
Understanding the Role of Endothelial Cells in Glioblastoma: Mechanisms and Novel Treatments
 , and
, and
Abstract
1. Introduction
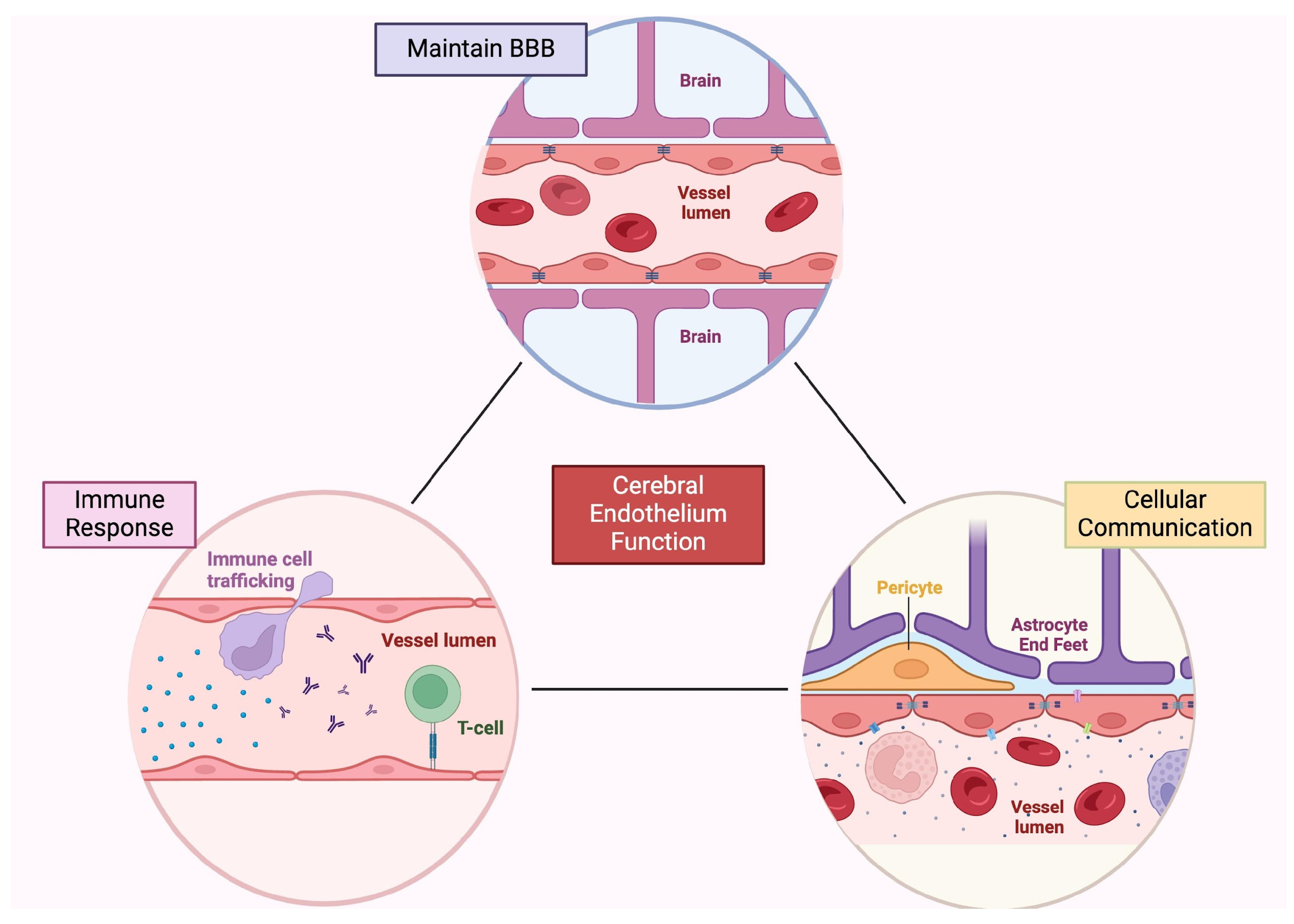
2. Cells of the Central Nervous System
2.1. Endothelial Cells
2.2. Neurons
2.3. Mural Cells
2.4. Astrocytes
2.5. Malignant Transformation
2.6. Tumor Vasculature
2.7. Structural Communication between TCs and ECs
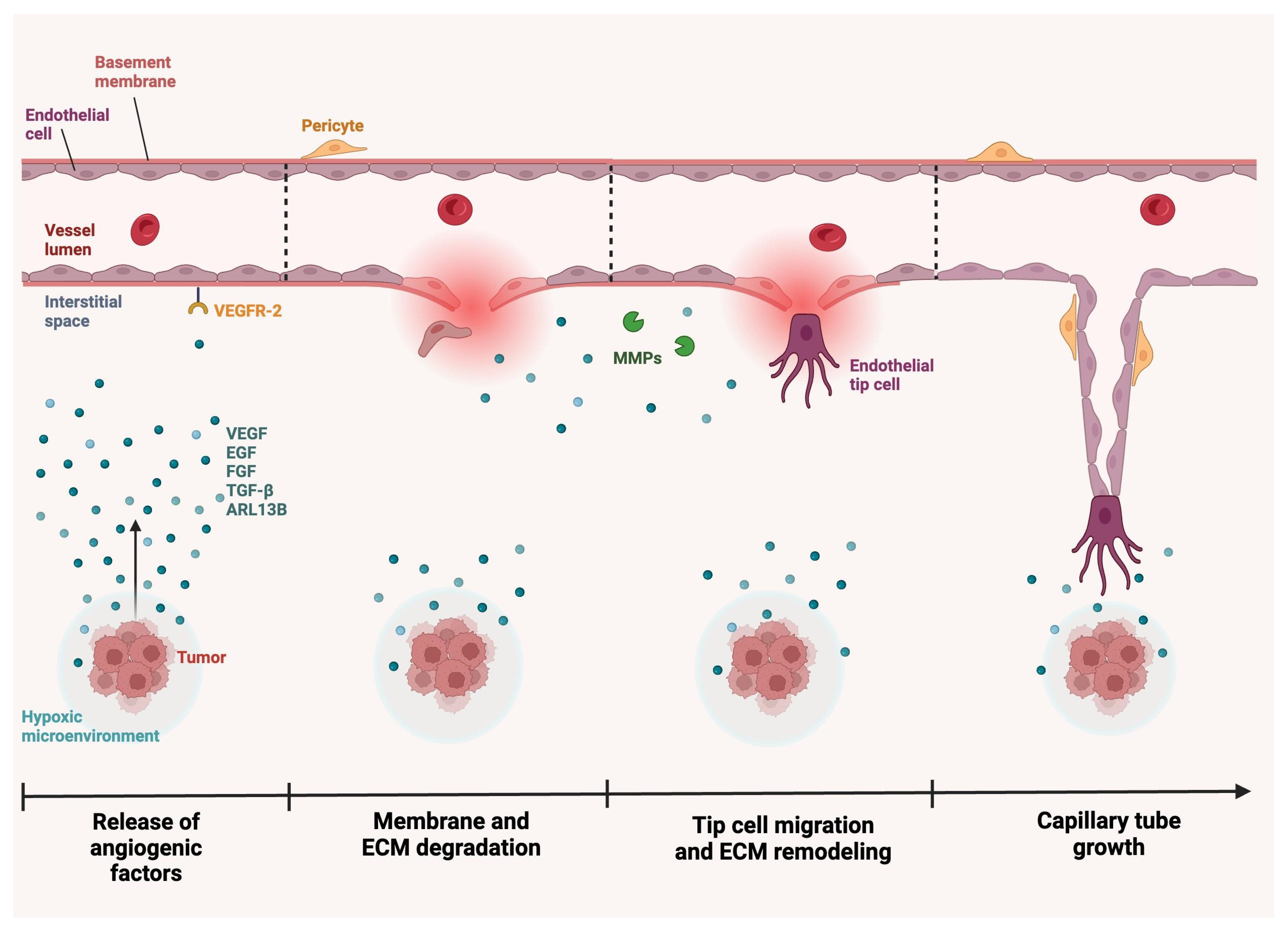
3. Angiogenesis in Glioblastoma
3.1. Angiogenesis Mechanism
3.2. Anti-Angiogenic Therapies in Glioblastoma
3.3. Other Methods to Target Angiogenesis
4. Alternative Interventions
4.1. Immunotherapy for Glioblastoma
4.2. Nanoparticles and Other Novel Technologies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grochans, S.; Cybulska, A.M.; Siminska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Klein, D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Iba, T.; Takakura, N. Mechanisms of new blood-vessel formation and proliferative heterogeneity of endothelial cells. Int. Immunol. 2020, 32, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Coomber, B.L.; Stewart, P.A. Morphometric analysis of CNS microvascular endothelium. Microvasc. Res. 1985, 30, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Guo, S.; Kim, W.J.; Lok, J.; Lee, S.R.; Besancon, E.; Luo, B.H.; Stins, M.F.; Wang, X.; Dedhar, S.; Lo, E.H. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 7582–7587. [Google Scholar] [CrossRef]
- Venkat, P.; Chopp, M.; Chen, J. New insights into coupling and uncoupling of cerebral blood flow and metabolism in the brain. Croat. Med. J. 2016, 57, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.J.; Gutterman, D.D. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation 2013, 20, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Zhang, Z.G.; Wang, L.; Zhang, R.L.; Zhang, L.; Morris, D.; Gregg, S.R.; Wu, Z.; Jiang, A.; Lu, M.; et al. Coupling of angiogenesis and neurogenesis in cultured endothelial cells and neural progenitor cells after stroke. J. Cereb. Blood Flow. Metab. 2008, 28, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Shalabi, S.; Belayachi, A.; Larrivee, B. Involvement of neuronal factors in tumor angiogenesis and the shaping of the cancer microenvironment. Front. Immunol. 2024, 15, 1284629. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Rege, S.V.; Ramanathan, A.; Wang, Y.; Ahuja, A.; Lazic, D.; Tsai, P.S.; Zhao, Z.; Zhou, Y.; et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci. 2017, 20, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nikolakopoulou, A.M.; Sweeney, M.D.; Lazic, D.; Zhao, Z.; Zlokovic, B.V. Acute Ablation of Cortical Pericytes Leads to Rapid Neurovascular Uncoupling. Front. Cell Neurosci. 2020, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Nikolakopoulou, A.M.; Montagne, A.; Kisler, K.; Dai, Z.; Wang, Y.; Huuskonen, M.T.; Sagare, A.P.; Lazic, D.; Sweeney, M.D.; Kong, P.; et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat. Neurosci. 2019, 22, 1089–1098. [Google Scholar] [CrossRef]
- Gastfriend, B.D.; Snyder, M.E.; Holt, H.E.; Daneman, R.; Palecek, S.P.; Shusta, E.V. Notch3 directs differentiation of brain mural cells from human pluripotent stem cell-derived neural crest. Sci. Adv. 2024, 10, eadi1737. [Google Scholar] [CrossRef]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.; Chapouly, C.; Andrique, L.; Bikfalvi, A.; Daubon, T. The Normal and Brain Tumor Vasculature: Morphological and Functional Characteristics and Therapeutic Targeting. Front. Physiol. 2021, 12, 622615. [Google Scholar] [CrossRef] [PubMed]
- Birbrair, A.; Zhang, T.; Wang, Z.M.; Messi, M.L.; Olson, J.D.; Mintz, A.; Delbono, O. Type-2 pericytes participate in normal and tumoral angiogenesis. Am. J. Physiol. Cell Physiol. 2014, 307, C25–C38. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Castro, B.; Robel, S.; Mishra, A. Astrocyte Endfeet in Brain Function and Pathology: Open Questions. Annu. Rev. Neurosci. 2023, 46, 101–121. [Google Scholar] [CrossRef]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood-Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 7146. [Google Scholar] [CrossRef]
- Hosli, L.; Zuend, M.; Bredell, G.; Zanker, H.S.; Porto de Oliveira, C.E.; Saab, A.S.; Weber, B. Direct vascular contact is a hallmark of cerebral astrocytes. Cell Rep. 2022, 39, 110599. [Google Scholar] [CrossRef] [PubMed]
- Bindocci, E.; Savtchouk, I.; Liaudet, N.; Becker, D.; Carriero, G.; Volterra, A. Three-dimensional Ca(2+) imaging advances understanding of astrocyte biology. Science 2017, 356, eaai8185. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonniere, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef]
- Guo, X.; Gu, L.; Li, Y.; Zheng, Z.; Chen, W.; Wang, Y.; Wang, Y.; Xing, H.; Shi, Y.; Liu, D.; et al. Histological and molecular glioblastoma, IDH-wildtype: A real-world landscape using the 2021 WHO classification of central nervous system tumors. Front. Oncol. 2023, 13, 1200815. [Google Scholar] [CrossRef] [PubMed]
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System tumours: A practical update on what neurosurgeons need to know-a minireview. Acta Neurochir. 2022, 164, 2453–2464. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, S.; Song, C.; Zha, Y.; Li, L. The prognostic value of MGMT promoter status by pyrosequencing assay for glioblastoma patients’ survival: A meta-analysis. World J. Surg. Oncol. 2016, 14, 261. [Google Scholar] [CrossRef]
- Gupta, R.K.; Niklasson, M.; Bergstrom, T.; Segerman, A.; Betsholtz, C.; Westermark, B. Tumor-specific migration routes of xenotransplanted human glioblastoma cells in mouse brain. Sci. Rep. 2024, 14, 864. [Google Scholar] [CrossRef] [PubMed]
- Mair, D.B.; Ames, H.M.; Li, R. Mechanisms of invasion and motility of high-grade gliomas in the brain. Mol. Biol. Cell 2018, 29, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Verhaak, R.G.; Canoll, P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert. Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Sugiarto, S.; Persson, A.I.; Munoz, E.G.; Waldhuber, M.; Lamagna, C.; Andor, N.; Hanecker, P.; Ayers-Ringler, J.; Phillips, J.; Siu, J.; et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell 2011, 20, 328–340. [Google Scholar] [CrossRef]
- Guarnaccia, L.; Navone, S.E.; Trombetta, E.; Cordiglieri, C.; Cherubini, A.; Crisa, F.M.; Rampini, P.; Miozzo, M.; Fontana, L.; Caroli, M.; et al. Angiogenesis in human brain tumors: Screening of drug response through a patient-specific cell platform for personalized therapy. Sci. Rep. 2018, 8, 8748. [Google Scholar] [CrossRef]
- Cuypers, A.; Truong, A.K.; Becker, L.M.; Saavedra-Garcia, P.; Carmeliet, P. Tumor vessel co-option: The past & the future. Front. Oncol. 2022, 12, 965277. [Google Scholar] [CrossRef]
- Dor, Y.; Porat, R.; Keshet, E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am. J. Physiol.-Cell Physiol. 2001, 280, C1367–C1374. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.A.; Eberhart, C.G. Molecular pathways: Not a simple tube--the many functions of blood vessels. Clin. Cancer Res. 2015, 21, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Geng, Z.; Wang, S.; Yu, Z.; Liu, T.; Guan, S.; Du, S.; Zhu, C. The driving mechanism and targeting value of mimicry between vascular endothelial cells and tumor cells in tumor progression. Biomed. Pharmacother. 2023, 165, 115029. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhang, D.; Wu, J.Y.; Xing, K.; Yeo, E.; Li, C.; Zhang, L.; Holland, E.; Yao, L.; Qin, L.; et al. Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci. Transl. Med. 2020, 12, eaay7522. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cortes, M.; Delgado-Bellido, D.; Oliver, F.J. Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much”. Front. Oncol. 2019, 9, 803. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Scully, S.; Bentley, B.; Yan, W.; Taylor, S.L.; Oh, D.; Moral, L.; Shao, R. Glioblastoma-derived tumor cells induce vasculogenic mimicry through Flk-1 protein activation. J. Biol. Chem. 2012, 287, 24821–24831. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayan, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef]
- Chepied, A.; Daoud-Omar, Z.; Meunier-Balandre, A.C.; Laird, D.W.; Mesnil, M.; Defamie, N. Involvement of the Gap Junction Protein, Connexin43, in the Formation and Function of Invadopodia in the Human U251 Glioblastoma Cell Line. Cells 2020, 9, 117. [Google Scholar] [CrossRef]
- McCutcheon, S.; Spray, D.C. Glioblastoma-Astrocyte Connexin 43 Gap Junctions Promote Tumor Invasion. Mol. Cancer Res. 2022, 20, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, C.; D’Alessio, A.; Tamagnone, L. Message in a Bottle: Endothelial Cell Regulation by Extracellular Vesicles. Cancers 2022, 14, 1969. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ma, X.; Wang, J.; Zhao, Y.; Wang, Y.; Bihl, J.C.; Chen, Y.; Jiang, C. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget 2017, 8, 36137–36148. [Google Scholar] [CrossRef] [PubMed]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Hyenne, V.; Ghoroghi, S.; Collot, M.; Bons, J.; Follain, G.; Harlepp, S.; Mary, B.; Bauer, J.; Mercier, L.; Busnelli, I.; et al. Studying the Fate of Tumor Extracellular Vesicles at High Spatiotemporal Resolution Using the Zebrafish Embryo. Dev. Cell 2019, 48, 554–572. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef]
- Kretschmer, M.; Rudiger, D.; Zahler, S. Mechanical Aspects of Angiogenesis. Cancers 2021, 13, 4987. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Al-Ostoot, F.H.; Salah, S.; Khamees, H.A.; Khanum, S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021, 28, 100422. [Google Scholar] [CrossRef] [PubMed]
- Geindreau, M.; Bruchard, M.; Vegran, F. Role of Cytokines and Chemokines in Angiogenesis in a Tumor Context. Cancers 2022, 14, 2446. [Google Scholar] [CrossRef] [PubMed]
- Peleli, M.; Moustakas, A.; Papapetropoulos, A. Endothelial-Tumor Cell Interaction in Brain and CNS Malignancies. Int. J. Mol. Sci. 2020, 21, 7371. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Rao, J.S. Antiangiogenic therapy in brain tumors. Expert. Rev. Neurother. 2008, 8, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Chen, F.; Zhang, Y.; Cai, W. New radiotracers for imaging of vascular targets in angiogenesis-related diseases. Adv. Drug Deliv. Rev. 2014, 76, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Dwivedi, S.K.D.; Bhattacharya, R.; Mukherjee, P.; Rao, G. VEGF signaling: Role in angiogenesis and beyond. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189079. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Nawaz, M.I. Molecular mechanism of VEGF and its role in pathological angiogenesis. J. Cell Biochem. 2022, 123, 1938–1965. [Google Scholar] [CrossRef]
- Patel, S.A.; Nilsson, M.B.; Le, X.; Cascone, T.; Jain, R.K.; Heymach, J.V. Molecular Mechanisms and Future Implications of VEGF/VEGFR in Cancer Therapy. Clin. Cancer Res. 2023, 29, 30–39. [Google Scholar] [CrossRef]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef]
- D’Alessio, A.; Proietti, G.; Lama, G.; Biamonte, F.; Lauriola, L.; Moscato, U.; Vescovi, A.; Mangiola, A.; Angelucci, C.; Sica, G. Analysis of angiogenesis related factors in glioblastoma, peritumoral tissue and their derived cancer stem cells. Oncotarget 2016, 7, 78541–78556. [Google Scholar] [CrossRef] [PubMed]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Batara, D.C.R.; Choi, M.C.; Shin, H.U.; Kim, H.; Kim, S.H. Friend or Foe: Paradoxical Roles of Autophagy in Gliomagenesis. Cells 2021, 10, 1411. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Liu, J.S.; Feng, M.; Li, J.M.; Lu, S.; Yang, M.; Cao, B.R.; Lang, J.Y.; Zhu, X.D. Comprehensively investigating the expression levels and the prognostic role of transforming growth factor beta-induced (TGFBI) in glioblastoma multiforme. Transl. Cancer Res. 2020, 9, 6487–6504. [Google Scholar] [CrossRef] [PubMed]
- Darland, D.C.; D’Amore, P.A. TGF beta is required for the formation of capillary-like structures in three-dimensional cocultures of 10T1/2 and endothelial cells. Angiogenesis 2001, 4, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.J.; Blobe, G.C. Dichotomous roles of TGF-beta in human cancer. Biochem. Soc. Trans. 2016, 44, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, I.; Schroeder, J.J.; Weiss, T.; Gramatzki, D.; Weller, M. A tumor-promoting role for soluble TbetaRIII in glioblastoma. Mol. Cell Biochem. 2021, 476, 2963–2973. [Google Scholar] [CrossRef]
- Murakami, M.; Simons, M. Fibroblast growth factor regulation of neovascularization. Curr. Opin. Hematol. 2008, 15, 215–220. [Google Scholar] [CrossRef]
- Allahmoradi, H.; Asghari, S.M.; Ahmadi, A.; Assareh, E.; Nazari, M. Anti-tumor and anti-metastatic activity of the FGF2 118-126 fragment dependent on the loop structure. Biochem. J. 2022, 479, 1285–1302. [Google Scholar] [CrossRef]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Loilome, W.; Joshi, A.D.; ap Rhys, C.M.; Piccirillo, S.; Vescovi, A.L.; Gallia, G.L.; Riggins, G.J. Glioblastoma cell growth is suppressed by disruption of Fibroblast Growth Factor pathway signaling. J. Neurooncol. 2009, 94, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Zahra, F.T.; Gupta, N.; Tullar, P.E.; Srivastava, S.K.; Mikelis, C.M. Low Dose of Penfluridol Inhibits VEGF-Induced Angiogenesis. Int. J. Mol. Sci. 2020, 21, 755. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Choi, Y.E.; Kim, E.S.; Han, Y.H.; Park, M.J.; Bae, I.H. miR-29b attenuates tumorigenicity and stemness maintenance in human glioblastoma multiforme by directly targeting BCL2L2. Oncotarget 2015, 6, 18429–18444. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Chong, K.; Lee, J.; Kim, C.; Kim, J.H.; Choi, K.; Choi, C. Differential dependency of human glioblastoma cells on vascular endothelial growth factor-A signaling via neuropilin-1. Int. J. Oncol. 2022, 61, 122. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Shi, Y.; Park, S.H.; Jeon, S.M.; Zhang, C.; Park, Y.Y.; Liu, R.; Li, J.; Cho, W.S.; Du, L.; et al. Mutual regulation between phosphofructokinase 1 platelet isoform and VEGF promotes glioblastoma tumor growth. Cell Death Dis. 2022, 13, 1002. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yan, Q.; Liao, B.; Zhao, L.; Xiong, S.; Wang, J.; Zou, D.; Pan, J.; Wu, L.; Deng, Y.; et al. The HIF1alpha/HIF2alpha-miR210-3p network regulates glioblastoma cell proliferation, dedifferentiation and chemoresistance through EGF under hypoxic conditions. Cell Death Dis. 2020, 11, 992. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Duda, D.G.; Emblem, K.E.; Taylor, J.W.; Gerstner, E.R.; Loeffler, J.S.; Batchelor, T.T.; Jain, R.K. Lessons from anti-vascular endothelial growth factor and anti-vascular endothelial growth factor receptor trials in patients with glioblastoma. J. Clin. Oncol. 2015, 33, 1197–1213. [Google Scholar] [CrossRef]
- Zafar, M.I.; Zheng, J.; Kong, W.; Ye, X.; Gou, L.; Regmi, A.; Chen, L.L. The role of vascular endothelial growth factor-B in metabolic homoeostasis: Current evidence. Biosci. Rep. 2017, 37, BSR20171089. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Chen, R.; Sun, G.; Liu, X.; Lin, X.; He, C.; Xing, L.; Liu, L.; Jensen, L.D.; Kumar, A.; et al. VEGF-B prevents excessive angiogenesis by inhibiting FGF2/FGFR1 pathway. Signal Transduct. Target. Ther. 2023, 8, 305. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xie, X.; Wang, T.; Xu, L.; Zhai, Z.; Wu, H.; Deng, L.; Lu, Q.; Chen, Z.; Yang, X.; et al. ARL13B promotes angiogenesis and glioma growth by activating VEGFA-VEGFR2 signaling. Neuro Oncol. 2023, 25, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Minh, L.B.; Dutra-Clarke, M.; Breunig, J.J.; Sarkisian, M.R. Glioma cell proliferation is enhanced in the presence of tumor-derived cilia vesicles. Cilia 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.N.; Hida, K.; Bielenberg, D.R.; Klagsbrun, M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006, 66, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Mehta, V.B.; Besner, G.E. HB-EGF promotes angiogenesis in endothelial cells via PI3-kinase and MAPK signaling pathways. Growth Factors 2007, 25, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Cribaro, G.P.; Saavedra-Lopez, E.; Romarate, L.; Mitxitorena, I.; Diaz, L.R.; Casanova, P.V.; Roig-Martinez, M.; Gallego, J.M.; Perez-Valles, A.; Barcia, C. Three-dimensional vascular microenvironment landscape in human glioblastoma. Acta Neuropathol. Commun. 2021, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Hasbum, A.; Quintanilla, J.; Jr, J.A.A.; Ding, M.H.; Levy, A.; Chew, S.A. Strategies to better treat glioblastoma: Antiangiogenic agents and endothelial cell targeting agents. Future Med. Chem. 2021, 13, 393–418. [Google Scholar] [CrossRef]
- Li, C.H.; Liao, P.L.; Yang, Y.T.; Huang, S.H.; Lin, C.H.; Cheng, Y.W.; Kang, J.J. Minocycline accelerates hypoxia-inducible factor-1 alpha degradation and inhibits hypoxia-induced neovasculogenesis through prolyl hydroxylase, von Hippel-Lindau-dependent pathway. Arch. Toxicol. 2014, 88, 659–671. [Google Scholar] [CrossRef]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef]
- Halatsch, M.E.; Dwucet, A.; Schmidt, C.J.; Muhlnickel, J.; Heiland, T.; Zeiler, K.; Siegelin, M.D.; Kast, R.E.; Karpel-Massler, G. In Vitro and Clinical Compassionate Use Experiences with the Drug-Repurposing Approach CUSP9v3 in Glioblastoma. Pharmaceuticals 2021, 14, 1241. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Kast, R.E.; Karpel-Massler, G.; Mayer, B.; Zolk, O.; Schmitz, B.; Scheuerle, A.; Maier, L.; Bullinger, L.; Mayer-Steinacker, R.; et al. A phase Ib/IIa trial of 9 repurposed drugs combined with temozolomide for the treatment of recurrent glioblastoma: CUSP9v3. Neurooncol. Adv. 2021, 3, vdab075. [Google Scholar] [CrossRef] [PubMed]
- Oprita, A.; Dobrescu, M.A.; Manea, E.V.; Popescu, S.O.; Sevastre, A.S.; Pirvu, A.S.; Buzatu, I.M.; Tache, D.E. In vitro evaluation of Axitinib and Sorafenib treatment in glioblastoma cell viability and morphology. Rom. J. Morphol. Embryol. 2023, 64, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Carra, E.; Barbieri, F.; Marubbi, D.; Pattarozzi, A.; Favoni, R.E.; Florio, T.; Daga, A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle 2013, 12, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef] [PubMed]
- Riedel, M.; Struve, N.; Muller-Goebel, J.; Kocher, S.; Petersen, C.; Dikomey, E.; Rothkamm, K.; Kriegs, M. Sorafenib inhibits cell growth but fails to enhance radio- and chemosensitivity of glioblastoma cell lines. Oncotarget 2016, 7, 61988–61995. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A.; et al. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 2013, 15, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Nghiemphu, P.L.; Ebiana, V.A.; Wen, P.; Gilbert, M.; Abrey, L.E.; Lieberman, F.; DeAngelis, L.M.; Robins, H.I.; Yung, W.K.A.; Chang, S.; et al. Phase I study of sorafenib and tipifarnib for recurrent glioblastoma: NABTC 05-02. J. Neurooncol. 2018, 136, 79–86. [Google Scholar] [CrossRef]
- Wei, J.; Xia, Y.; Meng, F.; Ni, D.; Qiu, X.; Zhong, Z. Small, Smart, and LDLR-Specific Micelles Augment Sorafenib Therapy of Glioblastoma. Biomacromolecules 2021, 22, 4814–4822. [Google Scholar] [CrossRef]
- Zhang, A.B.; Mozaffari, K.; Aguirre, B.; Li, V.; Kubba, R.; Desai, N.C.; Wei, D.; Yang, I.; Wadehra, M. Exploring the Past, Present, and Future of Anti-Angiogenic Therapy in Glioblastoma. Cancers 2023, 15, 830. [Google Scholar] [CrossRef]
- Yalamarty, S.S.K.; Filipczak, N.; Li, X.; Subhan, M.A.; Parveen, F.; Ataide, J.A.; Rajmalani, B.A.; Torchilin, V.P. Mechanisms of Resistance and Current Treatment Options for Glioblastoma Multiforme (GBM). Cancers 2023, 15, 2116. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central cancer treatment group trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef] [PubMed]
- Sumorek-Wiadro, J.; Zajac, A.; Langner, E.; Skalicka-Wozniak, K.; Maciejczyk, A.; Rzeski, W.; Jakubowicz-Gil, J. Antiglioma Potential of Coumarins Combined with Sorafenib. Molecules 2020, 25, 5192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, J.; Xu, T. Inhibition of eukaryotic initiation factor 4E by tomivosertib suppresses angiogenesis, growth, and survival of glioblastoma and enhances chemotherapy’s efficacy. Fundam. Clin. Pharmacol. 2023, 37, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; Nguyen, A.; Chandra, A.; Sidorov, M.K.; Yagnik, G.; Rick, J.; Han, S.W.; Chen, W.; Flanigan, P.M.; Schneidman-Duhovny, D.; et al. Cross-activating c-Met/beta1 integrin complex drives metastasis and invasive resistance in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E8685–E8694. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef] [PubMed]
- DeLay, M.; Jahangiri, A.; Carbonell, W.S.; Hu, Y.L.; Tsao, S.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Aghi, M.K. Microarray analysis verifies two distinct phenotypes of glioblastomas resistant to antiangiogenic therapy. Clin. Cancer Res. 2012, 18, 2930–2942. [Google Scholar] [CrossRef]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef]
- Zhao, C.; Gomez, G.A.; Zhao, Y.; Yang, Y.; Cao, D.; Lu, J.; Yang, H.; Lin, S. ETV2 mediates endothelial transdifferentiation of glioblastoma. Signal Transduct. Target. Ther. 2018, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liu, T.; Ma, P.; Mitteer, R.A., Jr.; Zhang, Z.; Kim, H.J.; Yeo, E.; Zhang, D.; Cai, P.; Li, C.; et al. c-Met-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J. Clin. Investig. 2016, 126, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shan, X.; Meng, X.; Gu, T.; Guo, L.; An, X.; Jiang, Q.; Ge, H.; Ning, X. Novel Integrin alphavbeta3-Specific Ligand for the Sensitive Diagnosis of Glioblastoma. Mol. Pharm. 2019, 16, 3977–3984. [Google Scholar] [CrossRef] [PubMed]
- Echavidre, W.; Picco, V.; Faraggi, M.; Montemagno, C. Integrin-alphavbeta3 as a Therapeutic Target in Glioblastoma: Back to the Future? Pharmaceutics 2022, 14, 1053. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Rosen, V.; Wozney, J.M.; Wang, E.A.; Cordes, P.; Celeste, A.; McQuaid, D.; Kurtzberg, L. Purification and molecular cloning of a novel group of BMPs and localization of BMP mRNA in developing bone. Connect. Tissue Res. 1989, 20, 313–319. [Google Scholar] [CrossRef]
- Dyer, L.A.; Pi, X.; Patterson, C. The role of BMPs in endothelial cell function and dysfunction. Trends Endocrinol. Metab. 2014, 25, 472–480. [Google Scholar] [CrossRef]
- Zhang, L.; Ye, Y.; Long, X.; Xiao, P.; Ren, X.; Yu, J. BMP signaling and its paradoxical effects in tumorigenesis and dissemination. Oncotarget 2016, 7, 78206–78218. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhao, J.; Brochmann, E.J.; Wang, J.C.; Murray, S.S. Bone morphogenetic protein-2 and tumor growth: Diverse effects and possibilities for therapy. Cytokine Growth Factor. Rev. 2017, 34, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Golan-Cancela, I.; Caja, L. The TGF-beta Family in Glioblastoma. Int. J. Mol. Sci. 2024, 25, 67. [Google Scholar] [CrossRef]
- Xu, C.; Hu, X.; Fan, Y.; Zhang, L.; Gao, Z.; Cai, C. Wif1 Mediates Coordination of Bone Morphogenetic Protein and Wnt Signaling in Neural and Glioma Stem Cells. Cell Transpl. 2022, 31, 9636897221134540. [Google Scholar] [CrossRef]
- Ye, L.; Jiang, W.G. Bone morphogenetic proteins in tumour associated angiogenesis and implication in cancer therapies. Cancer Lett. 2016, 380, 586–597. [Google Scholar] [CrossRef]
- Wiley, D.M.; Jin, S.W. Bone Morphogenetic Protein functions as a context-dependent angiogenic cue in vertebrates. Semin. Cell Dev. Biol. 2011, 22, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Dalmo, E.; Johansson, P.; Niklasson, M.; Gustavsson, I.; Nelander, S.; Westermark, B. Growth-Inhibitory Activity of Bone Morphogenetic Protein 4 in Human Glioblastoma Cell Lines Is Heterogeneous and Dependent on Reduced SOX2 Expression. Mol. Cancer Res. 2020, 18, 981–991. [Google Scholar] [CrossRef]
- Herrera, B.; Garcia-Alvaro, M.; Cruz, S.; Walsh, P.; Fernandez, M.; Roncero, C.; Fabregat, I.; Sanchez, A.; Inman, G.J. BMP9 is a proliferative and survival factor for human hepatocellular carcinoma cells. PLoS ONE 2013, 8, e69535. [Google Scholar] [CrossRef]
- Kaye, J.; Mondal, A.; Foty, R.; Jia, D.; Langenfeld, J. Bone morphogenetic protein receptor inhibitors suppress the growth of glioblastoma cells. Mol. Cell Biochem. 2022, 477, 1583–1595. [Google Scholar] [CrossRef]
- Yasinjan, F.; Xing, Y.; Geng, H.; Guo, R.; Yang, L.; Liu, Z.; Wang, H. Immunotherapy: A promising approach for glioma treatment. Front. Immunol. 2023, 14, 1255611. [Google Scholar] [CrossRef]
- Brown, C.E.; Hibbard, J.C.; Alizadeh, D.; Blanchard, M.S.; Natri, H.M.; Wang, D.; Ostberg, J.R.; Aguilar, B.; Wagner, J.R.; Paul, J.A.; et al. Locoregional delivery of IL-13Ralpha2-targeting CAR-T cells in recurrent high-grade glioma: A phase 1 trial. Nat. Med. 2024, 30, 1001–1012. [Google Scholar] [CrossRef]
- Bagley, S.J.; Logun, M.; Fraietta, J.A.; Wang, X.; Desai, A.S.; Bagley, L.J.; Nabavizadeh, A.; Jarocha, D.; Martins, R.; Maloney, E.; et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Ralpha2 in recurrent glioblastoma: Phase 1 trial interim results. Nat. Med. 2024, 30, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- You, W.C.; Lee, H.D.; Pan, H.C.; Chen, H.C. Re-irradiation combined with bevacizumab for recurrent glioblastoma beyond bevacizumab failure: Survival outcomes and prognostic factors. Sci. Rep. 2023, 13, 9442. [Google Scholar] [CrossRef] [PubMed]
- Theruvath, J.; Menard, M.; Smith, B.A.H.; Linde, M.H.; Coles, G.L.; Dalton, G.N.; Wu, W.; Kiru, L.; Delaidelli, A.; Sotillo, E.; et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med. 2022, 28, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Asija, S.; Chatterjee, A.; Yadav, S.; Chekuri, G.; Karulkar, A.; Jaiswal, A.K.; Goda, J.S.; Purwar, R. Combinatorial approaches to effective therapy in glioblastoma (GBM): Current status and what the future holds. Int. Rev. Immunol. 2022, 41, 582–605. [Google Scholar] [CrossRef]
- Zhou, S.; Huang, Y.; Chen, Y.; Liu, Y.; Xie, L.; You, Y.; Tong, S.; Xu, J.; Jiang, G.; Song, Q.; et al. Reprogramming systemic and local immune function to empower immunotherapy against glioblastoma. Nat. Commun. 2023, 14, 435. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Wang, A.; Yuan, Y.; Zhu, B.; Long, H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp. Hematol. Oncol. 2022, 11, 24. [Google Scholar] [CrossRef]
- Cui, X.; Ma, C.; Vasudevaraja, V.; Serrano, J.; Tong, J.; Peng, Y.; Delorenzo, M.; Shen, G.; Frenster, J.; Morales, R.T.; et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. eLife 2020, 9, e52253. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Pandit, R.; Chen, L.; Gotz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2020, 165–166, 1–14. [Google Scholar] [CrossRef]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Roda, F.; Caraffi, R.; Picciolini, S.; Tosi, G.; Vandelli, M.A.; Ruozi, B.; Bedoni, M.; Ottonelli, I.; Duskey, J.T. Recent Advances on Surface-Modified GBM Targeted Nanoparticles: Targeting Strategies and Surface Characterization. Int. J. Mol. Sci. 2023, 24, 2496. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, J.J.; Cunnane, E.M.; Ross, A.M.; Duskey, J.T.; Tosi, G.; Grabrucker, A.M. Drug delivery across the blood-brain barrier: Recent advances in the use of nanocarriers. Nanomedicine 2020, 15, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Blanco, J.; Sanz-Arriazu, L.; Lorenzoni, R.; Blanco-Prieto, M.J. Glioblastoma chemotherapeutic agents used in the clinical setting and in clinical trials: Nanomedicine approaches to improve their efficacy. Int. J. Pharm. 2020, 581, 119283. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ji, X.; He, D.; Zhang, R.; Liu, Q.; Xin, T. Nanoscale Drug Delivery Systems in Glioblastoma. Nanoscale Res. Lett. 2022, 17, 27. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Yang, J.J.; ZhuGe, D.L.; Lin, M.T.; Zhu, Q.Y.; Jin, B.H.; Tong, M.Q.; Shen, B.X.; Xiao, J.; Zhao, Y.Z. Glioma-Targeted Delivery of a Theranostic Liposome Integrated with Quantum Dots, Superparamagnetic Iron Oxide, and Cilengitide for Dual-Imaging Guiding Cancer Surgery. Adv. Healthc. Mater. 2018, 7, e1701130. [Google Scholar] [CrossRef] [PubMed]
- Shabani, L.; Abbasi, M.; Amini, M.; Amani, A.M.; Vaez, A. The brilliance of nanoscience over cancer therapy: Novel promising nanotechnology-based methods for eradicating glioblastoma. J. Neurol. Sci. 2022, 440, 120316. [Google Scholar] [CrossRef]
- Fang, C.; Wang, K.; Stephen, Z.R.; Mu, Q.; Kievit, F.M.; Chiu, D.T.; Press, O.W.; Zhang, M. Temozolomide nanoparticles for targeted glioblastoma therapy. ACS Appl. Mater. Interfaces 2015, 7, 6674–6682. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Factor | Receptor | Mechanism |
|---|---|---|
| VEGF-A | VEGFR-1 and VEGFR-2 | Stimulates EC migration, proliferation, survival, nitric oxide production, and angiogenic response |
| ARL13B | VEGFR-2 | Binds the intracellular domain of VEGFR2 on ECs to promote VEGFA-VEGFR2 signaling |
| TGF-β | TβRII | Acts on both TCs and ECs to induce neovascularization and causes ECM remodeling using MMPs |
| FGF2 | FGFR | Stimulates EC VEGF production and proliferation |
| EGF | EGFR | Promotes EC migration and capillary tube formation |
| ETV2 | Conserved ETS motifs in the ZRS | Transdifferentiates GBM stem cells to an endothelial lineage |
| Pleiotrophin | ALK1 | Causes VEGF deposition at blood vessels, induces EC proliferation |
| PDFG-B | PDGF receptor β | Stimulates VEGF production in ECs |
| Ang-1 | EC-specific Tie receptors | Acts on ECs to stimulate angiogenesis and EC survival |
| Agent | Target | Combination Therapies | NCT Trial Number | Phase | Status | Outcome (in Experimental Group Relative to Control) | Center or Company Name |
|---|---|---|---|---|---|---|---|
| Bevacizumab | VEGF | Temozolomide | NCT00590681 | Phase II | Completed Sept. 2014 | Prolonged PFS, no improvement in OS | University of Chicago, Genentech, Inc., Oceanside, CA, USA |
| Sorafenib | NCT00621686 | Phase II | Completed Feb. 2014 | No improvement in outcome | Alliance for Clinical Trials in Oncology, NCI, Bethesda, MD, USA | ||
| Erlotinib | NCT00671970 | Phase II | Completed Apr. 2010 | Similar PFS and radiographic response | Duke University, Genentech, Inc., Oceanside, CA, USA | ||
| Rindopepimut (CDX-110) | NCT01498328 | Phase II | Completed May 2016 | Improved PFS, ORR, and ability to discontinue steroids for ≥6 months | Celldex Therapeutics, Hampton, NJ, USA | ||
| Poly-ICLC | NCT02754362 | Phase II | Completed June 2019 | Pending | NYU Langone Health, MOUNT SINAI HOSPITAL, New York, NY, USA | ||
| Optune | NCT01925573 | Interventional | Completed Aug. 2019 | Pending | University of Maryland, Baltimore, NovoCure Ltd., Portsmouth, NH, USA | ||
| Trebananib | NCT01609790 | Interventional | Completed May 2022 | Shortened PFS | NCI, NRG Oncology, Columbus, OH, USA | ||
| Ascorbic Acid | NCT02833701 | Phase I | Completed Mar. 2019 | Pending | University of Nebraska, NCI, Bethesda, MD, USA | ||
| Retifanlimab + hypofractionated radiotherapy | NCT06160206 | Phase II | Ongoing | Pending | Academic and Community Cancer Research United, NCI, Bethesda, MD, USA | ||
| VB-111 | Ad-PPE-Fas-c | NA | NCT04406272 | Phase II | Ongoing | Pending | Dana-Farber Cancer Institute, VBL Therapeutics, New York, NY, USA |
| Apatinib | VEGFR-2 | Temozolomide | NCT04814329 | Observational | Ongoing | Pending | Beijing Sanbo Brain Hospital, Beijing, China |
| Anlotinib | EGFR | NA | NCT04004975 | Phase II | Completed July 2021 | Pending | Shandong Cancer Hospital and Institute, Jinan, Shandong, China |
| Temozolomide | NCT04547855 | Phase II | Ongoing | Pending | The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, China | ||
| Glasdegib | Sonic hedgehog receptor smoothened (SMO) | Temozolomide | NCT03466450 | Phase II | Completed Nov. 2023 | Pending | Hospital del Mar, Barcelona, Catalonia, Spain |
| Temozolomide | DNA (alkylating agent) | Radiation therapy + bevacizumab | NCT00884741 | Phase III | Completed Mar. 2013 | No improvement in OS | Providence Hospital, Portland, OR, USA |
| Napabucasin (BBI608) | STAT3 | Temozolomide | NCT02315534 | Phase II | Completed June 2019 | Pending | Laura and Isaac Perlmutter Cancer Center, New York, NY, USA |
| Thrombospondin-1 analog (ABT 510) | CD36 receptor found on ECs | Radiation | NCT00584883 | Phase I | Completed July 2008 | Pending | University of Alabama at Birmingham, Birmingham, AL, USA |
| Cediranib | (VEGFR)-1, VEGFR-2, VEGFR-3 | Olapirib | NCT02974621 | Phase II | Completed Dec. 2022 | Pending | UC San Diego Moores Cancer Center, San Diego, CA, USA |
| TTAC-0001 (Taniburimab) | VEGFR-2 | NA | NCT03033524 | Phase II | Completed June 2017 | Pending | PharmAbcine, Yuseong-gu, Daejeon, Republic of Korea |
| Bevacizumab | NCT03856099 | Phase II | Completed July 2022 | Pending | Stanford Advanced Medical Center, Palo Alto, CA, USA | ||
| Pembrolizumab | NCT03722342 | Phase I | Completed Sept. 2022 | Pending | Austin Hospital, Heidelberg, VIC, Australia | ||
| Recombinant Human Endostatin | Broad-spectrum angiogenesis inhibitor | Temozolomide + Irinotecan | NCT04267978 | Phase II | Ongoing | Pending | Beijing Sanbo Brain Hospital, Beijing, China |
| EGFR Bi-armed Activated T-cells (BATs) | EGFR | Temozolomide + radiation | NCT03344250 | Phase I | Ongoing | Prolonged OS and PFS | University of Virginia, Charlottesville, VA, USA |
| Nanoparticles | variable | Radiotherapy + Temozolomide | NCT04881032 | Phase I/II | Ongoing | Pending | CHU de Brest, Brest, Brittany, France |
| hrBMP4 | VEGF | NA | NCT02869243 | Phase I | Completed June 2021 | Reduction in tumor growth; 2/15 with complete regression and extended survival | Tel Aviv Sourasky Medical Center, Tel Aviv-Yafo, Israel |
| Erolotinib | EGFR | Sorafenib | NCT00445588 | Phase II | Completed Apr. 2026 | No significant survival improvement | University of Alabama at Birmingham, Birmingham, AL, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hovis, G.; Chandra, N.; Kejriwal, N.; Hsieh, K.J.-Y.; Chu, A.; Yang, I.; Wadehra, M. Understanding the Role of Endothelial Cells in Glioblastoma: Mechanisms and Novel Treatments. Int. J. Mol. Sci. 2024, 25, 6118. https://doi.org/10.3390/ijms25116118
Hovis G, Chandra N, Kejriwal N, Hsieh KJ-Y, Chu A, Yang I, Wadehra M. Understanding the Role of Endothelial Cells in Glioblastoma: Mechanisms and Novel Treatments. International Journal of Molecular Sciences. 2024; 25(11):6118. https://doi.org/10.3390/ijms25116118
Chicago/Turabian StyleHovis, Gabrielle, Neha Chandra, Nidhi Kejriwal, Kaleb Jia-Yi Hsieh, Alison Chu, Isaac Yang, and Madhuri Wadehra. 2024. "Understanding the Role of Endothelial Cells in Glioblastoma: Mechanisms and Novel Treatments" International Journal of Molecular Sciences 25, no. 11: 6118. https://doi.org/10.3390/ijms25116118
APA StyleHovis, G., Chandra, N., Kejriwal, N., Hsieh, K. J.-Y., Chu, A., Yang, I., & Wadehra, M. (2024). Understanding the Role of Endothelial Cells in Glioblastoma: Mechanisms and Novel Treatments. International Journal of Molecular Sciences, 25(11), 6118. https://doi.org/10.3390/ijms25116118