 2024, 19, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.G.; Francis, N.; Hill, R.; Waters, D.; Blanchard, C.; Santhakumar, A.B. Dietary Polyphenols and Gene Expression in Molecular Pathways Associated with Type 2 Diabetes Mellitus: A Review. Int. J. Mol. Sci. 2019, 21, 140. [Google Scholar] [CrossRef]
- Ding, S.; Jiang, H.; Fang, J. Regulation of Immune Function by Polyphenols. J. Immunol. Res. 2018, 2018, 1264074. [Google Scholar] [CrossRef]
- Chen, Z.; Hao, W.; Gao, C.; Zhou, Y.; Zhang, C.; Zhang, J.; Wang, R.; Wang, Y.; Wang, S. A polyphenol-assisted IL-10 mRNA delivery system for ulcerative colitis. Acta Pharm. Sin. B 2022, 12, 3367–3382. [Google Scholar] [CrossRef]
- Arafet, K.; Serrano-Aparicio, N.; Lodola, A.; Mulholland, A.J.; González, F.V.; Świderek, K.; Moliner, V. Mechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2021, 12, 1433–1444. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Jimi, E.; Huang, F.; Nakatomi, C. NF-κB Signaling Regulates Physiological and Pathological Chondrogenesis. Int. J. Mol. Sci. 2019, 20, 6275. [Google Scholar] [CrossRef]
- Marrugo, J.; Hernández, L.; Villalba, V. Prevalence of self-reported food allergy in Cartagena (Colombia) population. Allergol. Immunopathol. 2008, 36, 320–324. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF- B family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Huang, S.; Kim, K.; Musgrave, G.M.; Sharp, M.; Sinha, J.; Stansbury, J.W.; Musgrave, C.B.; Bowman, C.N. Determining Michael acceptor reactivity from kinetic, mechanistic, and computational analysis for the base-catalyzed thiol-Michael reaction. Polym. Chem. 2021, 12, 3619–3628. [Google Scholar] [CrossRef]
- Moreno, R.; Sobotzik, J.-M.; Schultz, C.; Schmitz, M.L. Specification of the NF-κB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKKε. Nucleic Acids Res. 2010, 38, 6029–6044. [Google Scholar] [CrossRef]
- Piesche, M.; Roos, J.; Kühn, B.; Fettel, J.; Hellmuth, N.; Brat, C.; Maucher, I.V.; Awad, O.; Matrone, C.; Steffensen, S.G.C.; et al. The Emerging Therapeutic Potential of Nitro Fatty Acids and Other Michael Acceptor-Containing Drugs for the Treatment of Inflammation and Cancer. Front. Pharmacol. 2020, 11, 1297. [Google Scholar] [CrossRef]
- Sugii, S.; Olson, P.; Sears, D.D.; Saberi, M.; Atkins, A.R.; Barish, G.D.; Hong, S.-H.; Castro, G.L.; Yin, Y.-Q.; Nelson, M.C.; et al. PPARγ activation in adipocytes is sufficient for systemic insulin sensitization. Proc. Natl. Acad. Sci. USA 2009, 106, 22504–22509. [Google Scholar] [CrossRef]
- Sobolev, V.V.; Tchepourina, E.; Korsunskaya, I.M.; Geppe, N.A.; Chebysheva, S.N.; Soboleva, A.G.; Mezentsev, A. The Role of Transcription Factor PPAR-γ in the Pathogenesis of Psoriasis, Skin Cells, and Immune Cells. Int. J. Mol. Sci. 2022, 23, 9708. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Wu, J.-S.; Tsai, H.-D.; Huang, C.-Y.; Chen, J.-J.; Sun, G.Y.; Lin, T.-N. Peroxisome Proliferator-activated receptor gamma (PPAR-γ) and neurodegenerative disorders. Mol. Neurobiol. 2012, 46, 114–124. [Google Scholar] [CrossRef]
- Govindarajulu, M.; Pinky, P.D.; Bloemer, J.; Ghanei, N.; Suppiramaniam, V.; Amin, R. Signaling Mechanisms of Selective PPARγ Modulators in Alzheimer’s Disease. PPAR Res. 2018, 2018, 10675. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Patial, V. Peroxisome proliferator-activated receptor gamma and its natural agonists in the treatment of kidney diseases. Front. Pharmacol. 2022, 13, 991059. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Kim, H.I.; Bang, Y.; Choi, H.J. Peroxisome proliferator-activated receptor gamma: A novel therapeutic target for cognitive impairment and mood disorders that functions via the regulation of adult neurogenesis. Arch. Pharmacal Res. 2021, 44, 553–563. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Chi, T.; Wang, M.; Wang, X.; Yang, K.; Xie, F.; Liao, Z.; Wei, P. PPAR-γ Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 737776. [Google Scholar] [CrossRef]
- Filippone, A.; Casili, G.; Scuderi, S.A.; Mannino, D.; Lanza, M.; Campolo, M.; Paterniti, I.; Capra, A.P.; Colarossi, C.; Bonasera, A.; et al. Sodium Propionate Contributes to Tumor Cell Growth Inhibition through PPAR-γ Signaling. Cancers 2022, 15, 217. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Tošić, I.; Frank, D.A. STAT3 as a mediator of oncogenic cellular metabolism: Pathogenic and therapeutic implications. Neoplasia 2021, 23, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, W.; Liang, L.; Zhou, Y.; Li, Y. The regulatory relationship between transcription factor STAT3 and noncoding RNA. Cell. Mol. Biol. Lett. 2024, 29, 4. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Stade, K.; Ford, C.S.; Guthrie, C.; Weis, K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 1997, 90, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, L.; Chen, L.; Gong, B.; Jia, D.; Sun, Q. Nuclear transport proteins: Structure, function, and disease relevance. Signal Transduct. Target. Ther. 2023, 8, 425. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Zhou, Q.; Burstein, E.; Yang, S.; Jia, D. Inhibiting cancer cell hallmark features through nuclear export inhibition. Signal Transduct. Target. Ther. 2016, 1, 16010. [Google Scholar] [CrossRef]
- Boons, E.; Vanstreels, E.; Jacquemyn, M.; Nogueira, T.C.; Neggers, J.E.; Vercruysse, T.; Oord, J.v.D.; Tamir, S.; Shacham, S.; Landesman, Y.; et al. Human Exportin-1 is a Target for Combined Therapy of HIV and AIDS Related Lymphoma. EBioMedicine 2015, 2, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Braggio, E.; Kortuem, K.M.; Egan, J.B.; Zhu, Y.X.; Xin, C.S.; E Tiedemann, R.; E Palmer, S.; Garbitt, V.M.; McCauley, D.; et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia 2013, 27, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-T.; Landesman, Y.; Acharya, C.; Calle, Y.; Zhong, M.Y.; Cea, M.; Tannenbaum, D.; Cagnetta, A.; Reagan, M.R.; Munshi, A.; et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia 2014, 28, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Ishizawa, J.; Ruvolo, V.; Dilip, A.; Quintás-Cardama, A.; McDonnell, T.J.; Neelapu, S.S.; Kwak, L.W.; Shacham, S.; Kauffman, M.; et al. Induction of p53-mediated transcription and apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma. Cancer Sci. 2014, 105, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, M.; Tamayo, A.T.; Shacham, S.; Kauffman, M.; Lee, J.; Zhang, L.; Ou, Z.; Li, C.; Sun, L.; et al. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp. Hematol. 2013, 41, 67–78.e64. [Google Scholar] [CrossRef] [PubMed]
- Lapalombella, R.; Sun, Q.; Williams, K.; Tangeman, L.; Jha, S.; Zhong, Y.; Goettl, V.; Mahoney, E.; Berglund, C.; Gupta, S.; et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 2012, 120, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- E Conway, A.; Haldeman, J.M.; Wechsler, D.S.; Lavau, C.P. A critical role for CRM1 in regulating HOXA gene transcription in CALM-AF10 leukemias. Leukemia 2015, 29, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Kornblau, S.M.; Ruvolo, V.; Dilip, A.; Duvvuri, S.; Davis, R.E.; Zhang, M.; Wang, Z.; Coombes, K.R.; Zhang, N.; et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013, 121, 4166–4174. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gery, S.; Sun, H.; Shacham, S.; Kauffman, M.; Koeffler, H.P. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2014, 74, 487–495. [Google Scholar] [CrossRef]
- Zhou, F.; Qiu, W.; Yao, R.; Xiang, J.; Sun, X.; Liu, S.; Lv, J.; Yue, L. CRM1 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Med. Oncol. 2013, 30, 726. [Google Scholar] [CrossRef]
- VAN DER Watt, P.J.; Zemanay, W.; Govender, D.; Hendricks, D.T.; Parker, M.I.; Leaner, V.D. Elevated expression of the nuclear export protein, Crm1 (exportin 1), associates with human oesophageal squamous cell carcinoma. Oncol. Rep. 2014, 32, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Kauffman, M.; Shacham, S.; Landesman, Y.; Yang, J.; Evans, C.P.; Weiss, R.H. CRM1 blockade by selective inhibitors of nuclear export attenuates kidney cancer growth. J. Urol. 2013, 189, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- van der Watt, P.J.; Maske, C.P.; Hendricks, D.T.; Parker, M.I.; Denny, L.; Govender, D.; Birrer, M.J.; Leaner, V.D. The Karyopherin proteins, Crm1 and Karyopherin β1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int. J. Cancer 2009, 124, 1829–1840. [Google Scholar] [CrossRef]
- Liu, X.; Chong, Y.; Liu, H.; Han, Y.; Niu, M. Novel reversible selective inhibitor of CRM1 for targeted therapy in ovarian cancer. J. Ovarian Res. 2015, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Noske, A.; Weichert, W.; Niesporek, S.; Röske, A.; Buckendahl, A.; Koch, I.; Sehouli, J.; Dietel, M.; Denkert, C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer 2008, 112, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-Y.; Yue, L.; Qiu, W.-S.; Wang, L.-W.; Zhou, X.-H.; Sun, Y.-J. Prognostic value of CRM1in pancreas cancer. Clin. Investig. Med. 2009, 32, E315. [Google Scholar] [CrossRef]
- Shen, A.; Wang, Y.; Zhao, Y.; Zou, L.; Sun, L.; Cheng, C. Expression of crm1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery 2009, 65, 153–160, discussion 159–160. [Google Scholar] [CrossRef]
- Ho, Y.; Yao, Y.; Dong, Y.; Lin, F.; Zhao, H.; Shen, Z.; Chen, P.; Sun, Y.-J.; Tang, L.-N.; Zheng, S.-E. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol. Rep. 2009, 21, 229–235. [Google Scholar] [CrossRef]
- Gao, W.; Lu, C.; Chen, L.; Keohavong, P. Overexpression of CRM1: A Characteristic Feature in a Transformed Phenotype of Lung Carcinogenesis and a Molecular Target for Lung Cancer Adjuvant Therapy. J. Thorac. Oncol. 2015, 10, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Zhi, P.; You, J.; Gao, J.-Q. Metformin synergistically suppress tumor growth with doxorubicin and reverse drug resistance by inhibiting the expression and function of P-glycoprotein in MCF7/ADR cells and xenograft models. Oncotarget 2018, 9, 2158–2174. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene— The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2021, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable” MYC. EBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef]
- Donati, G.; Amati, B. MYC and therapy resistance in cancer: Risks and opportunities. Mol. Oncol. 2022, 16, 3828–3854. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kambayashi, H.; Yamamoto, A.; Onishi, I.; Sugita, K.; Matsumura, M.; Ishibashi, S.; Ikeda, M.; Yamamoto, K.; Kitagawa, M.; et al. Efficient Identification of the MYC Regulator with the Use of the CRISPR Library and Context-Matched Database Screenings. Int. J. Mol. Sci. 2022, 23, 7723. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Cheung, A.F.; Simkevich, C.P.; Tam, W.; Ren, X.; Mateyak, M.K.; Sedivy, J.M. A large scale genetic analysis of c-myc-regulated gene expression patterns. J. Biol. Chem. 2003, 278, 12563–12573. [Google Scholar] [CrossRef] [PubMed]
- Blancato, J.; Singh, B.; Liu, A.; Liao, D.J.; Dickson, R.B. Correlation of amplification and overexpression of the c-myc oncogene in high-grade breast cancer: FISH, in situ hybridisation and immunohistochemical analyses. Br. J. Cancer 2004, 90, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Oncogenes and Cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. c-Myc Target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef]
- Amati, B.; Brooks, M.W.; Levy-Strumpf, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, Y. Receptor tyrosine kinases: Biological functions and anticancer targeted therapy. Medcomm 2023, 4, e446. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Verheul, H.M.W. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.D.; Adams, V.R.; Beardslee, T.; Medina, P. Afatinib for the treatment of EGFR mutation-positive NSCLC: A review of clinical findings. J. Oncol. Pharm. Pract. 2020, 26, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.; White, Y. Neratinib for the Treatment of Early-Stage HER2-Positive Breast Cancer. J. Adv. Pract. Oncol. 2018, 9, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Papaetis, G.S.; Syrigos, K.N. Sunitinib: A multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. BioDrugs 2009, 23, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Subiding, T.; Wang, X.; Lei, X. Research progress of covalent inhibitors. Chin. J. Org. Chem. 2018, 38, 2296. [Google Scholar] [CrossRef]
- Pan, Z.; Scheerens, H.; Li, S.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of selective irreversible inhibitors for bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Wang, X.; Dong, H.; Min, W.; Hao, H.; Yang, P. Selective inhibition of CDK4/6: A safe and effective strategy for developing anticancer drugs. Acta Pharm. Sin. B 2021, 11, 30–54. [Google Scholar] [CrossRef]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Pevarello, P.; Barbacid, M.; Bischoff, J.R. CDK inhibitors in cancer therapy: What is next? Trends Pharmacol. Sci. 2008, 29, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Keen, N.; Taylor, S. Aurora-kinase inhibitors as anticancer agents. Nat. Rev. Cancer 2004, 4, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Gadea, B.B.; Ruderman, J.V. Aurora kinase inhibitor ZM447439 Blocks chromosome-induced spindle assembly, the completion of chromosome condensation, and the establishment of the spindle integrity checkpoint in Xenopus egg extracts. Mol. Biol. Cell 2005, 16, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Hauf, S.; Cole, R.W.; LaTerra, S.; Zimmer, C.; Schnapp, G.; Walter, R.; Heckel, A.; van Meel, J.; Rieder, C.L.; Peters, J.-M. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003, 161, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.A.; Baker, P.R.S.; Schopfer, F.J.; Woodcock, S.R.; Napolitano, A.; D’Ischia, M. Nitro-fatty Acid Formation and Signaling. J. Biol. Chem. 2008, 283, 15515–15519. [Google Scholar] [CrossRef] [PubMed]
- Koutoulogenis, G.S.; Kokotos, G. Nitro Fatty Acids (NO2-FAs): An Emerging Class of Bioactive Fatty Acids. Molecules 2021, 26, 7536. [Google Scholar] [CrossRef] [PubMed]
- Panati, K.; Thimmana, L.; Narala, V.R. Electrophilic nitrated fatty acids are potential therapeutic candidates for inflammatory and fibrotic lung diseases. Nitric Oxide 2020, 102, 28–38. [Google Scholar] [CrossRef]
- Khoo, N.K.H.; Li, L.; Salvatore, S.R.; Schopfer, F.J.; Freeman, B.A. Electrophilic fatty acid nitroalkenes regulate Nrf2 and NF-κB signaling:A medicinal chemistry investigation of structure-function relationships. Sci. Rep. 2018, 8, 2295. [Google Scholar] [CrossRef]
- Awwad, K.; Steinbrink, S.D.; Frömel, T.; Lill, N.; Isaak, J.; Häfner, A.-K.; Roos, J.; Hofmann, B.; Heide, H.; Geisslinger, G.; et al. Electrophilic fatty acid species inhibit 5-lipoxygenase and attenuate sepsis-induced pulmonary inflammation. Antioxid. Redox Signal. 2014, 20, 2667–2680. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.; Eaton, P. Redox Regulation of Soluble Epoxide Hydrolase—Implications for Cardiovascular Health and Disease. Cells 2022, 11, 1932. [Google Scholar] [CrossRef] [PubMed]
- Malik, E.M.; Müller, C.E. Anthraquinones As Pharmacological Tools and Drugs. Med. Res. Rev. 2016, 36, 705–748. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, A.; Hoshino, T.; Westley, J.W. Anthracycline antibiotics. Crit. Rev. Biotechnol. 1985, 3, 133–157. [Google Scholar] [CrossRef]
- Hutchinson, C.R.; Colombo, A.L. Genetic engineering of doxorubicin production in Streptomyces peucetius: A review. J. Ind. Microbiol. Biotechnol. 1999, 23, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Etchin, J.; Sanda, T.; Mansour, M.R.; Kentsis, A.; Montero, J.; Le, B.T.; Christie, A.L.; McCauley, D.; Rodig, S.J.; Kauffman, M.; et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br. J. Haematol. 2013, 161, 117–127. [Google Scholar] [CrossRef]
- Sun, H.; Hattori, N.; Chien, W.; Sun, Q.; Sudo, M.; E-Ling, G.L.; Ding, L.; Lim, S.L.; Shacham, S.; Kauffman, M.; et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br. J. Cancer 2014, 111, 281–291. [Google Scholar] [CrossRef]
- Parikh, K.; Cang, S.; Sekhri, A.; Liu, D. Selective inhibitors of nuclear export (SINE)–A novel class of anti-cancer agents. J. Hematol. Oncol. 2014, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25 (Suppl. S2), 41–59. [Google Scholar] [CrossRef]
- Dehelean, C.A.; Marcovici, I.; Soica, C.; Mioc, M.; Coricovac, D.; Iurciuc, S.; Cretu, O.M.; Pinzaru, I. Plant-Derived Anticancer Compounds as New Perspectives in Drug Discovery and Alternative Therapy. Molecules 2021, 26, 1109. [Google Scholar] [CrossRef]
- Chen, D.; Ma, Y.; Guo, Z.; Liu, L.; Yang, Y.; Wang, Y.; Pan, B.; Wu, L.; Hui, Y.; Yang, W. Two Natural Alkaloids Synergistically Induce Apoptosis in Breast Cancer Cells by Inhibiting STAT3 Activation. Molecules 2020, 25, 216. [Google Scholar] [CrossRef] [PubMed]
- Park, U.-H.; Jeong, H.-S.; Jo, E.-Y.; Park, T.; Yoon, S.K.; Kim, E.-J.; Jeong, J.-C.; Um, S.-J. Piperine, a component of black pepper, inhibits adipogenesis by antagonizing PPARγ activity in 3T3-L1 cells. J. Agric. Food Chem. 2012, 60, 3853–3860. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Ray, A.K.; Mishra, S.K. Molecular and pharmacological aspects of piperine as a potential molecule for disease prevention and management: Evidence from clinical trials. Beni-Suef Univ. J. Basic Appl. Sci. 2022, 11, 16. [Google Scholar] [CrossRef]
- Arcaro, C.A.; Gutierres, V.O.; Assis, R.P.; Moreira, T.F.; Costa, P.I.; Baviera, A.M.; Brunetti, I.L. Piperine, a natural bioenhancer, nullifies the antidiabetic and antioxidant activities of curcumin in streptozotocin-diabetic rats. PLoS ONE 2014, 9, e113993. [Google Scholar] [CrossRef]
- Zadorozhna, M.; Tataranni, T.; Mangieri, D. Piperine: Role in prevention and progression of cancer. Mol. Biol. Rep. 2019, 46, 5617–5629. [Google Scholar] [CrossRef] [PubMed]
- Djaldetti, M. Piperine: A Savor Inducer and a Cancer Reducer. OBM Hepatol. Gastroenterol. 2021, 5, 1–26. [Google Scholar] [CrossRef]
- Rather, R.A.; Bhagat, M. Cancer Chemoprevention and Piperine: Molecular Mechanisms and Therapeutic Opportunities. Front. Cell Dev. Biol. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-N.; Zhong, B.; Wang, Y. Mechanism Underlying Antitumor Effects of Sinomenine. Chin. J. Integr. Med. 2019, 25, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Liu, D.; Zhao, Y.; He, J.; Kang, H.; Dai, Z.; Wang, X.; Zhang, S.; Zan, Y. Sinomenine inhibits breast cancer cell invasion and migration by suppressing NF-κB activation mediated by IL-4/miR-324-5p/CUEDC2 axis. Biochem. Biophys. Res. Commun. 2015, 464, 705–710. [Google Scholar] [CrossRef]
- Malla, R.R.; Kumari, S.; Deepak, K.G.K.; Gavara, M.M.; Guganavath, S.; Rokkam, P. Chapter 7—Terpenoids as Potential Targeted Therapeutics of Pancreatic Cancer: Current Advances and Future Directions. In Breaking Tolerance to Pancreatic Cancer Unresponsiveness to Chemotherapy; Nagaraju, G.P., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 5, pp. 111–116. [Google Scholar]
- Ahmad, N.; Nordin, M.F.M.; Mokhtar, N.; Wahab, I.M.A.; Mohamad, M.; Liong, T.K.; Amir, S.N.K.M. Zingiber zerumbet: Pharmacological values of zerumbone and the extraction technology evolution. J. Teknol. 2023, 85, 21–30. [Google Scholar] [CrossRef]
- Kalantari, K.; Moniri, M.; Boroumand Moghaddam, A.; Abdul Rahim, R.; Bin Ariff, A.; Izadiyan, Z.; Mohamad, R. A Review of the Biomedical Applications of Zerumbone and the Techniques for Its Extraction from Ginger Rhizomes. Molecules 2017, 22, 1645. [Google Scholar] [CrossRef]
- Haque, M.A.; Jantan, I.; Arshad, L.; Bukhari, S.N.A. Exploring the immunomodulatory and anticancer properties of zerumbone. Food Funct. 2017, 8, 3410–3431. [Google Scholar] [CrossRef]
- Abdelwahab, S.I.; Abdul, A.B.; Mohan, S.; Taha, M.M.E.; Syam, S.; Ibrahim, M.Y.; Mariod, A.A. Zerumbone induces apoptosis in T-acute lymphoblastic leukemia cells. Leuk. Res. 2011, 35, 268–271. [Google Scholar] [CrossRef]
- Prasannan, R.; Kalesh, K.A.; Shanmugam, M.K.; Nachiyappan, A.; Ramachandran, L.; Nguyen, A.H.; Kumar, A.P.; Lakshmanan, M.; Ahn, K.S.; Sethi, G. Key cell signaling pathways modulated by zerumbone: Role in the prevention and treatment of cancer. Biochem. Pharmacol. 2012, 84, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, S.I.; Abdul, A.B.; Zain, Z.N.M.; Hadi, A.H.A. Zerumbone inhibits interleukin-6 and induces apoptosis and cell cycle arrest in ovarian and cervical cancer cells. Int. Immunopharmacol. 2012, 12, 594–602. [Google Scholar] [CrossRef]
- Jegannathan, S.D.; Arul, S.; Dayalan, H. Zerumbone, a Sesquiterpene, Controls Proliferation and Induces Cell Cycle Arrest in Human Laryngeal Carcinoma Cell Line Hep-2. Nutr. Cancer 2016, 68, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, N.E.; Abu, N.; Rahman, H.S.; Ky, H.; Ho, W.Y.; Lim, K.L.; How, C.W.; Rasedee, A.; Alitheen, N.B.; Yeap, S.K. Nanostructured lipid carrier improved in vivo anti-tumor and immunomodulatory effect of Zerumbone in 4T1 challenged mice. RSC Adv. 2015, 5, 22066–22074. [Google Scholar] [CrossRef]
- Kirana, C.; McIntosh, G.H.; Record, I.R.; Jones, G.P. Antitumor activity of extract of zingiber aromaticum and its bioactive sesquiterpenoid zerumbone. Nutr. Cancer 2003, 45, 218–225. [Google Scholar] [CrossRef]
- Abdelwahab, S.I.; Abdul, A.B.; Devi, N.; Taha, M.M.E.; Al-Zubairi, A.S.; Mohan, S.; Mariod, A.A. Regression of cervical intraepithelial neoplasia by zerumbone in female Balb/c mice prenatally exposed to diethylstilboestrol: Involvement of mitochondria-regulated apoptosis. Exp. Toxicol. Pathol. 2010, 62, 461–469. [Google Scholar] [CrossRef]
- Girisa, S.; Shabnam, B.; Monisha, J.; Fan, L.; Halim, C.E.; Arfuso, F.; Ahn, K.S.; Sethi, G.; Kunnumakkara, A.B. Potential of Zerumbone as an Anti-Cancer Agent. Molecules 2019, 24, 734. [Google Scholar] [CrossRef]
- Hu, H.; Bai, H.; Huang, L.; Yang, B.; Zhao, H. Eupalinolide J Inhibits Cancer Metastasis by Promoting STAT3 Ubiquitin-Dependent Degradation. Molecules 2023, 28, 3143. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xu, X.; Dai, L.; Wang, Y.; Yang, B.; Zhao, H.; Lou, C. Eupalinolide J induces apoptosis, cell cycle arrest, mitochondrial membrane potential disruption and DNA damage in human prostate cancer cells. J. Toxicol. Sci. 2020, 45, 15–23. [Google Scholar] [CrossRef]
- Ninkuu, V.; Zhang, L.; Yan, J.; Fu, Z.; Yang, T.; Zeng, H. Biochemistry of Terpenes and Recent Advances in Plant Protection. Int. J. Mol. Sci. 2021, 22, 5710. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Yuanhuacin and Related Anti-Inflammatory and Anticancer Daphnane Diterpenes from Genkwa Flos—An Overview. Biomolecules 2022, 12, 192. [Google Scholar] [CrossRef]
- Kang, J.I.; Hong, J.-Y.; Lee, H.-J.; Bae, S.Y.; Jung, C.; Park, H.J.; Lee, S.K. Anti-Tumor Activity of Yuanhuacine by Regulating AMPK/mTOR Signaling Pathway and Actin Cytoskeleton Organization in Non-Small Cell Lung Cancer Cells. PLoS ONE 2015, 10, e0144368. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.; Schwarz, S.; Perl, V.; Köwitsch, A.; Siewert, B.; Csuk, R. Incorporation of a Michael acceptor enhances the antitumor activity of triterpenoic acids. Eur. J. Med. Chem. 2015, 101, 391–399. [Google Scholar] [CrossRef]
- Ghosh, S. Chapter 12—Triterpenoids: Structural diversity, biosynthetic pathway, and bioactivity. In Studies in Natural Products Chemistry; Attaur, R., Ed.; Elsevier: New York, NY, USA, 2020; Volume 67, pp. 411–461. [Google Scholar]
- Jesus, J.A.; Lago, J.H.G.; Laurenti, M.D.; Yamamoto, E.S.; Passero, L.F.D. Antimicrobial activity of oleanolic and ursolic acids: An update. Evid. -Based Complement. Altern. Med. 2015, 2015, 620472. [Google Scholar] [CrossRef]
- Hu, H.; Straub, A.; Tian, Z.; Bassler, N.; Cheng, J.; Peter, K. Celastrol, a triterpene extracted from tripterygium wilfordii hook f, inhibits platelet activation. J. Cardiovasc. Pharmacol. 2009, 54, 240–245. [Google Scholar] [CrossRef]
- Liu, J.; Lee, J.; Hernandez, M.A.S.; Mazitschek, R.; Ozcan, U. Treatment of Obesity with Celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef]
- Lee, J.-H.; Choi, K.J.; Seo, W.D.; Jang, S.Y.; Kim, M.; Lee, B.W.; Kim, J.Y.; Kang, S.; Park, K.H.; Lee, Y.-S.; et al. Enhancement of radiation sensitivity in lung cancer cells by celastrol is mediated by inhibition of Hsp90. Int. J. Mol. Med. 2011, 27, 441–446. [Google Scholar] [CrossRef]
- Camargo, K.C.; de Aguilar, M.G.; Moraes, A.R.A.; de Castro, R.G.; Szczerbowski, D.; Miguel, E.L.M.; Oliveira, L.R.; Sousa, G.F.; Vidal, D.M.; Duarte, L.P. Pentacyclic Triterpenoids Isolated from Celastraceae: A Focus in the 13C-NMR Data. Molecules 2022, 27, 959. [Google Scholar] [CrossRef] [PubMed]
- Tiedemann, R.E.; Schmidt, J.; Keats, J.J.; Shi, C.-X.; Zhu, Y.X.; Palmer, S.E.; Mao, X.; Schimmer, A.D.; Stewart, A.K. Identification of a potent natural triterpenoid inhibitor of proteosome chymotrypsin-like activity and NF-κB with antimyeloma activity in vitro and in vivo. Blood 2009, 113, 4027–4037. [Google Scholar] [CrossRef]
- Wang, H.; Teriete, P.; Hu, A.; Raveendra-Panickar, D.; Pendelton, K.; Lazo, J.S.; Eiseman, J.; Holien, T.; Misund, K.; Oliynyk, G.; et al. Direct inhibition of c-Myc-Max heterodimers by celastrol and celastrol-inspired triterpenoids. Oncotarget 2015, 6, 32380–32395. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-X.; He, H.; Qiu, F. Natural withanolides: An overview. Nat. Prod. Rep. 2011, 28, 705–740. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Dai, C.; Fu, Y.; Loo, J.F.; Xia, D.; Gao, S.P.; Ma, Z.; Chen, Z. Physalin A exerts anti-tumor activity in non-small cell lung cancer cell lines by suppressing JAK/STAT3 signaling. Oncotarget 2016, 7, 9462–9476. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- He, H.; Zang, L.-H.; Feng, Y.-S.; Wang, J.; Liu, W.-W.; Chen, L.-X.; Kang, N.; Tashiro, S.-I.; Onodera, S.; Qiu, F.; et al. Physalin A induces apoptotic cell death and protective autophagy in HT1080 human fibrosarcoma cells. J. Nat. Prod. 2013, 76, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gu, J.; Zong, M.; Zhang, Q.; Li, H.; Li, D.; Mou, X.; Liu, P.; Liu, Y.; Qiu, F. Anti-inflammatory action of physalin A by blocking the activation of NF-κB signaling pathway. J. Ethnopharmacol. 2021, 267, 113490. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Leu, Y.-L.; Chang, Y.-L.; Wu, T.-S.; Kuo, P.-C.; Liao, Y.-R.; Teng, C.-M.; Pan, S.-L. Physalin f induces cell apoptosis in human renal carcinoma cells by targeting NF-kappaB and generating reactive oxygen species. PLoS ONE 2012, 7, e40727. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Su, A.; Mi, L.; Zhang, Y.; He, T.; Qiu, Y.; Wei, T.; Li, Z.; Zhu, J.; Wu, W. Withaferin A: A Dietary Supplement with Promising Potential as an Anti-Tumor Therapeutic for Cancer Treatment—Pharmacology and Mechanisms. Drug Des. Dev. Ther. 2023, 17, 2909–2929. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; Hammers, H.; Bargagna-Mohan, P.; Zhan, X.; Herbstritt, C.; Ruiz, A.; Zhang, L.; Hanson, A.; Conner, B.; Rougas, J.; et al. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis 2004, 7, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Shiragannavar, V.D.; Gowda, N.G.S.; Kumar, D.P.; Mirshahi, F.; Santhekadur, P.K. Withaferin A Acts as a Novel Regulator of Liver X Receptor-alpha in HCC. Front. Oncol. 2020, 10, 628506. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Chakraborty, M.; Chandra, A.; Alam, M.P. Structure-activity relationship (SAR) and molecular dynamics study of withaferin-A fragment derivatives as potential therapeutic lead against main protease (Mpro) of SARS-CoV-2. J. Mol. Model. 2021, 27, 97. [Google Scholar] [CrossRef] [PubMed]
- Elekofehinti, O.O.; Iwaloye, O.; Olawale, F.; Ariyo, E.O. Saponins in Cancer Treatment: Current Progress and Future Prospects. Pathophysiology 2021, 28, 250–272. [Google Scholar] [CrossRef] [PubMed]
- Cuttriss, A.J.; Cazzonelli, C.I.; Wurtzel, E.T.; Pogson, B.J. Carotenoids. In Advances in Botanical Research; Rébeillé, F., Douce, R., Eds.; Academic Press: Cambridge, CA, USA, 2011; Volume 58, pp. 1–36. [Google Scholar]
- Sangeetha, R.K.; Bhaskar, N.; Baskaran, V. Comparative effects of β-carotene and fucoxanthin on retinol deficiency induced oxidative stress in rats. Mol. Cell. Biochem. 2009, 331, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ha, A.W.; Na, S.J.; Kim, W.K. Antioxidant effects of fucoxanthin rich powder in rats fed with high fat diet. Nutr. Res. Pract. 2013, 7, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.-N.; Jeon, S.-M.; Kim, H.-J.; Lee, M.-K.; Shin, S.-K.; Shin, Y.C.; Park, Y.-B.; Choi, M.-S. Fucoxanthin supplementation improves plasma and hepatic lipid metabolism and blood glucose concentration in high-fat fed C57BL/6N mice. Chem. Interact. 2010, 186, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, M.; Park, Y.; Shin, Y.; Choi, M. Beneficial effects of Undaria pinnatifida ethanol extract on diet-induced-insulin resistance in C57BL/6J mice. Food Chem. Toxicol. 2011, 49, 727–733. [Google Scholar] [CrossRef]
- Woo, M.; Jeon, S.; Shin, Y.C.; Lee, M.; Kang, M.A.; Choi, M. Anti-obese property of fucoxanthin is partly mediated by altering lipid-regulating enzymes and uncoupling proteins of visceral adipose tissue in mice. Mol. Nutr. Food Res. 2009, 53, 1603–1611. [Google Scholar] [CrossRef]
- Jeon, S.M.; Kim, H.J.; Woo, M.N.; Lee, M.K.; Shin, Y.C.; Park, Y.B.; Choi, M.S. Fucoxanthin-rich seaweed extract suppresses body weight gain and improves lipid metabolism in high-fat-fed C57BL/6J mice. Biotechnol. J. 2010, 5, 961–969. [Google Scholar] [CrossRef]
- Hu, X.; Li, Y.; Li, C.; Fu, Y.; Cai, F.; Chen, Q.; Li, D. Combination of fucoxanthin and conjugated linoleic acid attenuates body weight gain and improves lipid metabolism in high-fat diet-induced obese rats. Arch. Biochem. Biophys. 2012, 519, 59–65. [Google Scholar] [CrossRef]
- Maeda, H.; Hosokawa, M.; Sashima, T.; Takahashi, N.; Kawada, T.; Miyashita, K. Fucoxanthin and its metabolite, fucoxanthinol, suppress adipocyte differentiation in 3T3-L1 cells. Int. J. Mol. Med. 2006, 18, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, T.; Rajendran, P.; Nandakumar, N.; Balasubramanian, M.P.; Nishigaki, I. Cancer preventive efficacy of marine carotenoid fucoxanthin: Cell cycle arrest and apoptosis. Nutrients 2013, 5, 4978–4989. [Google Scholar] [CrossRef] [PubMed]
- Davinelli, S.; Nielsen, M.E.; Scapagnini, G. Astaxanthin in Skin Health, Repair, and Disease: A Comprehensive Review. Nutrients 2018, 10, 522. [Google Scholar] [CrossRef]
- Bjorklund, G.; Dadar, M.; Martins, N.; Chirumbolo, S.; Goh, B.H.; Smetanina, K.; Lysiuk, R. Brief Challenges on Medicinal Plants: An Eye-Opening Look at Ageing-Related Disorders. Basic Clin. Pharmacol. Toxicol. 2018, 122, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Yamashita, E.; Miki, W. Quenching activities of common hydrophilic and lipophilic antioxidants against singlet oxygen using chemiluminescence detection system. Carotenoid Sci. 2007, 11, 16–20. [Google Scholar]
- Naguib, Y.M.A. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Kowsalya, K.; Vidya, N.; Vijayalakshmi, V.; Arun, M. Super Nutritive Marine Astaxanthin, an Effectual Dietary Carotenoid for Neurodegenerative Diseases. Int. Res. J. Multidiscip. Technovation 2019, 1, 115–124. [Google Scholar] [CrossRef]
- Fakhri, S.; Abbaszadeh, F.; Dargahi, L.; Jorjani, M. Astaxanthin: A mechanistic review on its biological activities and health benefits. Pharmacol. Res. 2018, 136, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bahbah, E.I.; Ghozy, S.; Attia, M.S.; Negida, A.; Bin Emran, T.; Mitra, S.; Albadrani, G.M.; Abdel-Daim, M.M.; Uddin, S.; Simal-Gandara, J. Molecular Mechanisms of Astaxanthin as a Potential Neurotherapeutic Agent. Mar. Drugs 2021, 19, 201. [Google Scholar] [CrossRef]
- Wang, S.; Qi, X. The Putative Role of Astaxanthin in Neuroinflammation Modulation: Mechanisms and Therapeutic Potential. Front. Pharmacol. 2022, 13, 916653. [Google Scholar] [CrossRef]
- Kavitha, K.; Kowshik, J.; Kishore, T.K.K.; Baba, A.B.; Nagini, S. Astaxanthin inhibits NF-κB and Wnt/β-catenin signaling pathways via inactivation of Erk/MAPK and PI3K/Akt to induce intrinsic apoptosis in a hamster model of oral cancer. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 4433–4444. [Google Scholar] [CrossRef] [PubMed]
- Davinelli, S.; Saso, L.; D’angeli, F.; Calabrese, V.; Intrieri, M.; Scapagnini, G. Astaxanthin as a Modulator of Nrf2, NF-κB, and Their Crosstalk: Molecular Mechanisms and Possible Clinical Applications. Molecules 2022, 27, 502. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-Q.; Zhao, Y.-X.; Li, S.-Y.; Qiang, J.-W.; Ji, Y.-Z. Anti-Tumor Effects of Astaxanthin by Inhibition of the Expression of STAT3 in Prostate Cancer. Mar. Drugs 2020, 18, 415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, W.-E.; Hu, L.; Zhao, L.; Huang, J. Carotenoids inhibit proliferation and regulate expression of peroxisome proliferators-activated receptor gamma (PPARγ) in K562 cancer cells. Arch. Biochem. Biophys. 2011, 512, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Carbone, K.; Gervasi, F. An Updated Review of the Genus Humulus: A Valuable Source of Bioactive Compounds for Health and Disease Prevention. Plants 2022, 11, 3434. [Google Scholar] [CrossRef] [PubMed]
- Ayabe, T.; Fukuda, T.; Ano, Y. Improving Effects of Hop-Derived Bitter Acids in Beer on Cognitive Functions: A New Strategy for Vagus Nerve Stimulation. Biomolecules 2020, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Hazato, T.; Ashino, H.; Yamamoto, Y.; Iwasaki, E.; Tobe, H.; Yamamoto, K.; Yamamoto, S. Inhibition of angiogenesis by humulone, a bitter acid from beer hop. Biochem. Biophys. Res. Commun. 2001, 289, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Kundu, J.K.; Hwang, D.-M.; Na, H.-K.; Surh, Y.-J. Humulone inhibits phorbol ester-induced COX-2 expression in mouse skin by blocking activation of NF-κB and AP-1: IκB kinase and c-Jun-N-terminal kinase as respective potential upstream targets. Carcinogen 2007, 28, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Spampanato, C.; DE Maria, S.; Sarnataro, M.; Giordano, E.; Zanfardino, M.; Baiano, S.; Cartenì, M.; Morelli, F. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int. J. Oncol. 2012, 40, 935–941. [Google Scholar] [CrossRef]
- Lamy, V.; Roussi, S.; Chaabi, M.; Gossé, F.; Schall, N.; Lobstein, A.; Raul, F. Chemopreventive effects of lupulone, a hop β-acid, on human colon cancer-derived metastatic SW620 cells and in a rat model of colon carcinogenesis. Carcinogen 2007, 28, 1575–1581. [Google Scholar] [CrossRef]
- Chan, Y.A.; Podevels, A.M.; Kevany, B.M.; Thomas, M.G. Biosynthesis of polyketide synthase extender units. Nat. Prod. Rep. 2009, 26, 90–114. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, A.; Figadère, B.; Zafra-Polo, M.-C.; Barrachina, I.; Estornell, E.; Cortes, D. Acetogenins from Annonaceae: Recent progress in isolation, synthesis and mechanisms of action. Nat. Prod. Rep. 2005, 22, 269–303. [Google Scholar] [CrossRef] [PubMed]
- Neske, A.; Hidalgo, J.R.; Cabedo, N.; Cortes, D. Acetogenins from Annonaceae family. Their potential biological applications. Phytochemistry 2020, 174, 112332. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Herrera, N.; Perez-Plasencia, C.; Castro-Torres, V.A.; Martinez-Vazquez, M.; Gonzalez-Esquinca, A.R.; Zentella-Dehesa, A. Selective Acetogenins and Their Potential as Anticancer Agents. Front. Pharmacol. 2019, 10, 783. [Google Scholar] [CrossRef] [PubMed]
- Wahab, S.M.A.; Jantan, I.; Hague, A.; Arshad, L. Exploring the Leaves of Annona muricata L. as a Source of Potential Anti-inflammatory and Anticancer Agents. Front. Pharmacol. 2018, 9, 661. [Google Scholar] [CrossRef]
- Padma, P.; Pramod, N.; Thyagarajan, S.; Khosa, R. Effect of the extract of Annona muricata and Petunia nyctaginiflora on Herpes simplex virus. J. Ethnopharmacol. 1998, 61, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Apriyanto, D.R.; Aoki-Utsubo, C.; Hartati, S.; Dewi, B.E.; Hotta, H. Potential of Indonesian plants (Annona muricata, Garcinia latissima, and Garcinia celebica) against hepatitis C virus. Adv. Sci. Lett. 2018, 24, 6807–6810. [Google Scholar] [CrossRef]
- Le Donne, M.; Lentini, M.; Alibrandi, A.; Salimbeni, V.; Giuffre, G.; Mazzeo, F.; Triolo, O.; D’Anna, R. Antiviral activity of Ellagic acid and Annona Muricata in cervical HPV related pre-neoplastic lesions: A randomized trial. J. Funct. Foods 2017, 35, 549–554. [Google Scholar] [CrossRef]
- Betancur-Galvis, L.; Saez, J.; Granados, H.; Salazar, A.; Ossa, J. Antitumor and antiviral activity of Colombian medicinal plant extracts. Mem. Do Inst. Oswaldo Cruz 1999, 94, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Rakotomanga, M.; Razakantoanina, V.; Raynaud, S.; Loiseau, P.; Hocquemiller, R.; Jaureguiberry, G. Antiplasmodial activity of acetogenins and inhibitory effect on plasmodium falciparumadenylate translocase. J. Chemother. 2004, 16, 350–356. [Google Scholar] [CrossRef]
- Tormo, J.R.; Gallardo, T.; Aragón, R.; Cortes, D.; Estornell, E. Specific interactions of monotetrahydrofuranic annonaceous acetogenins as inhibitors of mitochondrial complex I. Chem. Biol. Interact. 1999, 122, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Ilango, S.; Sahoo, D.K.; Paital, B.; Kathirvel, K.; Gabriel, J.I.; Subramaniam, K.; Jayachandran, P.; Dash, R.K.; Hati, A.K.; Behera, T.R.; et al. A Review on Annona muricata and Its Anticancer Activity. Cancers 2022, 14, 4539. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, T.; Gunji, S.; Tsuji, H.; Beppu, T. Leptomycins A and B, new antifungal antibiotics. I. Taxonomy of the producing strain and their fermentation, purification and characterization. J. Antibiot. 1983, 36, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shao, C.; Cobos, E.; Singh, K.P.; Gao, W. Chemotherapeutic sensitization of leptomycin b resistant lung cancer cells by pretreatment with doxorubicin. PLoS ONE 2012, 7, e32895. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef] [PubMed]
- Wolff, B.; Sanglier, J.-J.; Wang, Y. Leptomycin B is an inhibitor of nuclear export: Inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 1997, 4, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Fukuda, M.; Nishida, E. Nuclear Export of Map Kinase (ERK) involves a map kinase kinase (Mek-Dependent) active transport mechanism. J. Cell Biol. 2000, 148, 849–856. [Google Scholar] [CrossRef]
- Huang, T.T.; Kudo, N.; Yoshida, M.; Miyamoto, S. A nuclear export signal in the N-terminal regulatory domain of IκBα controls cytoplasmic localization of inactive NF-κB/IκBα complexes. Proc. Natl. Acad. Sci. USA 2000, 97, 1014–1019. [Google Scholar] [CrossRef]
- Hietanen, S.; Lain, S.; Krausz, E.; Blattner, C.; Lane, D.P. Activation of p53 in cervical carcinoma cells by small molecules. Proc. Natl. Acad. Sci. USA 2000, 97, 8501–8506. [Google Scholar] [CrossRef]
- Jang, B.-C.; Muñoz-Najar, U.; Paik, J.-H.; Claffey, K.; Yoshida, M.; Hla, T. Leptomycin B, an inhibitor of the nuclear export receptor CRM1, inhibits COX-2 expression. J. Biol. Chem. 2003, 278, 2773–2776. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Polyphenols as Antioxidant/Pro-Oxidant Compounds and Donors of Reducing Species: Relationship with Human Antioxidant Metabolism. Processes 2023, 11, 2771. [Google Scholar] [CrossRef]
- Espíndola, K.M.M.; Ferreira, R.G.; Narvaez, L.E.M.; Rosario, A.C.R.S.; da Silva, A.H.M.; Silva, A.G.B.; Vieira, A.P.O.; Monteiro, M.C. Chemical and Pharmacological Aspects of Caffeic Acid and Its Activity in Hepatocarcinoma. Front. Oncol. 2019, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.A.O.; Freire, C.S.R.; Domingues, M.R.M.; Silvestre, A.J.D.; Neto, C.P. Characterization of phenolic components in polar extracts of eucalyptus globulus labill. bark by high-performance liquid chromatography–Mass spectrometry. J. Agric. Food Chem. 2011, 59, 9386–9393. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Karim, S.; Akram, M.R.; Khan, S.A.; Azhar, S.; Mumtaz, A.; Bin Asad, M.H.H. Caffeic acid phenethyl ester and therapeutic potentials. BioMed Res. Int. 2014, 2014, 145342. [Google Scholar] [CrossRef] [PubMed]
- Maier, V.; Metzler, D.; Huber, A. 3-0-Caffeoylshikimic acid (dactylifric acid) and its isomers, a new class of enzymic browning substrates. Biochem. Biophys. Res. Commun. 1964, 14, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Kitts, D.D. Role of Chlorogenic Acids in Controlling Oxidative and Inflammatory Stress Conditions. Nutrients 2015, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-H.; Chen, C.-C.; Kuo, Y.-H.; Fuh, L.-J.; Lan, W.-C.; Wang, T.-H.; Chiu, K.-C.; Nguyen, T.-H.V.; Hsia, S.-M.; Shieh, T.-M. Caffeic Acid Phenethyl Ester and Caffeamide Derivatives Suppress Oral Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2023, 24, 9819. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Xin, W.; Chen, L.; Fu, Y.; Qi, Y.; Ding, H.; Fang, L. Caffeic acid phenethyl ester suppresses metastasis of breast cancer cells by inactivating FGFR1 via MD2. PLoS ONE 2023, 18, e0289031. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-Y.; Jim, W.-T.; Su, L.-C.; Chung, C.-J.; Lin, C.-Y.; Huo, C.; Tseng, J.-C.; Huang, S.-H.; Lai, C.-J.; Chen, B.-C.; et al. Caffeic acid phenethyl ester is a potential therapeutic agent for oral cancer. Int. J. Mol. Sci. 2015, 16, 10748–10766. [Google Scholar] [CrossRef]
- Zhao, W.-X.; Wang, L.; Yang, J.-L.; Li, L.-Z.; Xu, W.-M.; Li, T. Caffeic acid phenethyl ester attenuates pro-inflammatory and fibrogenic phenotypes of LPS-stimulated hepatic stellate cells through the inhibition of NF-κB signaling. Int. J. Mol. Med. 2014, 33, 687–694. [Google Scholar] [CrossRef]
- Chung, T.-W.; Kim, S.-J.; Choi, H.-J.; Kwak, C.-H.; Song, K.-H.; Suh, S.-J.; Kim, K.-J.; Ha, K.-T.; Park, Y.-G.; Chang, Y.-C.; et al. CAPE suppresses VEGFR-2 activation, and tumor neovascularization and growth. J. Mol. Med. 2013, 91, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, M.; Li, Y.; Zhang, D.; Yang, Y.; Li, L. Natural Xanthine Oxidase Inhibitor 5-O-Caffeoylshikimic Acid Ameliorates Kidney Injury Caused by Hyperuricemia in Mice. Molecules 2021, 26, 7307. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, Y.; Li, Y.; Hu, Y.; Zhang, Q.; Huang, Y.; Shi, K.; Ran, C.; Hou, J.; Zhou, G.; et al. Chlorogenic Acid Decreases Malignant Characteristics of Hepatocellular Carcinoma Cells by Inhibiting DNMT1 Expression. Front. Pharmacol. 2020, 11, 867. [Google Scholar] [CrossRef] [PubMed]
- Ranjbary, A.G.; Bagherzadeh, A.; Sabbaghi, S.S.; Faghihi, A.; Karimi, D.N.; Naji, S.; Kardani, M. Chlorogenic acid induces apoptosis and cell-cycle arrest in colorectal cancer cells. Mol. Biol. Rep. 2023, 50, 9845–9857. [Google Scholar] [CrossRef] [PubMed]
- Yesil-Celiktas, O.; Sevimli, C.; Bedir, E.; Vardar-Sukan, F. Inhibitory effects of rosemary extracts, carnosic acid and rosmarinic acid on the growth of various human cancer cell lines. Plant Foods Hum. Nutr. 2010, 65, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, C.; Moorthy, N.S.N.; Ramasamy, S.; Vanam, U.; Manivannan, E.; Karunagaran, D.; Trivedi, P. Advances in chalcones with anticancer activities. Recent Pat. Anti-Cancer Drug Discov. 2015, 10, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A mini review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Batovska, D.I.; Todorova, I.T. Trends in utilization of the pharmacological potential of chalcones. Curr. Clin. Pharmacol. 2010, 5, 1–29. [Google Scholar] [CrossRef]
- Zhou, B.; Xing, C. Diverse Molecular Targets for Chalcones with Varied Bioactivities. Med. Chem. 2015, 5, 388–404. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A.O.; et al. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef]
- Jeon, K.-H.; Yu, H.-B.; Kwak, S.Y.; Kwon, Y.; Na, Y. Synthesis and topoisomerases inhibitory activity of heteroaromatic chalcones. Bioorganic Med. Chem. 2016, 24, 5921–5928. [Google Scholar] [CrossRef] [PubMed]
- Amolins, M.W.; Blagg, B.S.J. Natural product inhibitors of Hsp90: Potential leads for drug discovery. Mini-Rev. Med. Chem. 2009, 9, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V.; Singh, S.K. Perspectives of medicinally privileged chalcone based metal coordination compounds for biomedical applications. Eur. J. Med. Chem. 2019, 174, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Saxena, G.; Dixit, S.; Hamidullah; Singh, S.K.; Singh, S.K.; Arshad, M.; Konwar, R. Synthesis, characterization and biological activities of some Ru(II) complexes with substituted chalcones and their applications as chemotherapeutics against breast cancer. J. Mol. Struct. 2016, 1111, 90–99. [Google Scholar] [CrossRef]
- Gazdova, M.; Michalkova, R.; Kello, M.; Vilkova, M.; Kudlickova, Z.; Baloghova, J.; Mirossay, L.; Mojzis, J. Chalcone-Acridine Hybrid Suppresses Melanoma Cell Progression via G2/M Cell Cycle Arrest, DNA Damage, Apoptosis, and Modulation of MAP Kinases Activity. Int. J. Mol. Sci. 2022, 23, 12266. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.A.; Abuo-Rahma, G.E.-D.A. Molecular targets and anticancer activity of quinoline–chalcone hybrids: Literature review. RSC Adv. 2020, 10, 31139–31155. [Google Scholar] [CrossRef]
- Mongre, R.K.; Sharma, N.; Sodhi, S.S.; Ghosh, M.; Singh, A.K.; Kim, N.; Park, Y.H.; Shin, Y.G.; Kim, S.J.; Jiao, Z.J.; et al. Novel phyto-derivative BRM270 inhibits hepatocellular carcinoma cells proliferation by inducing G2/M phase cell cycle arrest and apoptosis in xenograft mice model. Biomed. Pharmacother. 2017, 87, 741–754. [Google Scholar] [CrossRef]
- Harish, V.; Haque, E.; Śmiech, M.; Taniguchi, H.; Jamieson, S.; Tewari, D.; Bishayee, A. Xanthohumol for Human Malignancies: Chemistry, Pharmacokinetics and Molecular Targets. Int. J. Mol. Sci. 2021, 22, 4478. [Google Scholar] [CrossRef]
- Siahmazgi, Z.G.; Irani, S.; Ghiaseddin, A.; Fallah, P.; Haghpanah, V. Xanthohumol hinders invasion and cell cycle progression in cancer cells through targeting MMP2, MMP9, FAK and P53 genes in three-dimensional breast and lung cancer cells culture. Cancer Cell Int. 2023, 23, 153. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; El Rayess, Y.; Rizk, A.A.; Sadaka, C.; Zgheib, R.; Zam, W.; Sestito, S.; Rapposelli, S.; Neffe-Skocińska, K.; Zielińska, D.; et al. Turmeric and Its Major Compound Curcumin on Health: Bioactive Effects and Safety Profiles for Food, Pharmaceutical, Biotechnological and Medicinal Applications. Front. Pharmacol. 2020, 11, 01021. [Google Scholar] [CrossRef]
- Fadus, M.C.; Lau, C.; Bikhchandani, J.; Lynch, H.T. Curcumin: An age-old anti-inflammatory and anti-neoplastic agent. J. Tradit. Complement. Med. 2017, 7, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Benameur, T.; Soleti, R.; Panaro, M.A.; La Torre, M.E.; Monda, V.; Messina, G.; Porro, C. Curcumin as Prospective Anti-Aging Natural Compound: Focus on Brain. Molecules 2021, 26, 4794. [Google Scholar] [CrossRef]
- El Nebrisi, E. Neuroprotective Activities of Curcumin in Parkinson’s Disease: A Review of the Literature. Int. J. Mol. Sci. 2021, 22, 11248. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Ao, M.; Dong, B.; Jiang, Y.; Yu, L.; Chen, Z.; Hu, C.; Xu, R. Anti-Inflammatory Effects of Curcumin in the Inflammatory Diseases: Status, Limitations and Countermeasures. Drug Des. Dev. Ther. 2021, 15, 4503–4525. [Google Scholar] [CrossRef]
- Mansouri, K.; Rasoulpoor, S.; Daneshkhah, A.; Abolfathi, S.; Salari, N.; Mohammadi, M.; Rasoulpoor, S.; Shabani, S. Clinical effects of curcumin in enhancing cancer therapy: A systematic review. BMC Cancer 2020, 20, 791. [Google Scholar] [CrossRef]
- Zoi, V.; Galani, V.; Tsekeris, P.; Kyritsis, A.P.; Alexiou, G.A. Radiosensitization and Radioprotection by Curcumin in Glioblastoma and Other Cancers. Biomedicines 2022, 10, 312. [Google Scholar] [CrossRef]
- Kou, H.; Huang, L.; Jin, M.; He, Q.; Zhang, R.; Ma, J. Effect of curcumin on rheumatoid arthritis: A systematic review and meta-analysis. Front. Immunol. 2023, 14, 1121655. [Google Scholar] [CrossRef]
- Marcu, M.G.; Jung, Y.J.; Lee, S.; Chung, E.J.; Lee, M.J.; Trepel, J.; Neckers, L. Curcumin is an Inhibitor of p300 Histone Acetylatransferase. Med. Chem. 2006, 2, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Krauthauser, C.; Maduskuie, V.; Fawcett, P.T.; Olson, J.M.; A Rajasekaran, S. Curcumin-induced HDAC inhibition and attenuation of medulloblastoma growth in vitro and in vivo. BMC Cancer 2011, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Aggarwal, B.B. Activation of Transcription Factor NF-κB Is Suppressed by Curcumin (Diferuloylmethane). J. Biol. Chem. 1995, 270, 24995–25000. [Google Scholar] [CrossRef] [PubMed]
- Salucci, S.; Bavelloni, A.; Stella, A.B.; Fabbri, F.; Vannini, I.; Piazzi, M.; Volkava, K.; Scotlandi, K.; Martinelli, G.; Faenza, I.; et al. The Cytotoxic Effect of Curcumin in Rhabdomyosarcoma Is Associated with the Modulation of AMPK, AKT/mTOR, STAT, and p53 Signaling. Nutrients 2023, 15, 740. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Duan, L.; Yang, F.; Yang, L.; Deng, Y.; Yu, Y.; Xu, Y.; Zhang, Y. Curcumin suppress inflammatory response in traumatic brain injury via p38/MAPK signaling pathway. Phytother. Res. 2022, 36, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Praditya, D.; Kirchhoff, L.; Brüning, J.; Rachmawati, H.; Steinmann, J.; Steinmann, E. Anti-infective Properties of the Golden Spice Curcumin. Front. Microbiol. 2019, 10, 912. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Moore, T.W.; Hu, F.; Brown, A.P.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Yoon, Y.; Shim, H.; Marcus, A.I.; et al. Inhibition of the NF-κB signaling pathway by the curcumin analog, 3,5-Bis(2-pyridinylmethylidene)-4-piperidone (EF31): Anti-inflammatory and anti-cancer properties. Int. Immunopharmacol. 2012, 12, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.H.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef]
- Fetoni, A.R.; Paciello, F.; Mezzogori, D.; Rolesi, R.; Eramo, S.L.M.; Paludetti, G.; Troiani, D. Molecular targets for anticancer redox chemotherapy and cisplatin-induced ototoxicity: The role of curcumin on pSTAT3 and Nrf-2 signalling. Br. J. Cancer 2015, 113, 1434–1444. [Google Scholar] [CrossRef]
- Pandey, A.; Vishnoi, K.; Mahata, S.; Tripathi, S.C.; Misra, S.P.; Misra, V.; Mehrotra, R.; Dwivedi, M.; Bharti, A.C. Berberine and Curcumin Target Survivin and STAT3 in Gastric Cancer Cells and Synergize Actions of Standard Chemotherapeutic 5-Fluorouracil. Nutr. Cancer 2015, 67, 1293–1304. [Google Scholar] [CrossRef]
- Bimonte, S.; Barbieri, A.; Palma, G.; Rea, D.; Luciano, A.; D’aiuto, M.; Arra, C.; Izzo, F. Dissecting the role of curcumin in tumour growth and angiogenesis in mouse model of human breast cancer. BioMed Res. Int. 2015, 2015, 878134. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra, M.L.F.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Wu, D.; Jiang, Y.; Prasad, K.N.; Lin, S.; Jiang, G.; He, J.; Zhao, M.; Luo, W.; Yang, B. Identification of flavonoids in litchi (Litchi chinensis Sonn.) leaf and evaluation of anticancer activities. J. Funct. Foods 2014, 6, 555–563. [Google Scholar] [CrossRef]
- Felice, M.R.; Maugeri, A.; De Sarro, G.; Navarra, M.; Barreca, D. Molecular Pathways Involved in the Anti-Cancer Activity of Flavonols: A Focus on Myricetin and Kaempferol. Int. J. Mol. Sci. 2022, 23, 4411. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Antioxidant Metabolism Pathways in Vitamins, Polyphenols, and Selenium: Parallels and Divergences. Int. J. Mol. Sci. 2024, 25, 2600. [Google Scholar] [CrossRef] [PubMed]
- Qattan, M.Y.; Khan, M.I.; Alharbi, S.H.; Verma, A.K.; Al-Saeed, F.A.; Abduallah, A.M.; Al Areefy, A.A. Therapeutic Importance of Kaempferol in the Treatment of Cancer through the Modulation of Cell Signalling Pathways. Molecules 2022, 27, 8864. [Google Scholar] [CrossRef] [PubMed]
- Pejčić, T.; Zeković, M.; Bumbaširević, U.; Kalaba, M.; Vovk, I.; Bensa, M.; Popović, L.; Tešić, M.; Tešić, Ž. The Role of Isoflavones in the Prevention of Breast Cancer and Prostate Cancer. Antioxidants 2023, 12, 368. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pathania, A.S.; Saxena, A.; Vishwakarma, R.; Ali, A.; Bhushan, S. The anticancer potential of flavonoids isolated from the stem bark of Erythrina suberosa through induction of apoptosis and inhibition of STAT signaling pathway in human leukemia HL-60 cells. Chem. Interact. 2013, 205, 128–137. [Google Scholar] [CrossRef]
- Gangopadhyay, A.; Chakraborty, S.; Jash, S.K.; Gorai, D. Cytotoxicity of natural flavones and flavonols against different cancer cells. J. Iran. Chem. Soc. 2022, 19, 1547–1573. [Google Scholar] [CrossRef]
- Cardenas, H.; Arango, D.; Nicholas, C.; Duarte, S.; Nuovo, G.J.; He, W.; Voss, O.H.; Gonzalez-Mejia, M.E.; Guttridge, D.C.; Grotewold, E.; et al. Dietary Apigenin Exerts Immune-Regulatory Activity in Vivo by Reducing NF-κB Activity, Halting Leukocyte Infiltration and Restoring Normal Metabolic Function. Int. J. Mol. Sci. 2016, 17, 323. [Google Scholar] [CrossRef]
- Angulo, P.; Kaushik, G.; Subramaniam, D.; Dandawate, P.; Neville, K.; Chastain, K.; Anant, S. Natural compounds targeting major cell signaling pathways: A novel paradigm for osteosarcoma therapy. J. Hematol. Oncol. 2017, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Afaq, F.; Mukhtar, H. Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene 2002, 21, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Han, X.; Cheng, W.; Ni, J.; Zhang, Y.; Lin, J.; Song, Z. Apigenin inhibits proliferation and invasion, and induces apoptosis and cell cycle arrest in human melanoma cells. Oncol. Rep. 2017, 37, 2277–2285. [Google Scholar] [CrossRef]
- Huang, C.; Wei, Y.-X.; Shen, M.-C.; Tu, Y.-H.; Wang, C.-C.; Huang, H.-C. Chrysin, Abundant in Morinda citrifolia Fruit Water–EtOAc Extracts, Combined with Apigenin Synergistically Induced Apoptosis and Inhibited Migration in Human Breast and Liver Cancer Cells. J. Agric. Food Chem. 2016, 64, 4235–4245. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Song, Y.; Yao, J.; Huang, K.; Zhu, X. Apigenin suppresses colorectal cancer cell proliferation, migration and invasion via inhibition of the Wnt/β-catenin signaling pathway. Oncol. Lett. 2016, 11, 3075–3080. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Hu, Q.; He, Z.; Chen, X.; Wang, J.; Yin, X.; Ma, X.; Zeng, J. Scutellaria baicalensis Georgi and Their Natural Flavonoid Compounds in the Treatment of Ovarian Cancer: A Review. Molecules 2023, 28, 5082. [Google Scholar] [CrossRef] [PubMed]
- Tai, M.C.; Tsang, S.Y.; Chang, L.Y.F.; Xue, H. Therapeutic potential of wogonin: A naturally occurring flavonoid. CNS Drug Rev. 2005, 11, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Seong, R.-K.; Kim, J.-A.; Shin, O.S. Wogonin, a flavonoid isolated from Scutellaria baicalensis, has anti-viral activities against influenza infection via modulation of AMPK pathways. Acta Virol. 2018, 62, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Banik, K.; Khatoon, E.; Harsha, C.; Rana, V.; Parama, D.; Thakur, K.K.; Bishayee, A.; Kunnumakkara, A.B. Wogonin and its analogs for the prevention and treatment of cancer: A systematic review. Phytother. Res. 2022, 36, 1854–1883. [Google Scholar] [CrossRef]
- Hui, K.M.; Huen, M.S.; Wang, H.Y.; Zheng, H.; Sigel, E.; Baur, R.; Ren, H.; Li, Z.W.; Wong, J.-F.; Xue, H. Anxiolytic effect of wogonin, a benzodiazepine receptor ligand isolated from Scutellaria baicalensis Georgi. Biochem. Pharmacol. 2002, 64, 1415–1424. [Google Scholar] [CrossRef]
- Kong, Z.; Shen, Q.; Jiang, J.; Deng, M.; Zhang, Z.; Wang, G. Wogonin improves functional neuroprotection for acute cerebral ischemia in rats by promoting angiogenesis via TGF-β1. Ann. Transl. Med. 2019, 7, 639. [Google Scholar] [CrossRef] [PubMed]
- Ruibin, J.; Bo, J.; Danying, W.; Chihong, Z.; Jianguo, F.; Linhui, G. Therapy Effects of Wogonin on Ovarian Cancer Cells. BioMed Res. Int. 2017, 2017, 9381513. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Xue, M.; Xiao, S.-S.; Wan, Y.-J.; Xu, D.-B. A Combination Therapy with Baicalein and Taxol Promotes Mitochondria-Mediated Cell Apoptosis: Involving in Akt/β-Catenin Signaling Pathway. DNA Cell Biol. 2016, 35, 646–656. [Google Scholar] [CrossRef]
- Singh, S.; Meena, A.; Luqman, S. Baicalin mediated regulation of key signaling pathways in cancer. Pharmacol. Res. 2021, 164, 105387. [Google Scholar] [CrossRef]
- Sajeev, A.; Hegde, M.; Girisa, S.; Devanarayanan, T.N.; Alqahtani, M.S.; Abbas, M.; Sil, S.K.; Sethi, G.; Chen, J.-T.; Kunnumakkara, A.B. Oroxylin A: A Promising Flavonoid for Prevention and Treatment of Chronic Diseases. Biomolecules 2022, 12, 1185. [Google Scholar] [CrossRef]
- Shi, X.; Chen, G.; Liu, X.; Qiu, Y.; Yang, S.; Zhang, Y.; Fang, X.; Zhang, C.; Liu, X. Scutellarein inhibits cancer cell metastasis in vitro and attenuates the development of fibrosarcoma in vivo. Int. J. Mol. Med. 2015, 35, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Chen, Z.; Yang, X.; Yan, Q.; Xu, M.; Liu, W.; He, Q.; Zhang, Y.; Cheng, W.; Zhao, W. Scutellarein induces apoptosis and inhibits proliferation, migration, and invasion in ovarian cancer via inhibition of EZH2/FOXO1 signaling. J. Biochem. Mol. Toxicol. 2021, 35, e22870. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Liu, Z.; Verwilst, P.; Koo, S.; Jangjili, P.; Kim, J.S.; Lin, W. Coumarin-Based Small-Molecule Fluorescent Chemosensors. Chem. Rev. 2019, 119, 10403–10519. [Google Scholar] [CrossRef] [PubMed]
- Maria João, M.; Lourdes, S.; Eugenio, U.; Orlando, A.A.; Enrique, M.; Estela Guardado, Y. Coumarins—An Important Class of Phytochemicals. In Phytochemicals; Rao, A.V., Leticia, G.R., Eds.; IntechOpen: Rijeka, Croatia, 2015; p. Ch. 5. [Google Scholar]
- Van Gorp, R.H.; Schurgers, L.J. New Insights into the Pros and Cons of the Clinical Use of Vitamin K Antagonists (VKAs) Versus Direct Oral Anticoagulants (DOACs). Nutrients 2015, 7, 9538–9557. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Cruz-Martins, N.; López-Jornet, P.; Lopez, E.P.-F.; Harun, N.; Yeskaliyeva, B.; Beyatli, A.; Sytar, O.; Shaheen, S.; Sharopov, F.; et al. Natural Coumarins: Exploring the Pharmacological Complexity and Underlying Molecular Mechanisms. Oxidative Med. Cell. Longev. 2021, 2021, 6492346. [Google Scholar] [CrossRef]
- Harborne, J.B. The natural coumarins: Occurrence, chemistry and biochemistry (book). Plant Cell Environ. 1982, 5, 435–436. [Google Scholar] [CrossRef]
- Hassanein, E.H.M.; Sayed, A.M.; Hussein, O.E.; Mahmoud, A.M. Coumarins as Modulators of the Keap1/Nrf2/ARE Signaling Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 1675957. [Google Scholar] [CrossRef] [PubMed]
- Al-Warhi, T.; Sabt, A.; Elkaeed, E.B.; Eldehna, W.M. Recent advancements of coumarin-based anticancer agents: An up-to-date review. Bioorg. Chem. 2020, 103, 104163. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.S.; Gupta, J.; Sharma, S.; Sahu, D. An insight into the therapeutic applications of coumarin compounds and their mechanisms of action. Eur. J. Pharm. Sci. 2020, 152, 105424. [Google Scholar] [CrossRef] [PubMed]
- Jameel, E.; Umar, T.; Kumar, J.; Hoda, N. Coumarin: A Privileged Scaffold for the Design and Development of Antineurodegenerative Agents. Chem. Biol. Drug Des. 2016, 87, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, L.C. Coumarin Derivatives in Inflammatory Bowel Disease. Molecules 2021, 26, 422. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Coumarin carbonic anhydrase inhibitors from natural sources. J. Enzym. Inhib. Med. Chem. 2020, 35, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.; Matos, M.J.; Uriarte, E.; Santana, L. Trending Topics on Coumarin and Its Derivatives in 2020. Molecules 2021, 26, 501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587. [Google Scholar] [CrossRef]
- Reddy, D.S.; Kongot, M.; Kumar, A. Coumarin hybrid derivatives as promising leads to treat tuberculosis: Recent developments and critical aspects of structural design to exhibit anti-tubercular activity. Tuberculosis 2021, 127, 102050. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Q.; Zhang, Y.; Liang, C. Coumarin-based derivatives with potential anti-HIV activity. Fitoterapia 2021, 150, 104863. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Zhang, A.; Yang, Y.; Yang, P. Coumarin-containing hybrids and their antibacterial activities. Arch. Pharm. 2020, 353, e1900380. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Fan, J.; Liu, L.; Liu, X.; Gao, F. Coumarin derivatives with anticancer activities: An update. Arch. Pharm. 2020, 353, e2000025. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef] [PubMed]
- Jarząb, A.; Grabarska, A.; Skalicka-Woźniak, K.; Stepulak, A. Pharmacological features of osthole. Postep. Hig. Med. Dosw. 2017, 71, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lu, J.-J.; Ding, J. Natural Products in Cancer Therapy: Past, Present and Future. Nat. Prod. Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hou, Y.; Qu, P.; Cai, Y. Editorial: Combating Cancer with Natural Products: What Would Non-Coding RNAs Bring? Front. Oncol. 2021, 11, 747586. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, J.; Yao, Y.; Wang, C.; Zhang, J.; Shu, X.; Sun, X.; Li, Y.; Liu, K.; Yuan, H.; et al. Covalent binding design strategy: A prospective method for discovery of potent targeted anticancer agents. Eur. J. Med. Chem. 2017, 142, 493–505. [Google Scholar] [CrossRef]
- Wilson, A.J.; Kerns, J.K.; Callahan, J.F.; Moody, C.J. Keap calm, and carry on covalently. J. Med. Chem. 2013, 56, 7463–7476. [Google Scholar] [CrossRef]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- H Johansson, M. Reversible Michael additions: Covalent inhibitors and prodrugs. Mini Rev. Med. Chem. 2012, 12, 1330–1344. [Google Scholar] [PubMed]
- Hearn, B.R.; Fontaine, S.D.; Schneider, E.L.; Kraemer, Y.; Ashley, G.W.; Santi, D.V. Attenuation of the reaction of michael acceptors with biologically important nucleophiles. Bioconj. Chem. 2021, 32, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.M.; Moreira, R. Michael acceptors as cysteine protease inhibitors. Mini-Rev. Med. Chem. 2007, 7, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Schlage, P.; Mező, G.; Orbán, E.; Bősze, S.; Manea, M. Anthracycline-GnRH derivative bioconjugates with different linkages: Synthesis, in vitro drug release and cytostatic effect. J. Control. Release 2011, 156, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, A.; Kumari, S.; Booth, V. Conjugates for use in peptide therapeutics: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0255753. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, L.-K.; Buevich, A.V.; Li, G.; Tang, H.; Vachal, P.; Colletti, S.L.; Shi, Z.-C. Chemoselective peptide modification via photocatalytic tryptophan β-position conjugation. J. Am. Chem. Soc. 2018, 140, 6797–6800. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, D.V.; Allevato, M.M.; Camargo, M.F.; Lesperance, J.; Quraishi, M.A.; Aguilera, J.; Franiak-Pietryga, I.; Scanderbeg, D.J.; Wang, Z.; Molinolo, A.A.; et al. Monomethyl auristatin antibody and peptide drug conjugates for trimodal cancer chemo-radio-immunotherapy. Nat. Commun. 2022, 13, 3869. [Google Scholar] [CrossRef] [PubMed]
- de la Rubia, J.; Alonso, R.; Clavero, M.E.; Askari, E.; García, A.; Antón, C.; Fernández, M.; Escalante, F.; García, A.; Rios-Tamayo, R.; et al. Belantamab Mafodotin in Patients with Relapsed/Refractory Multiple Myeloma: Results of the Compassionate Use or the Expanded Access Program in Spain. Cancers 2023, 15, 2964. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Pugh, S.L.; Wang, T.J.C.; Aldape, K.; Gan, H.K.; Preusser, M.; A Vogelbaum, M.; Sulman, E.P.; Won, M.; Zhang, P.; et al. Depatuxizumab mafodotin in EGFR-amplified newly diagnosed glioblastoma: A phase III randomized clinical trial. Neuro-Oncology 2023, 25, 339–350. [Google Scholar] [CrossRef]
- Cheng-Sánchez, I.; Moya-Utrera, F.; Porras-Alcalá, C.; López-Romero, J.M.; Sarabia, F. Antibody-Drug Conjugates Containing Payloads from Marine Origin. Mar. Drugs 2022, 20, 494. [Google Scholar] [CrossRef]
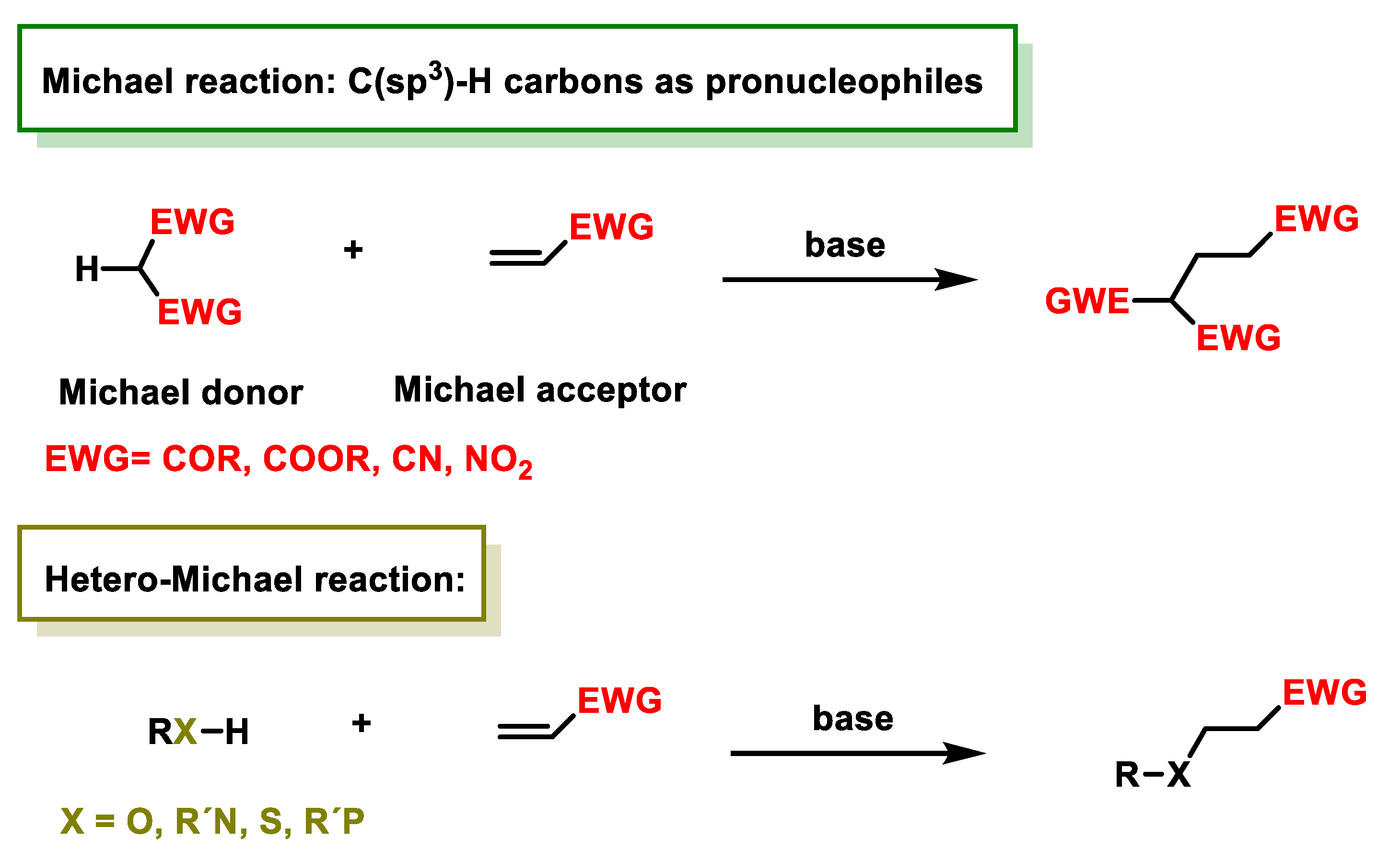
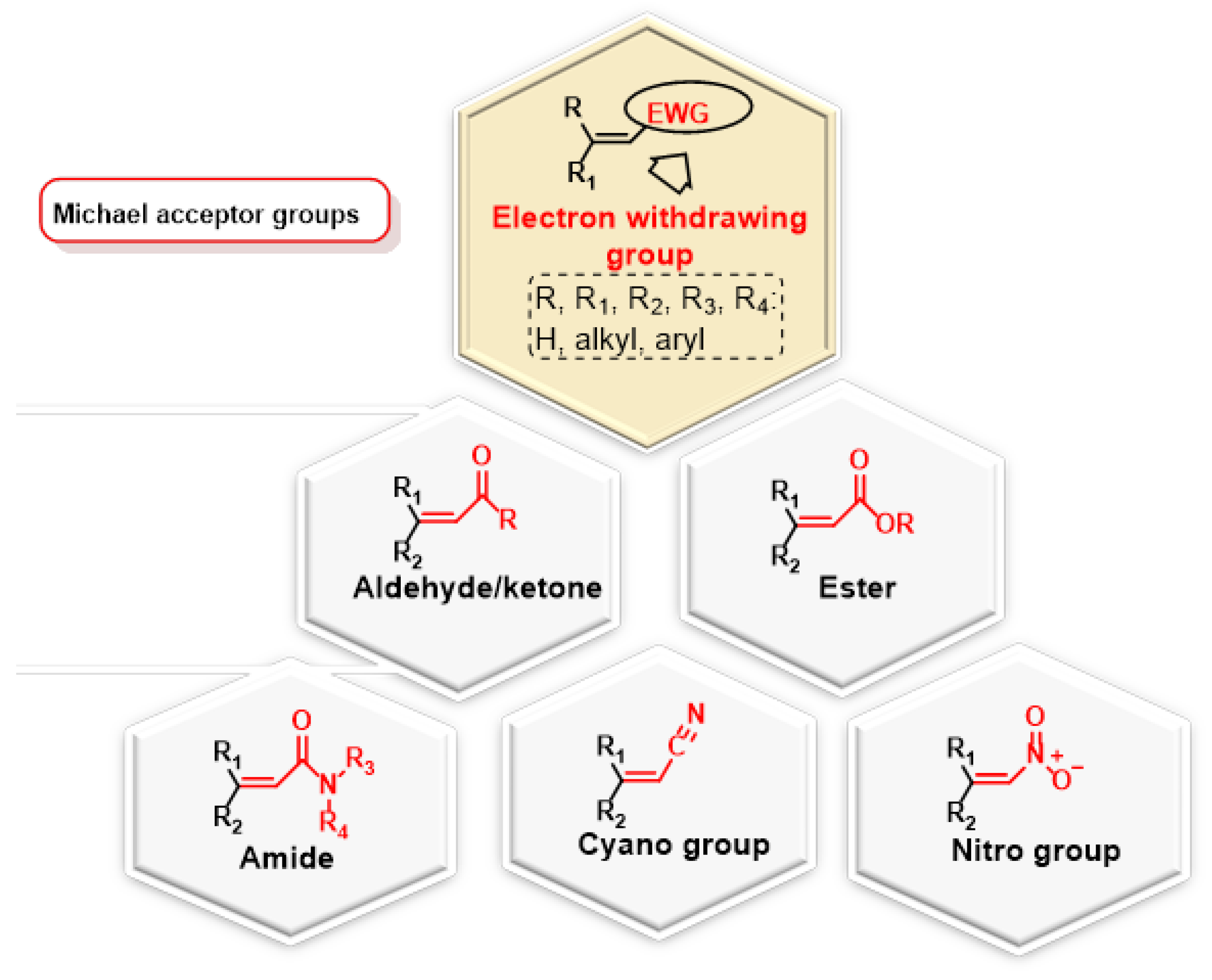

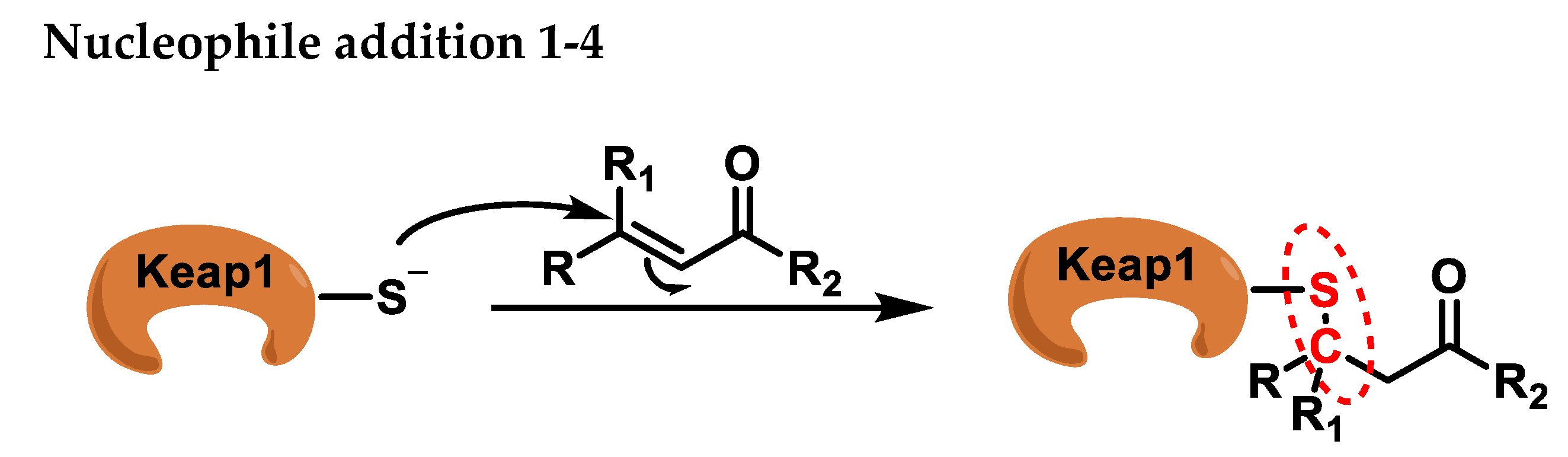

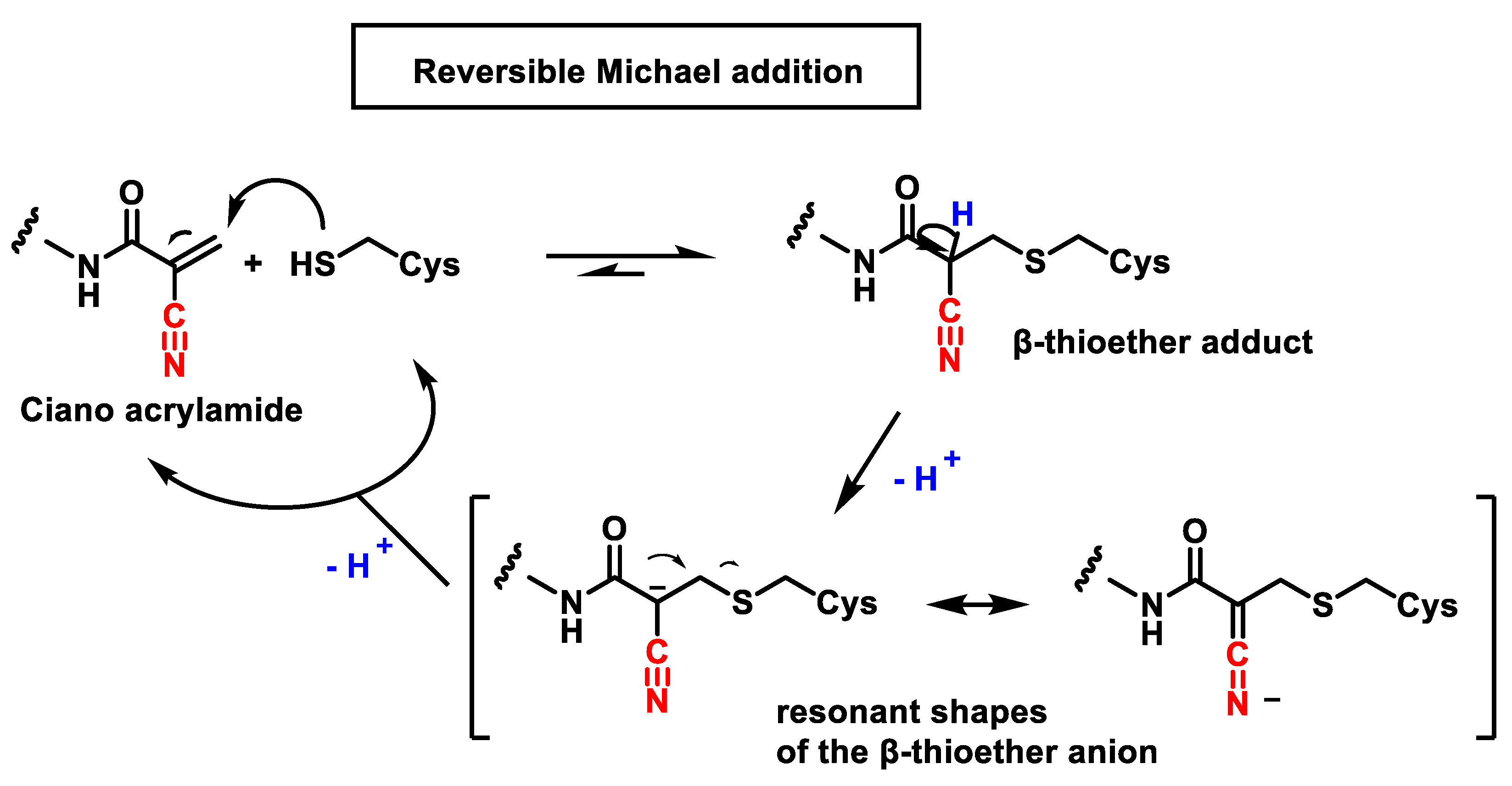
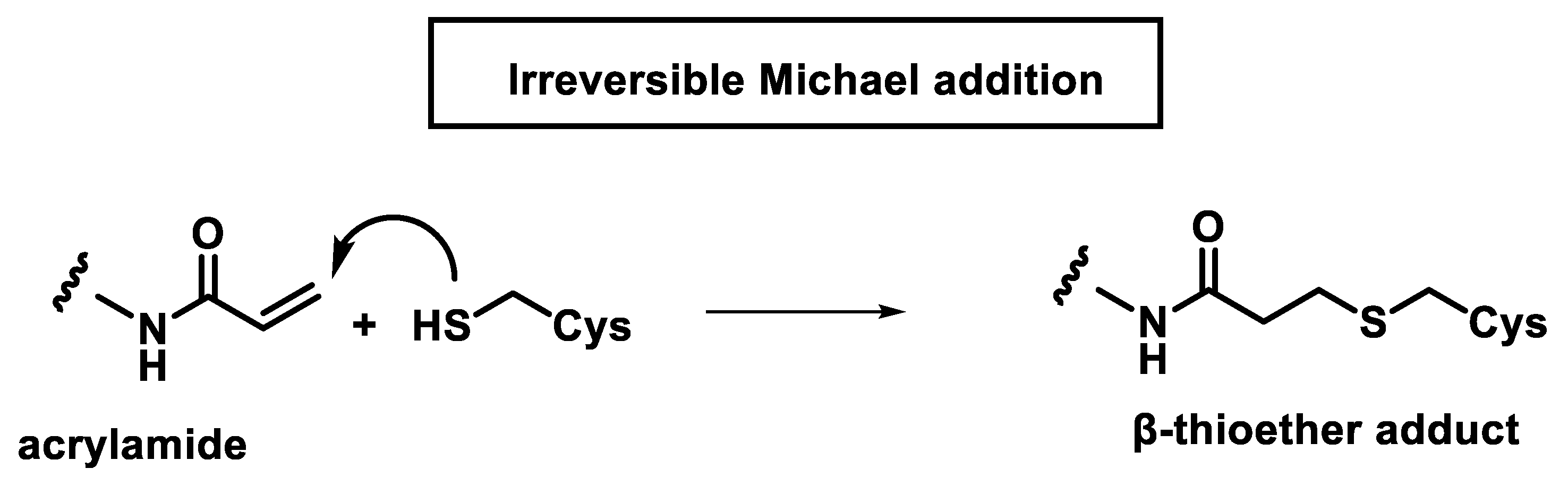
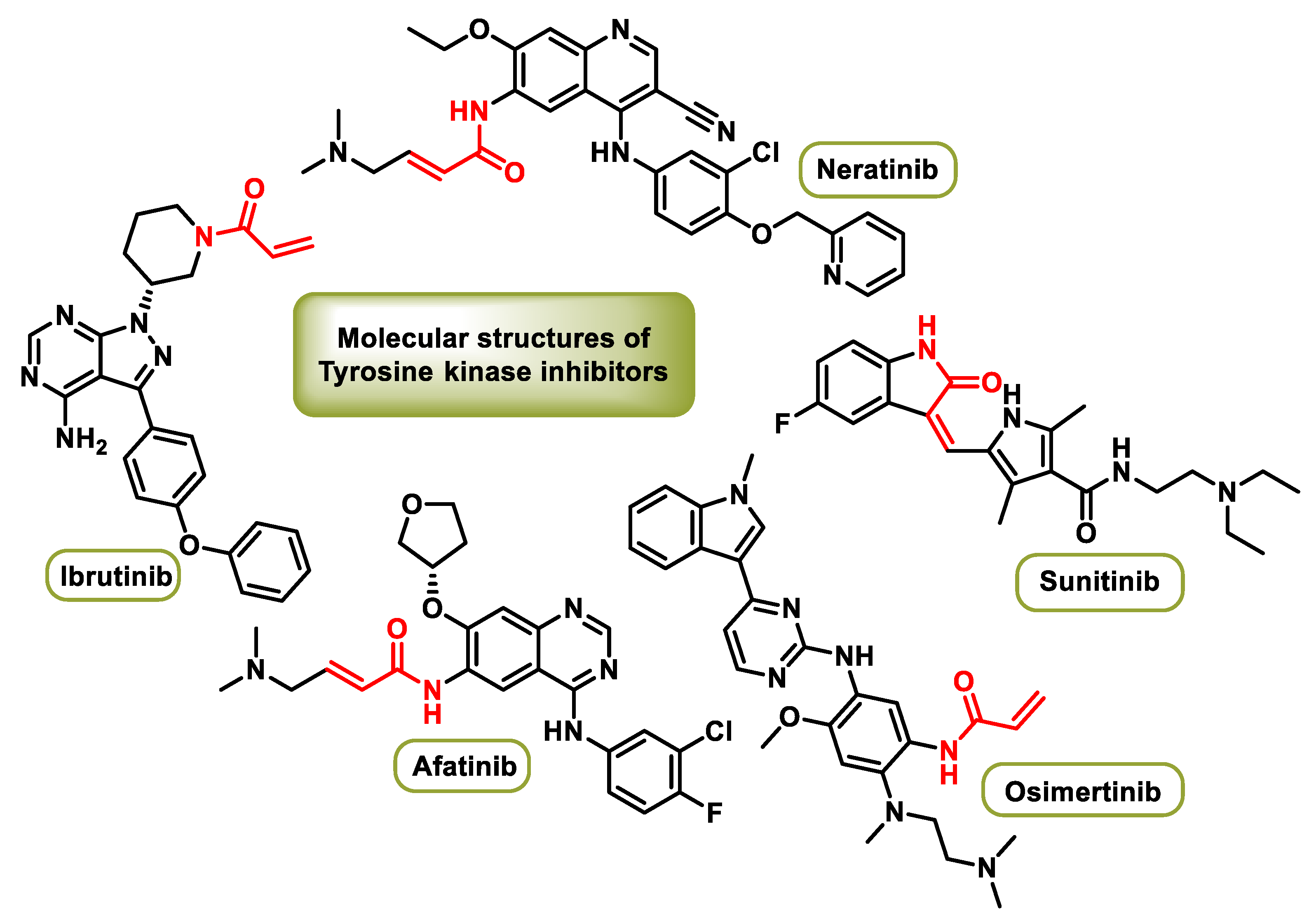

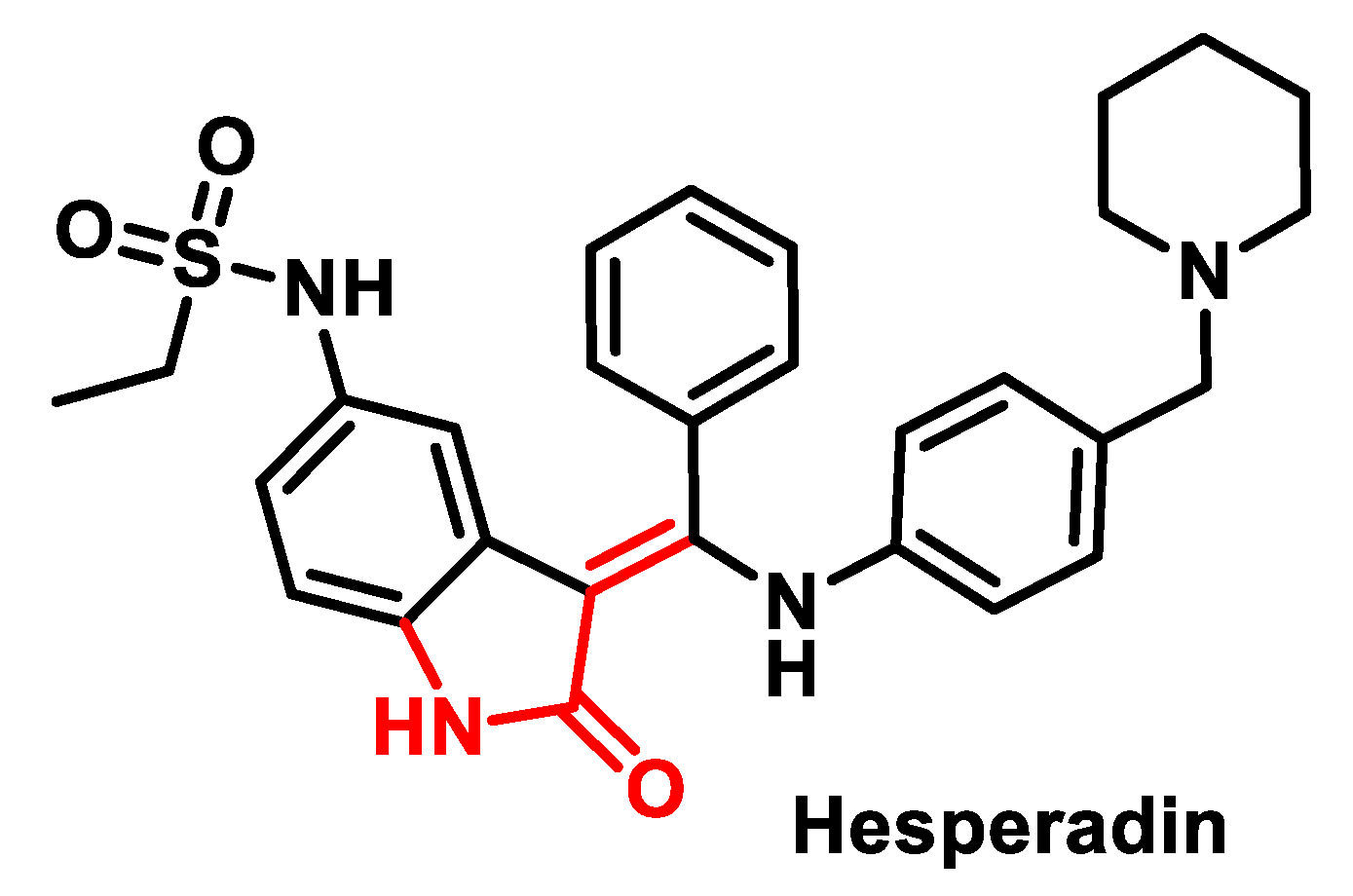
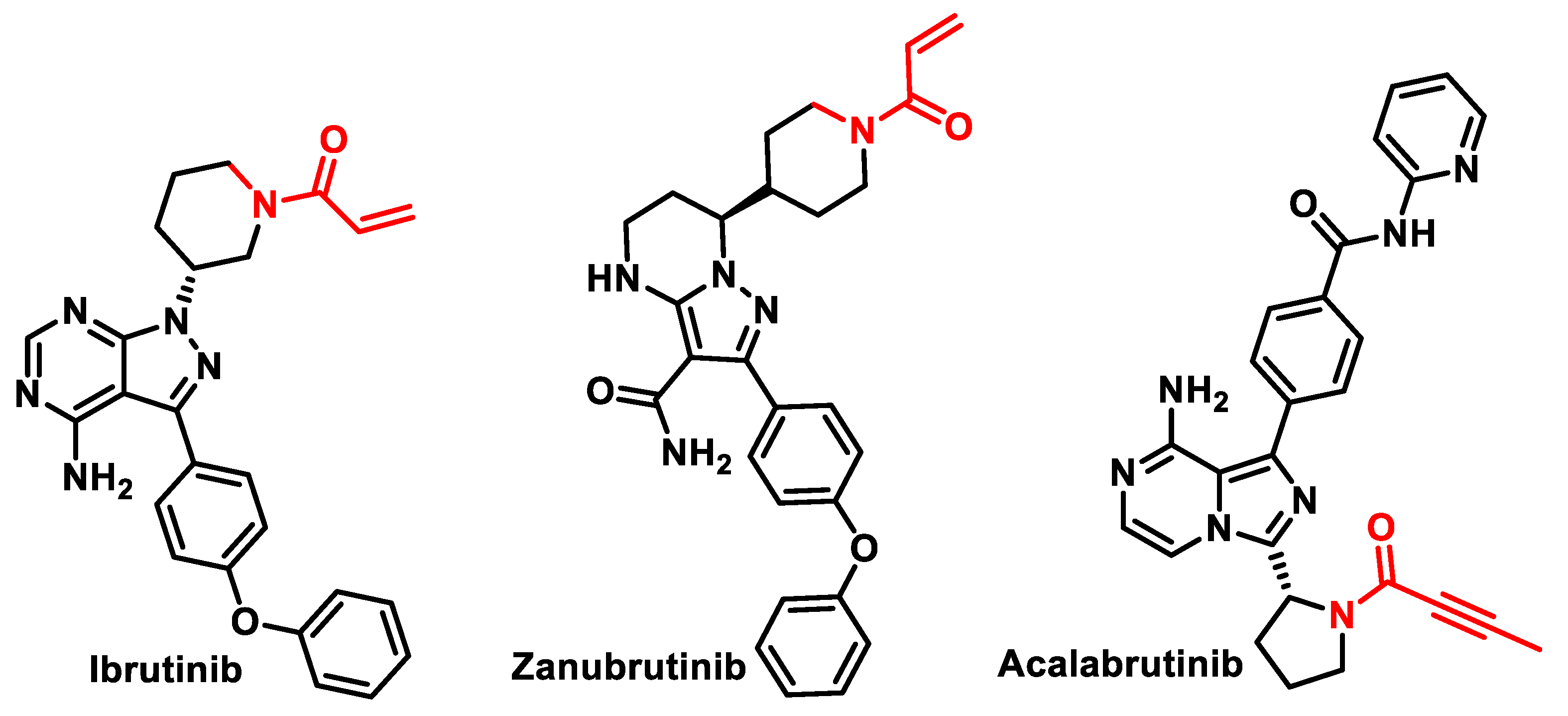
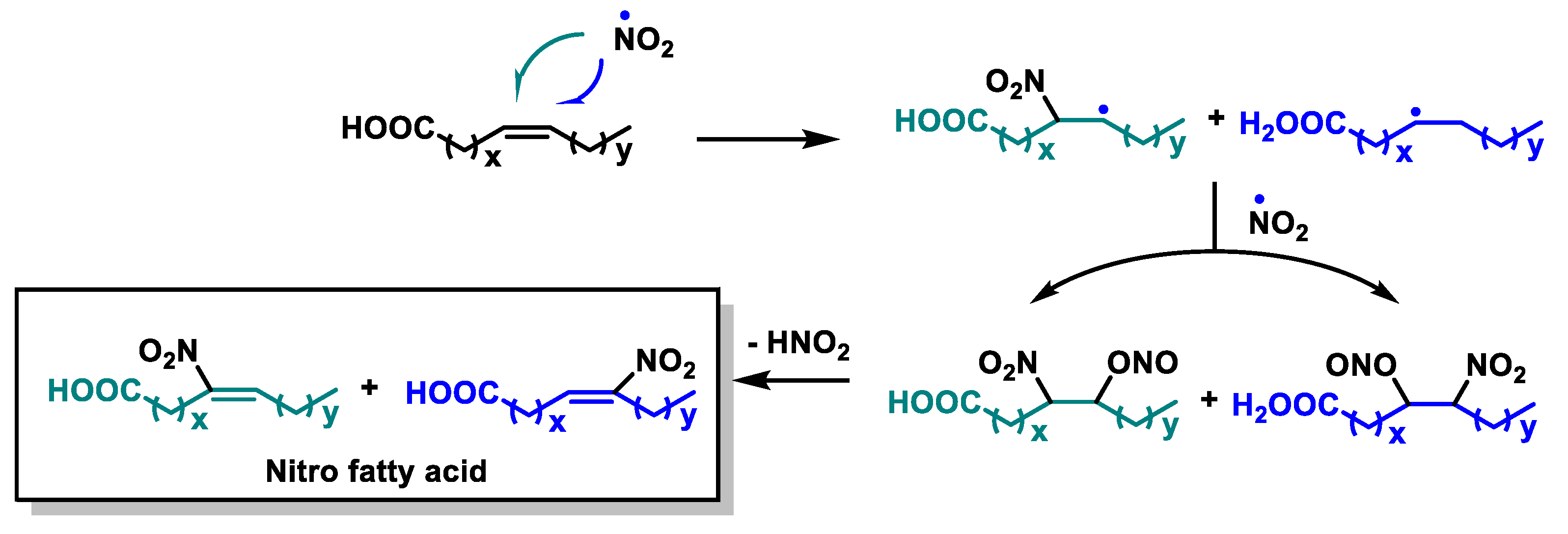

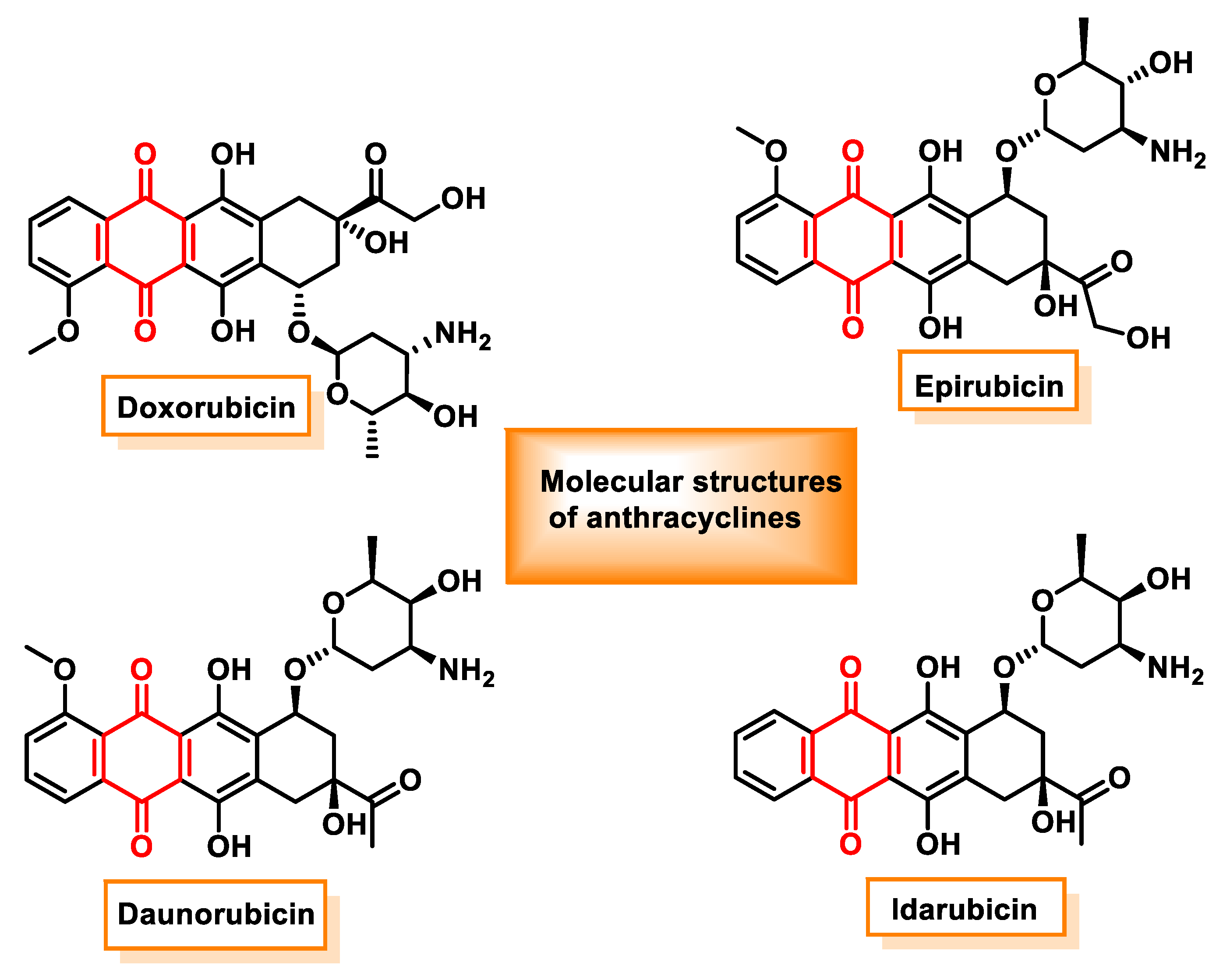
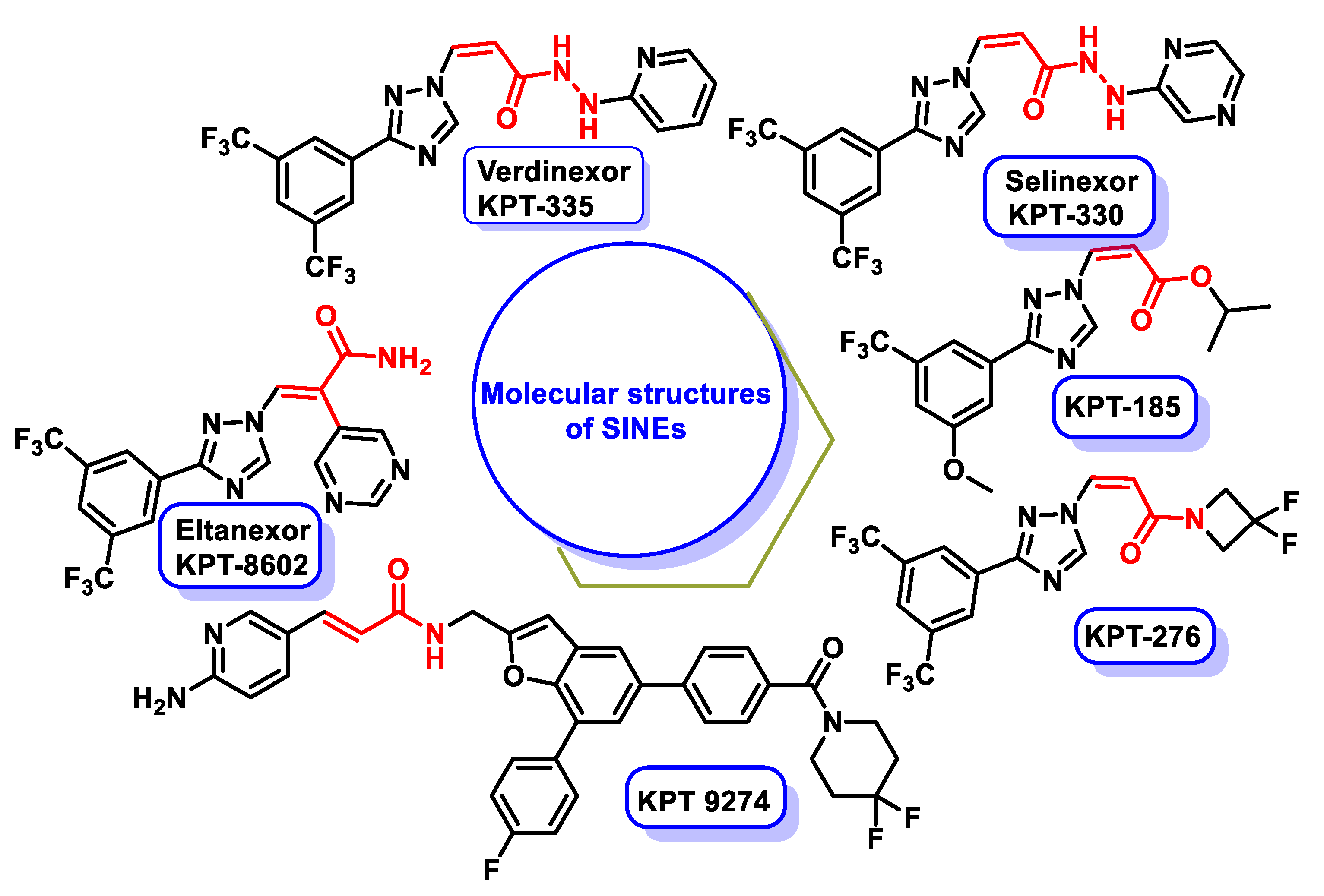

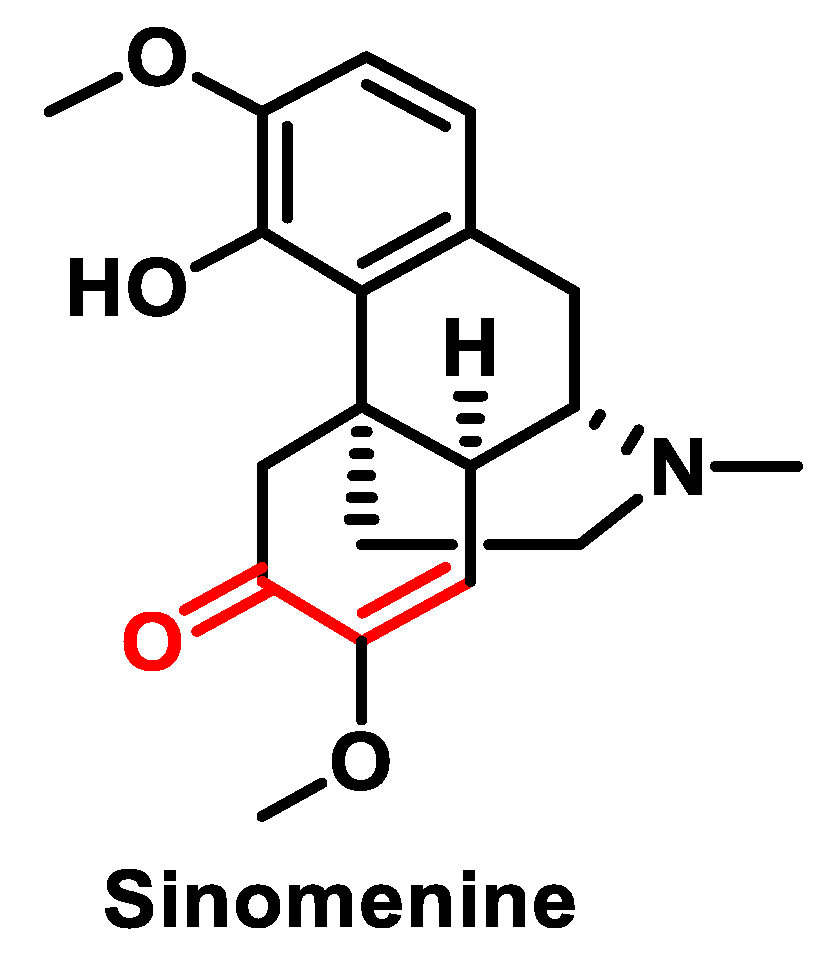

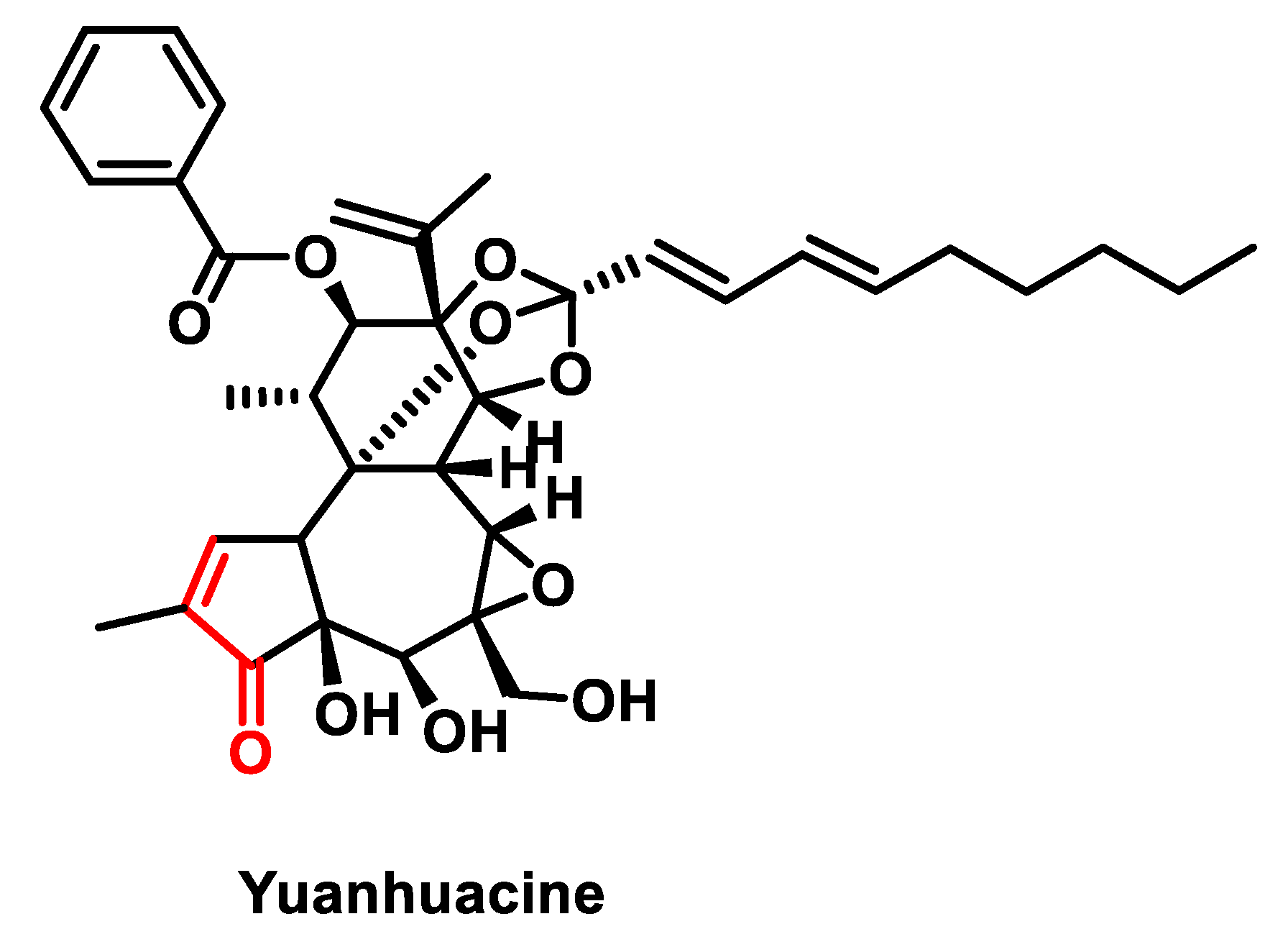
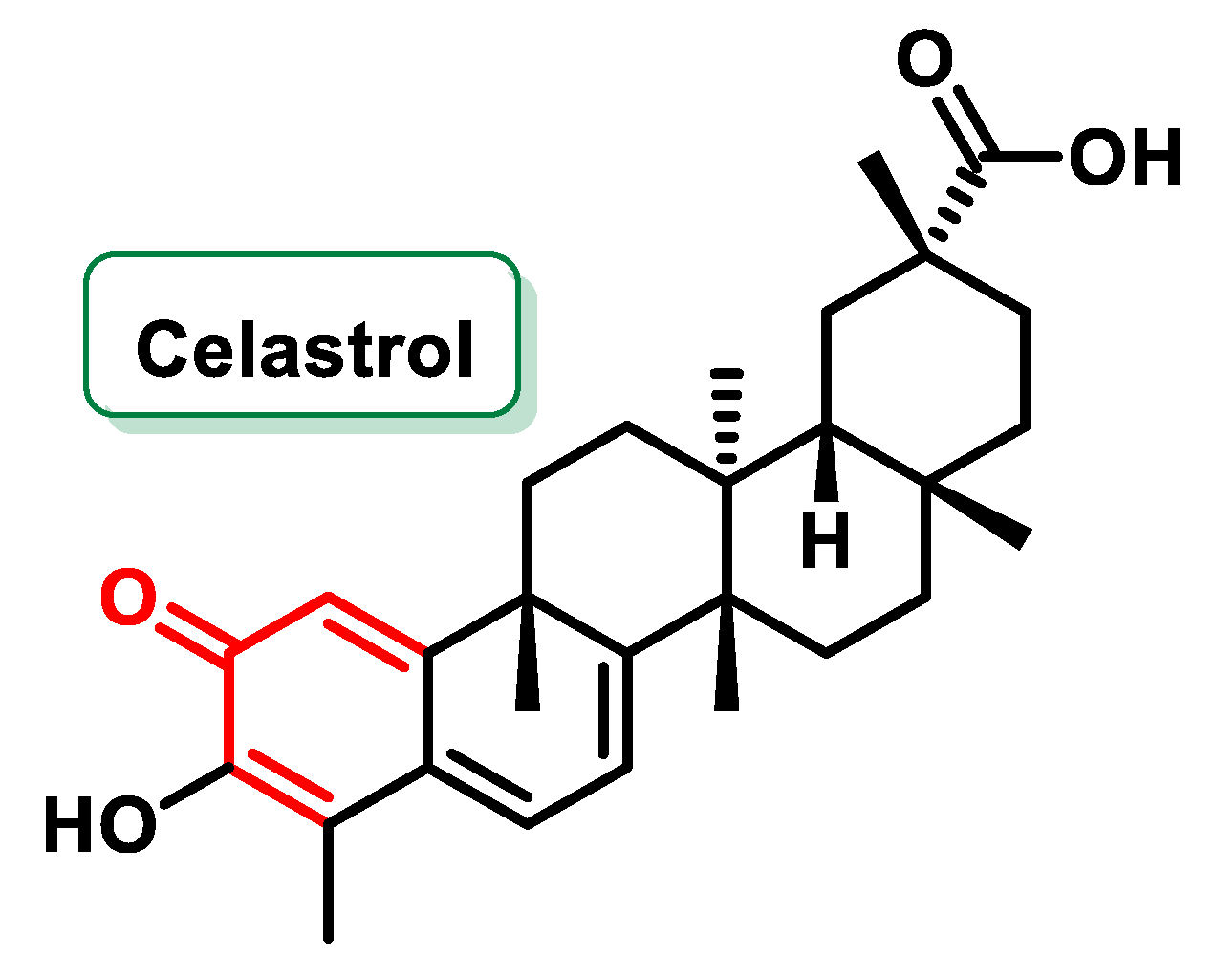
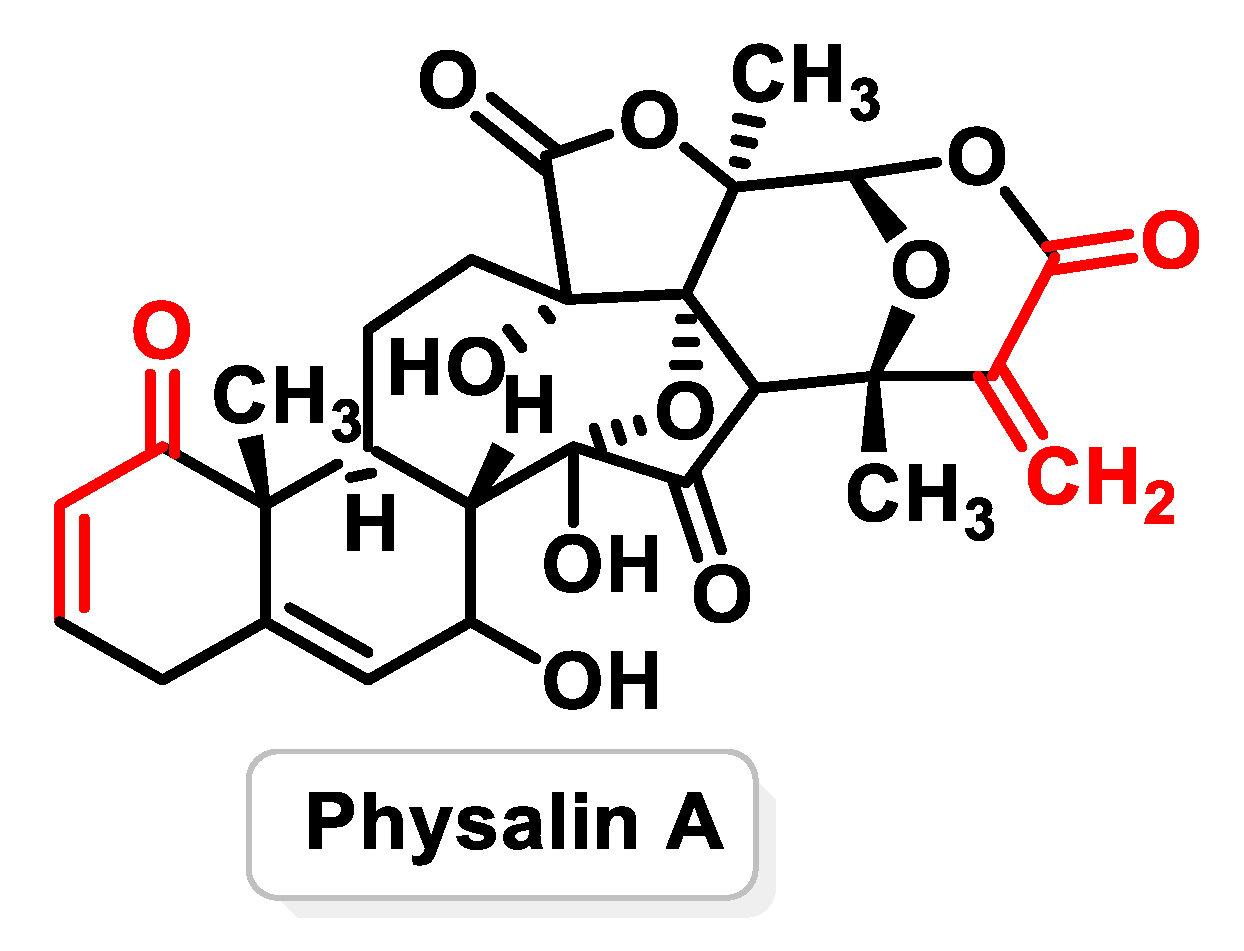
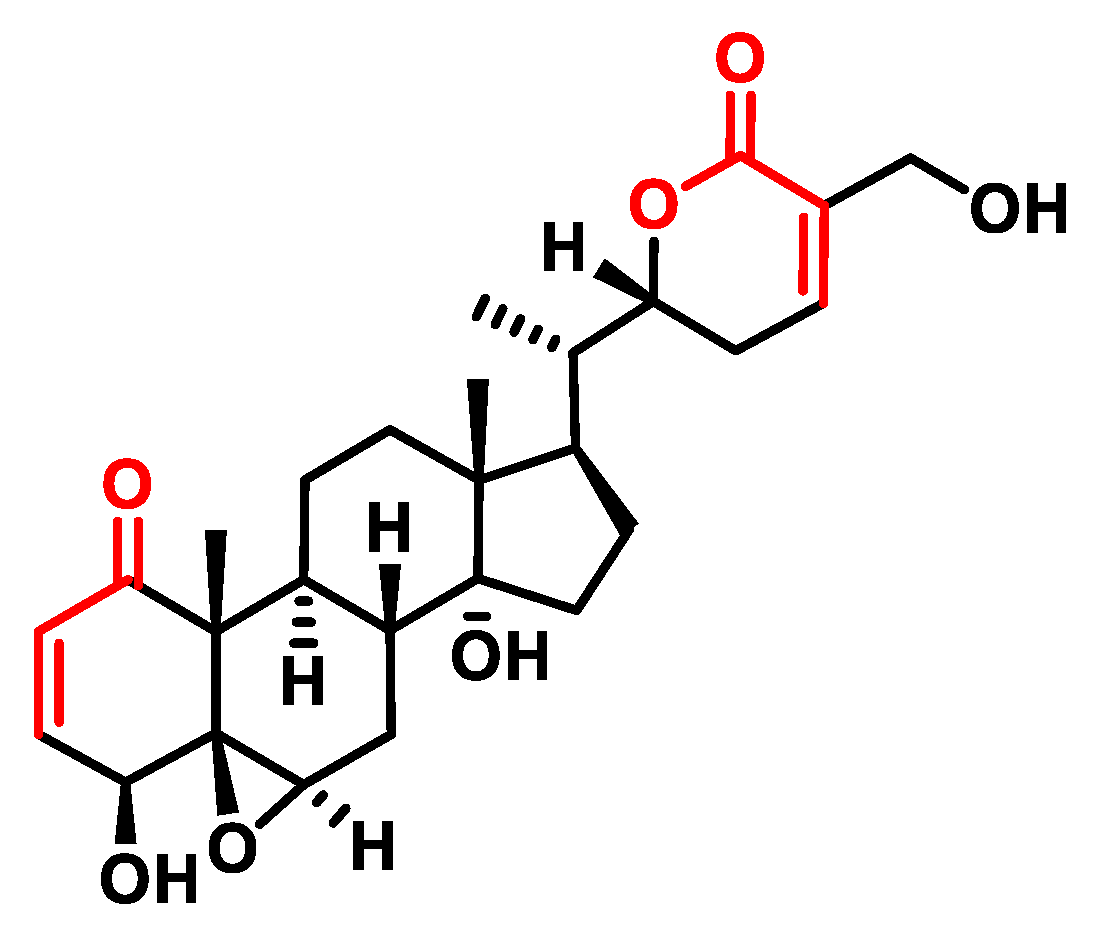
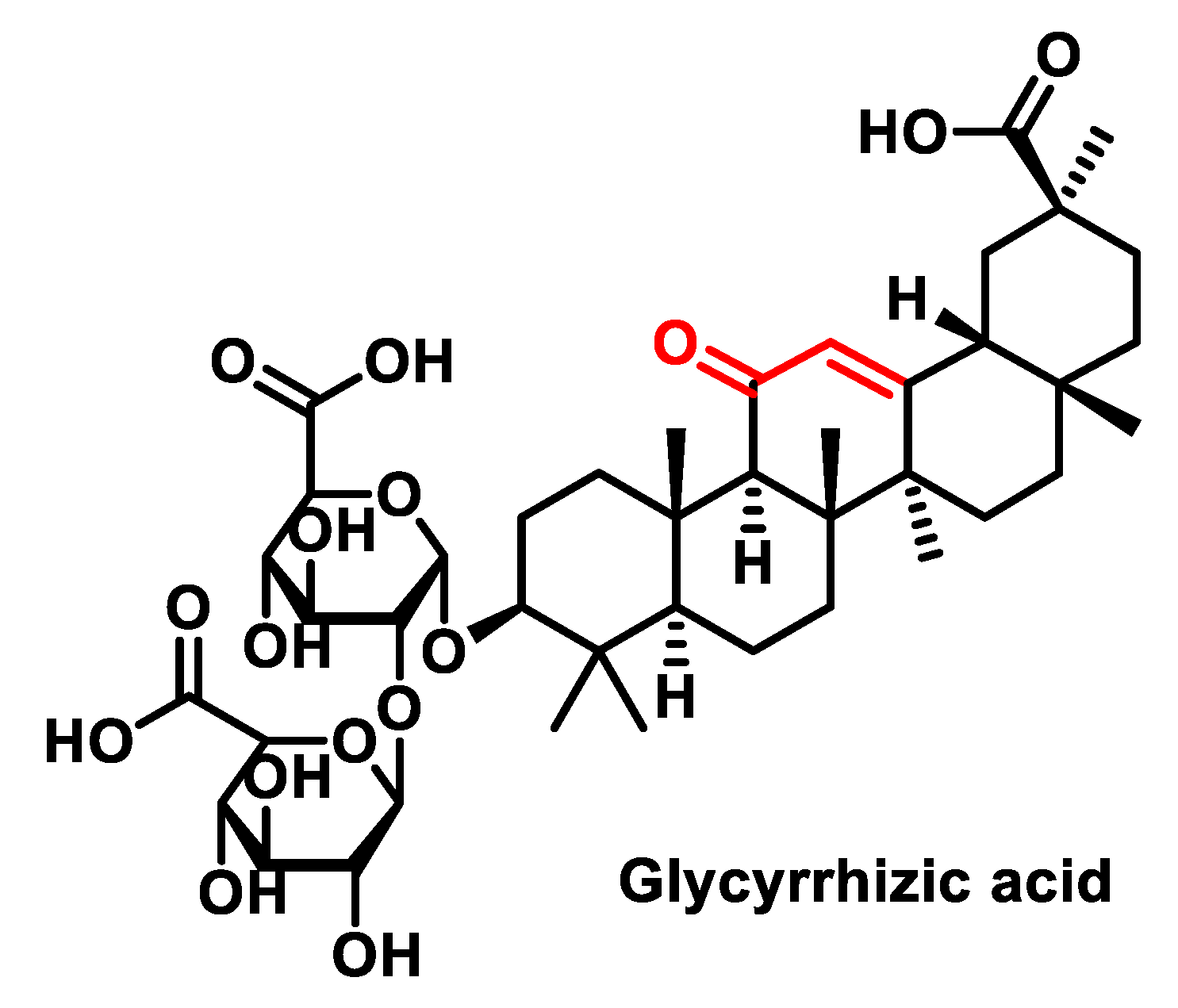
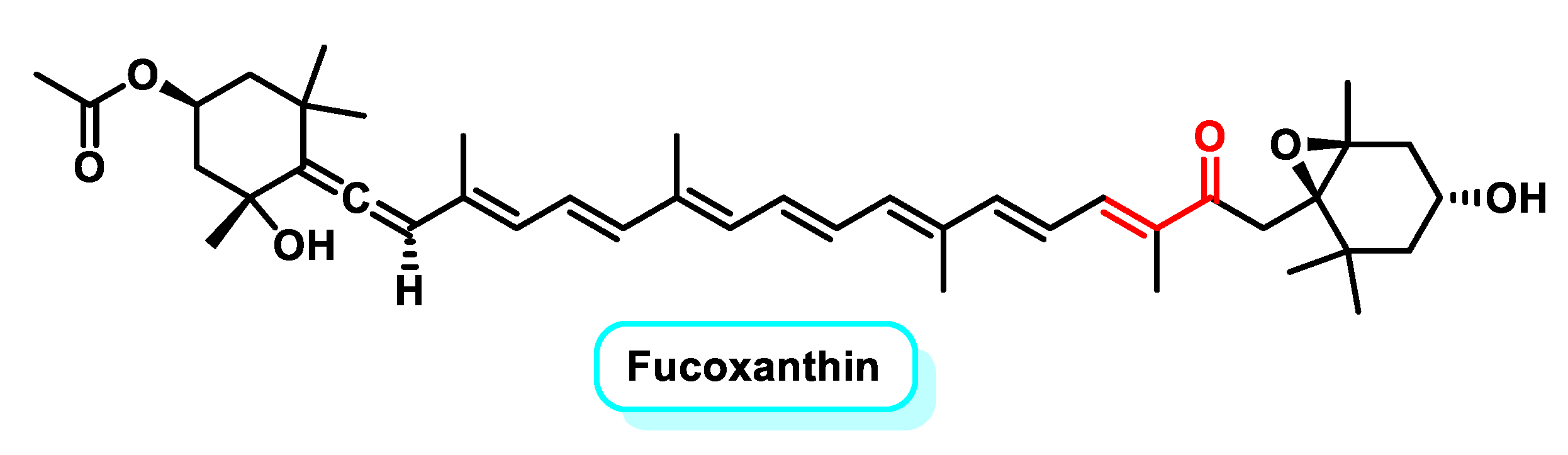
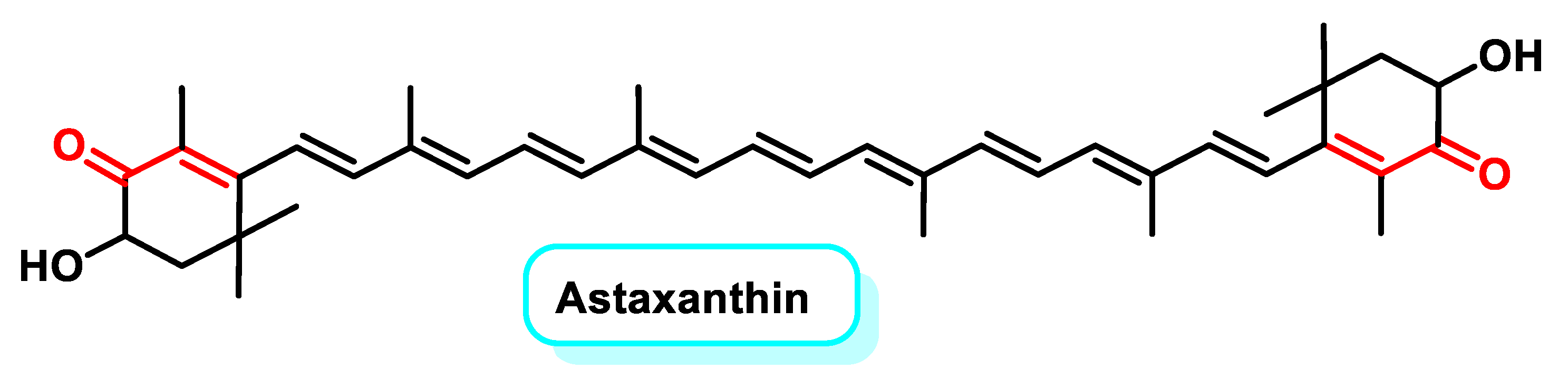
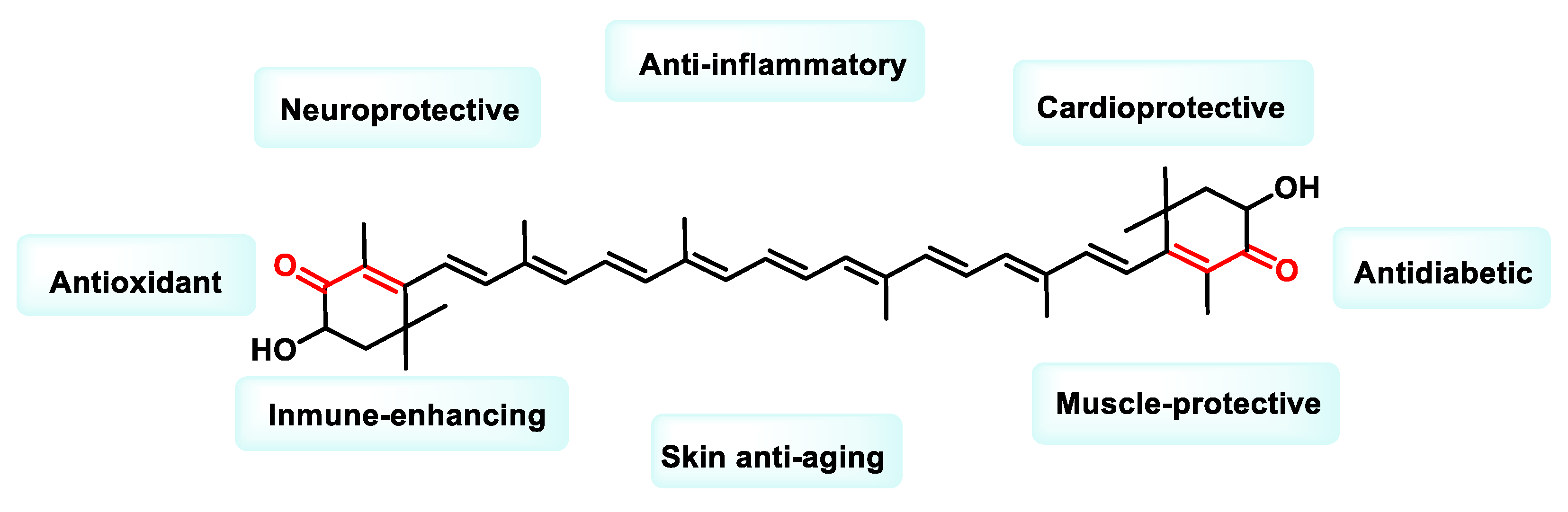
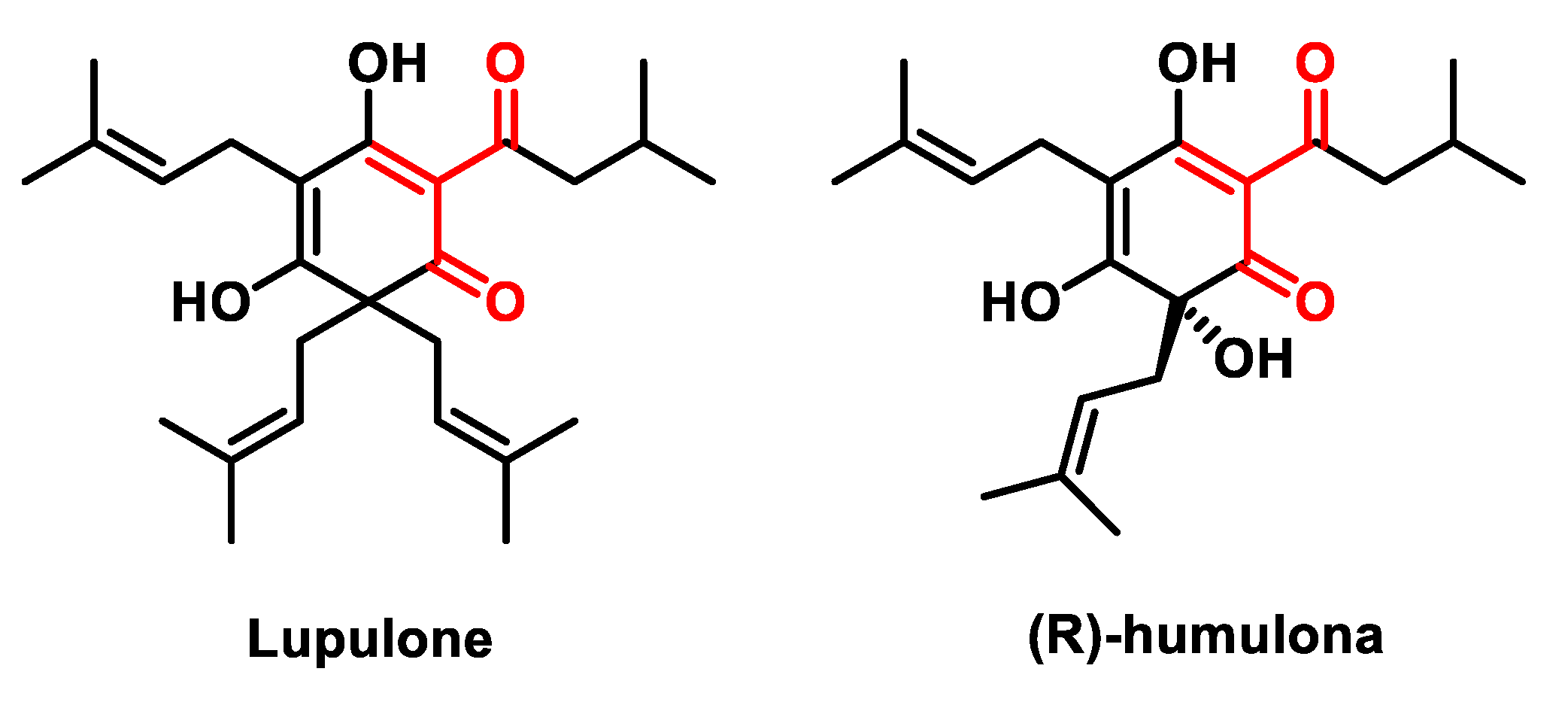
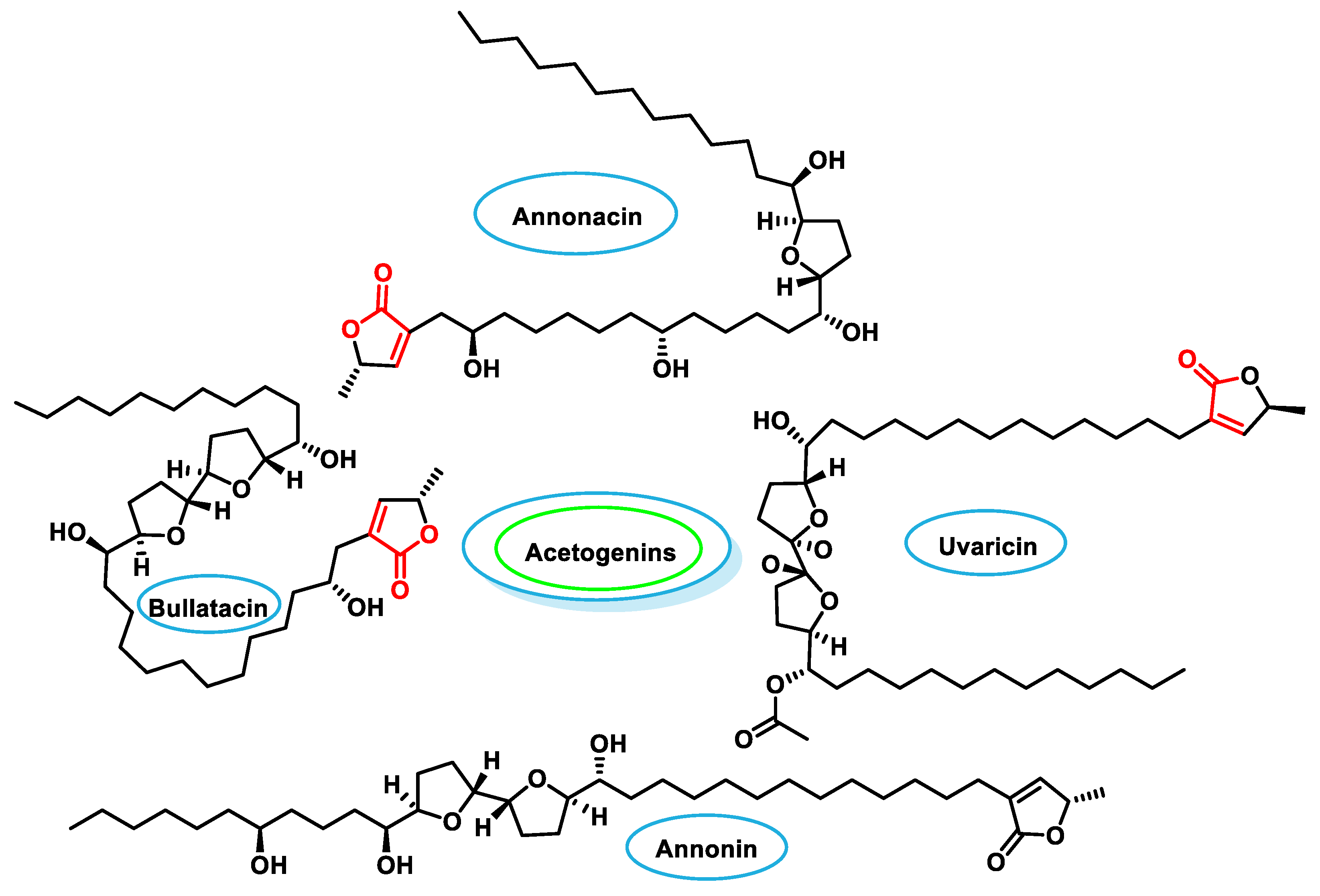
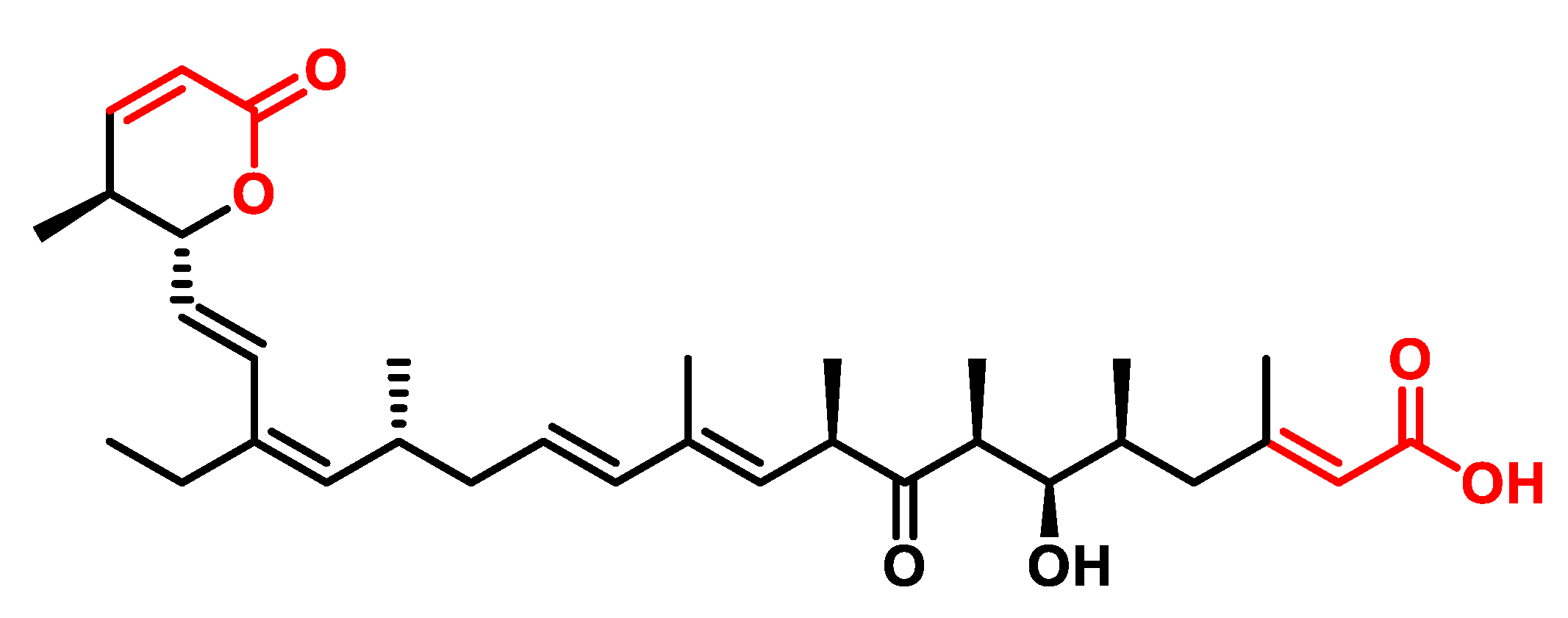

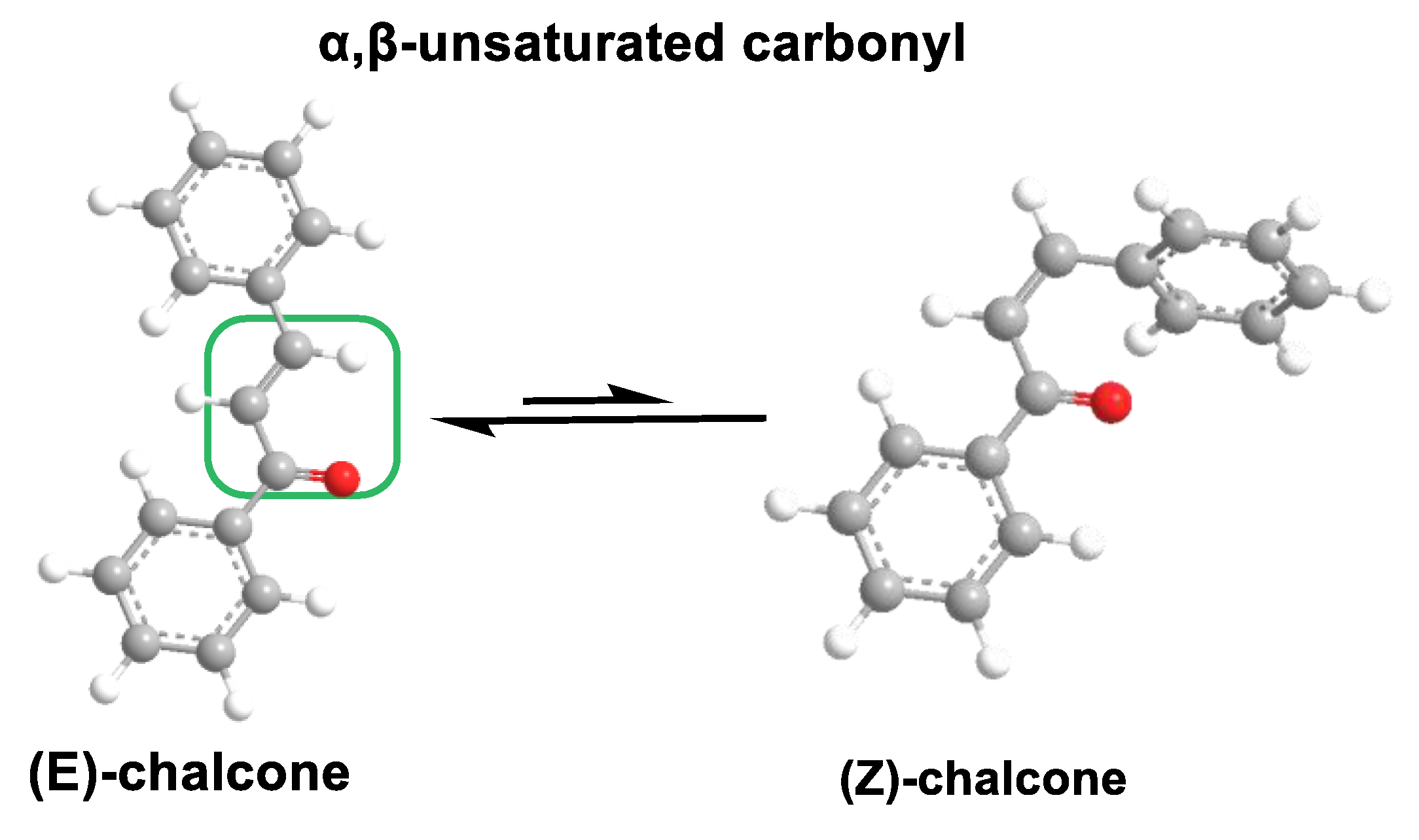
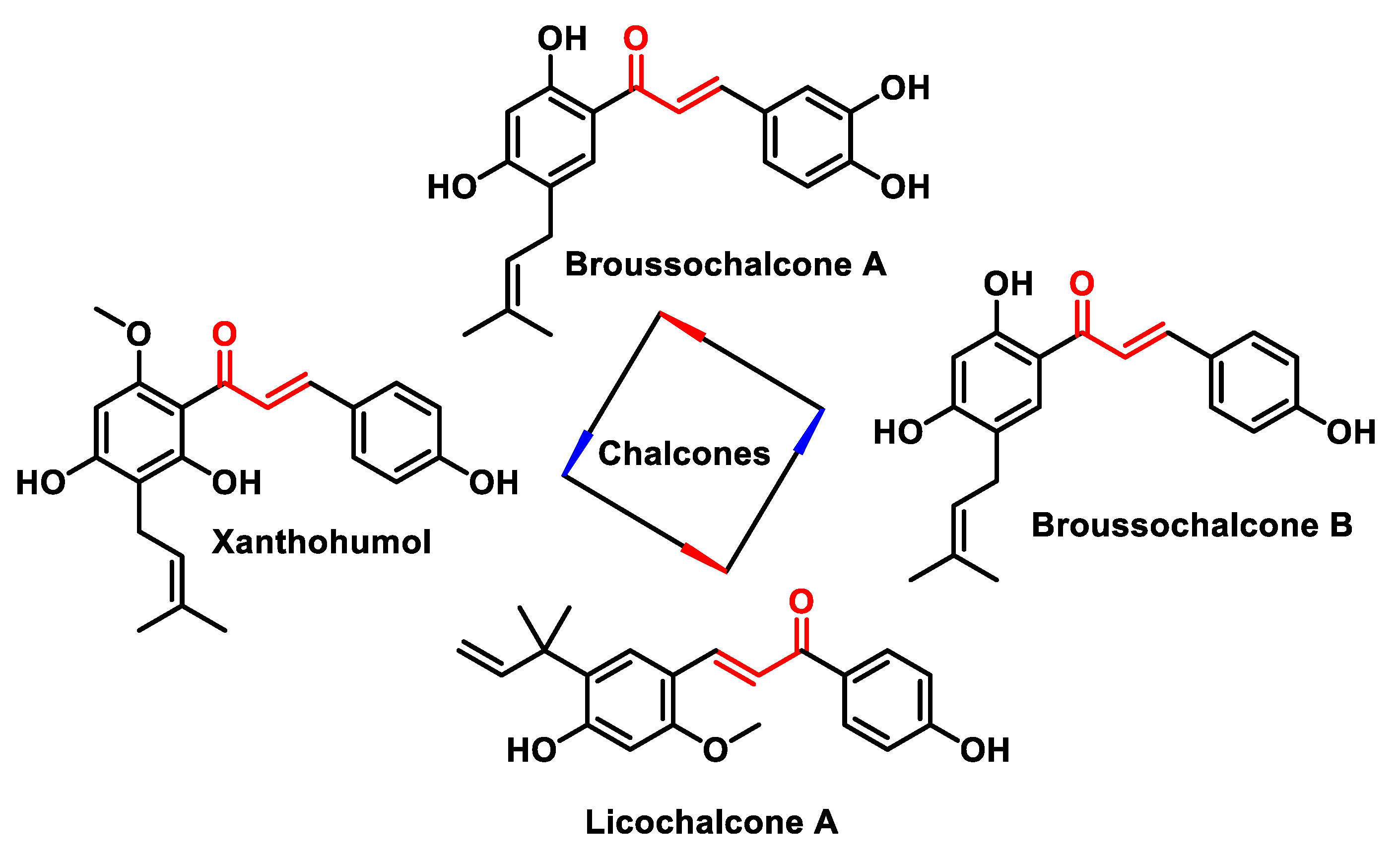
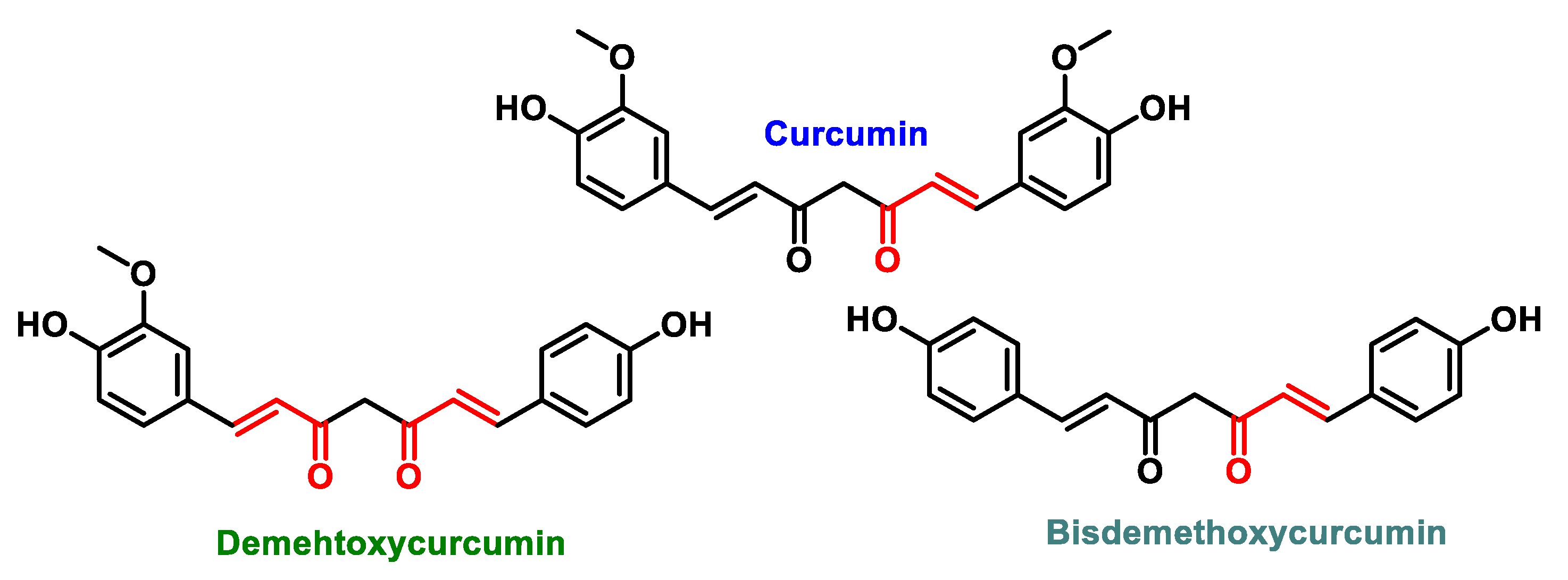
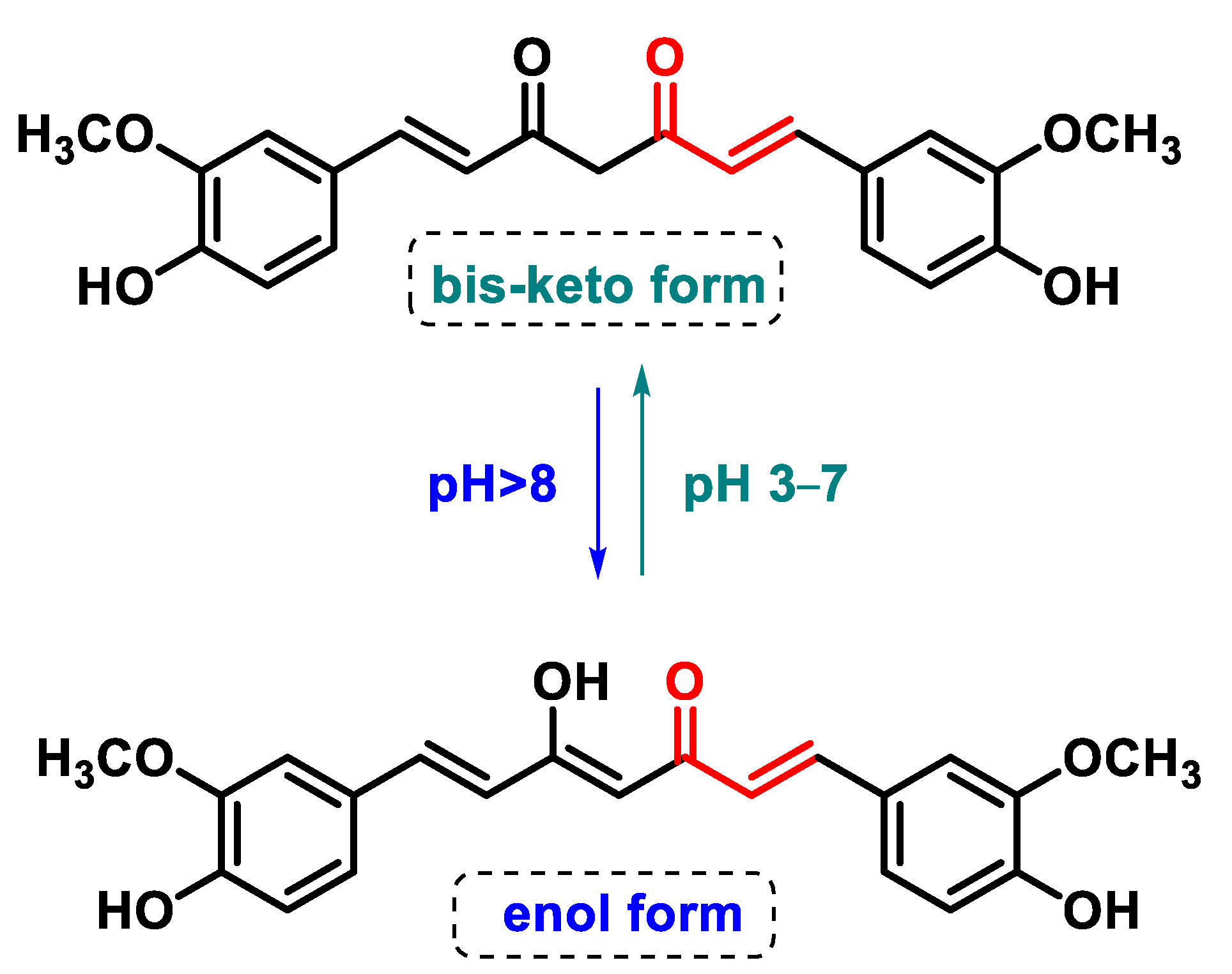
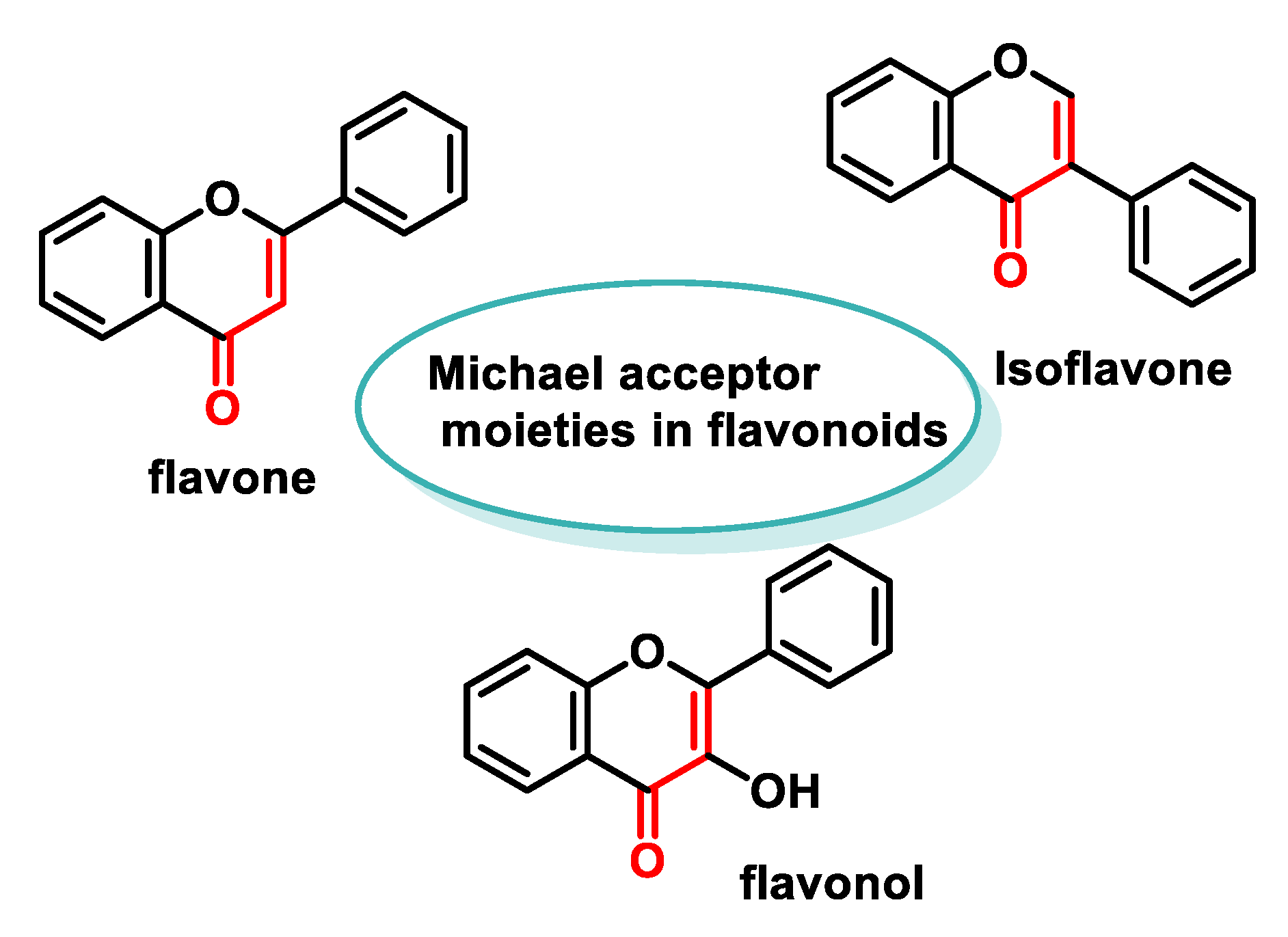


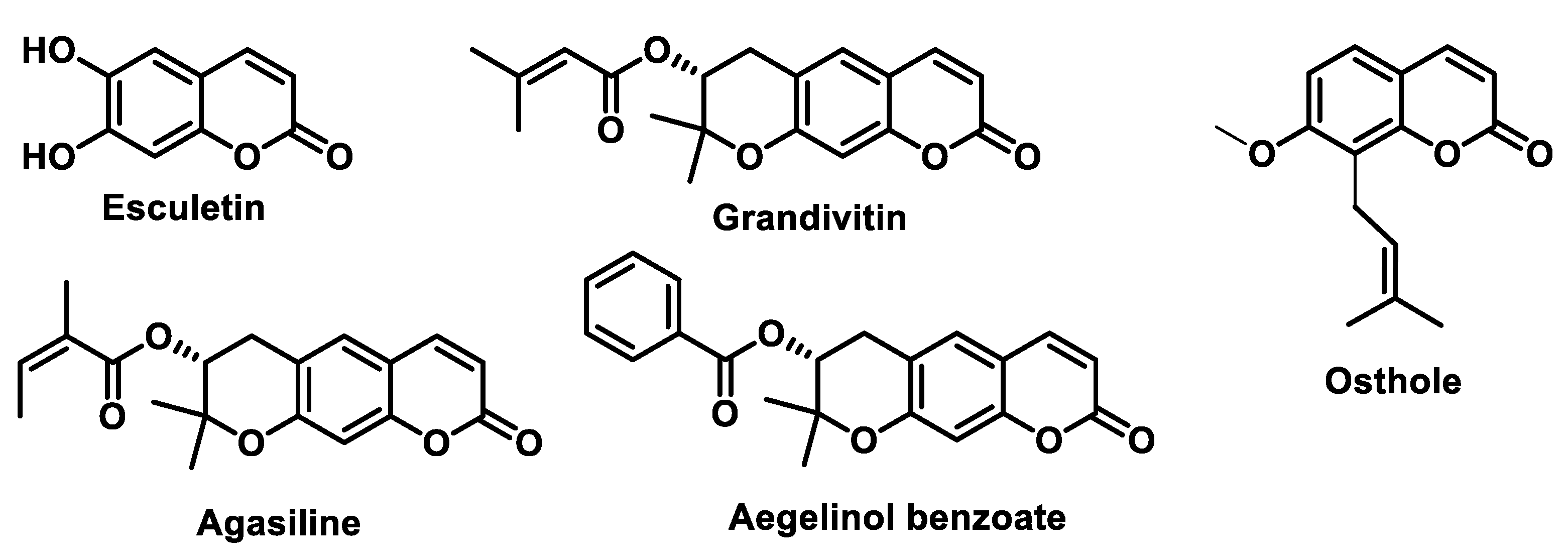
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrés, C.M.C.; Pérez de la Lastra, J.M.; Bustamante Munguira, E.; Andrés Juan, C.; Pérez-Lebeña, E. Michael Acceptors as Anti-Cancer Compounds: Coincidence or Causality? Int. J. Mol. Sci. 2024, 25, 6099. https://doi.org/10.3390/ijms25116099
Andrés CMC, Pérez de la Lastra JM, Bustamante Munguira E, Andrés Juan C, Pérez-Lebeña E. Michael Acceptors as Anti-Cancer Compounds: Coincidence or Causality? International Journal of Molecular Sciences. 2024; 25(11):6099. https://doi.org/10.3390/ijms25116099
Chicago/Turabian StyleAndrés, Celia María Curieses, José Manuel Pérez de la Lastra, Elena Bustamante Munguira, Celia Andrés Juan, and Eduardo Pérez-Lebeña. 2024. "Michael Acceptors as Anti-Cancer Compounds: Coincidence or Causality?" International Journal of Molecular Sciences 25, no. 11: 6099. https://doi.org/10.3390/ijms25116099
APA StyleAndrés, C. M. C., Pérez de la Lastra, J. M., Bustamante Munguira, E., Andrés Juan, C., & Pérez-Lebeña, E. (2024). Michael Acceptors as Anti-Cancer Compounds: Coincidence or Causality? International Journal of Molecular Sciences, 25(11), 6099. https://doi.org/10.3390/ijms25116099