
mTOR: Its Critical Role in Metabolic Diseases, Cancer, and the Aging Process


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
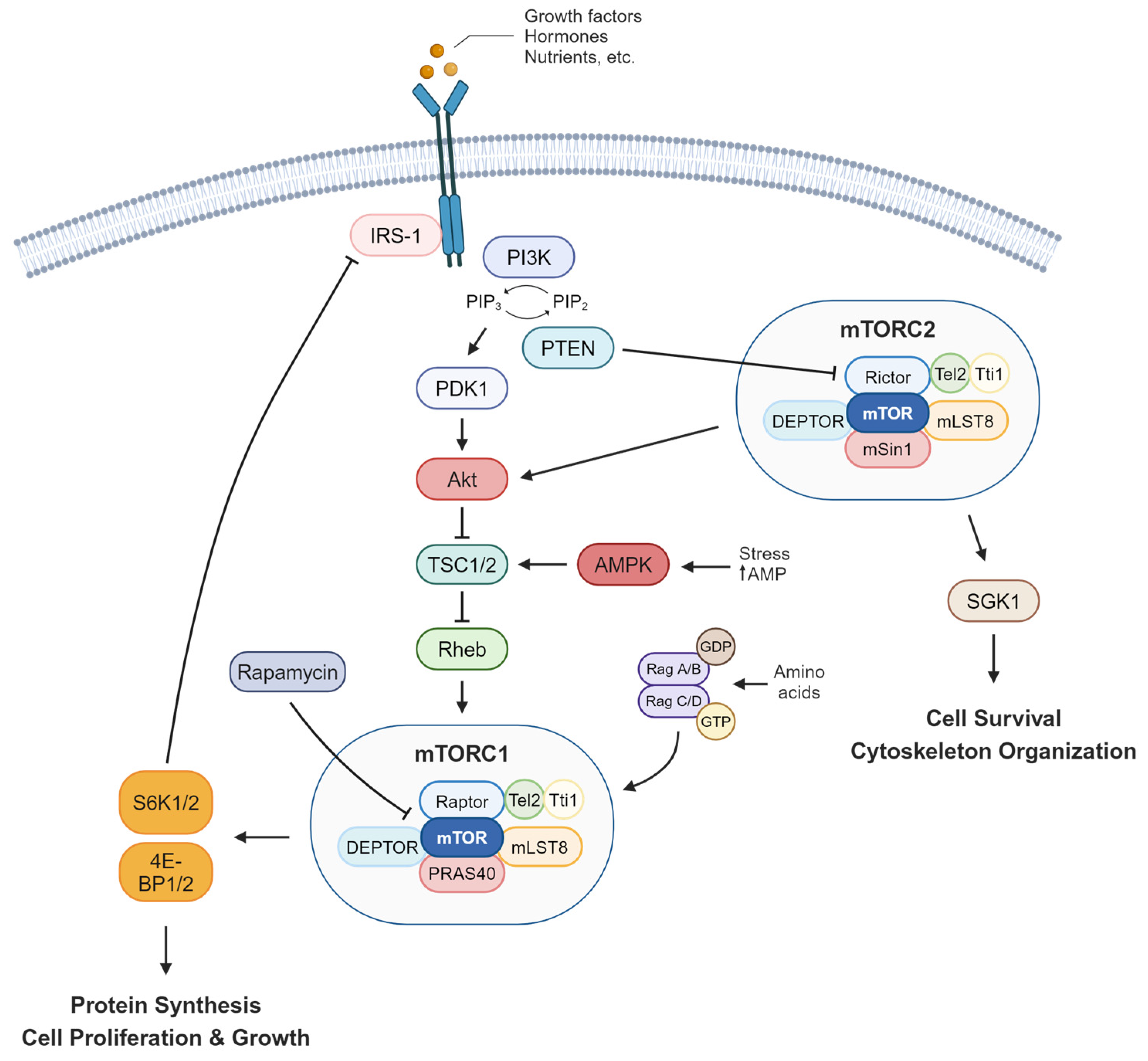
2. mTOR Signaling and Human Diseases
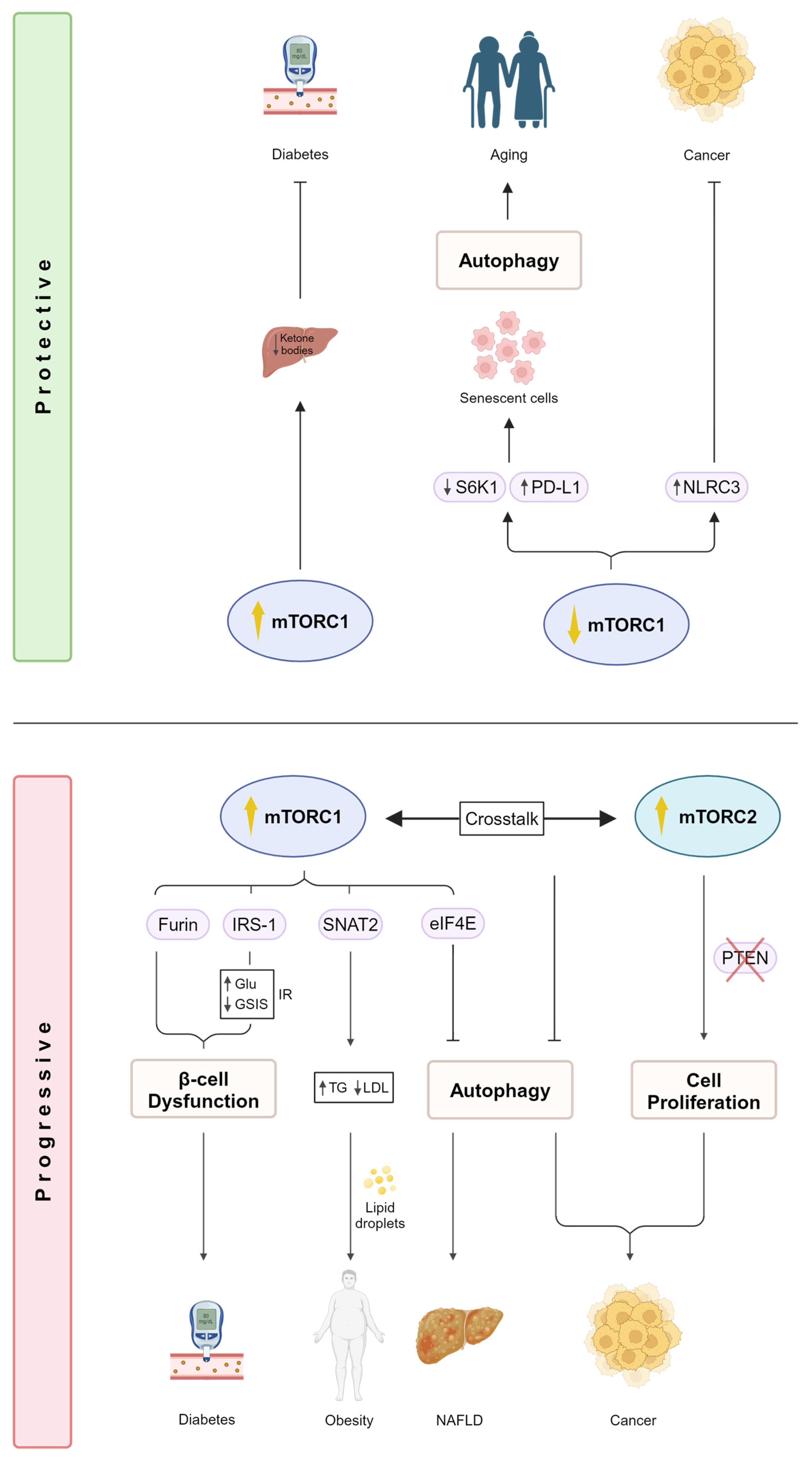
2.1. mTOR Regulation in Metabolic Diseases
2.2. mTOR Signaling Promotes Cancer
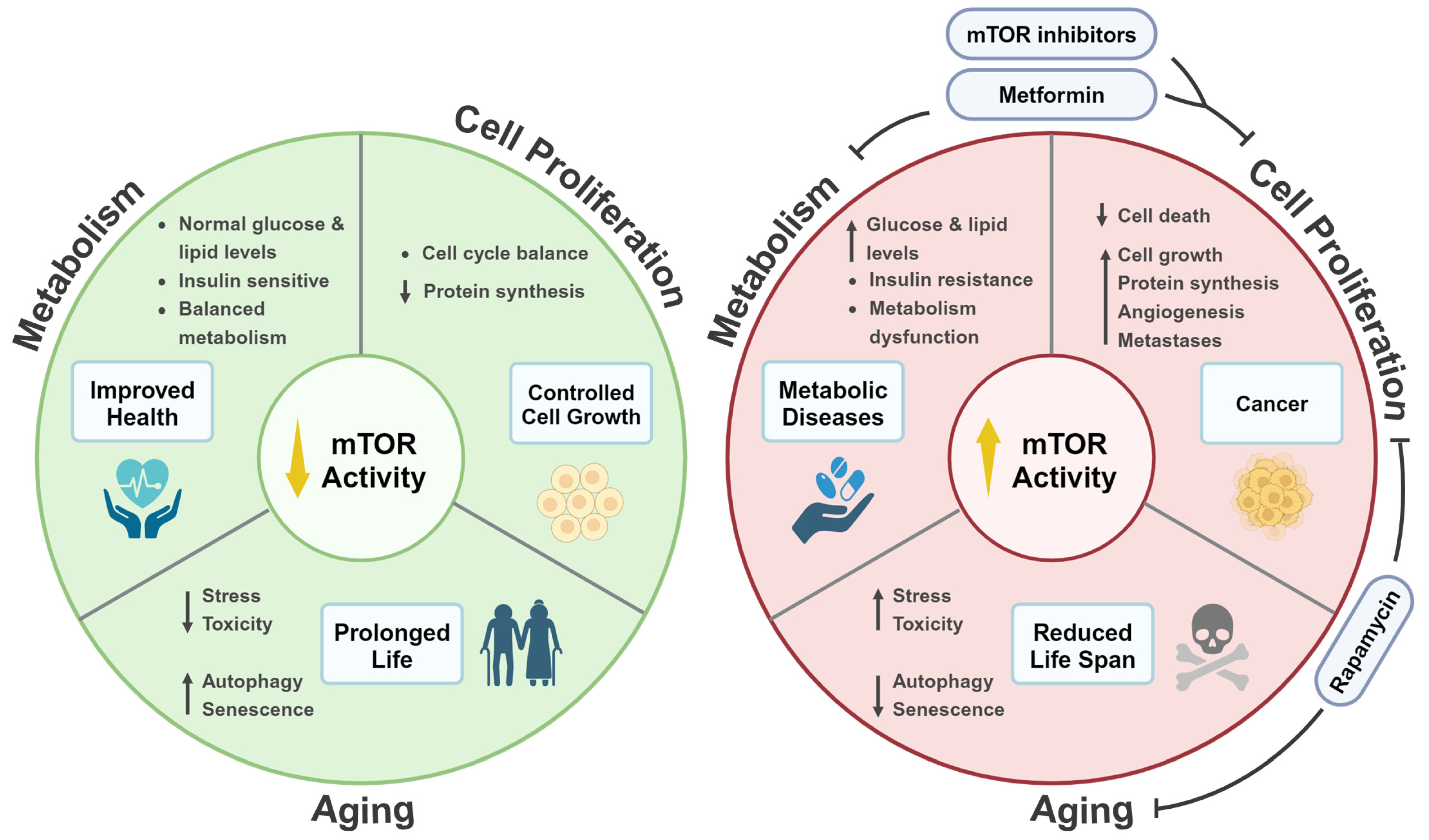
3. mTOR Signaling and Life Extension
3.1. The Role of mTOR Signaling in Aging Is Evolutionarily Conserved
3.2. Autophagy Promotes Life Span Extension in a mTOR-Dependent Manner
3.3. Mitochondrial Proteins Regulate Life Span via mTOR Signaling
4. Inhibitors of mTOR Signaling Pathways
4.1. First-Generation mTOR Inhibitors
4.2. Second-Generation mTOR Inhibitors
4.3. Third-Generation mTOR Inhibitors
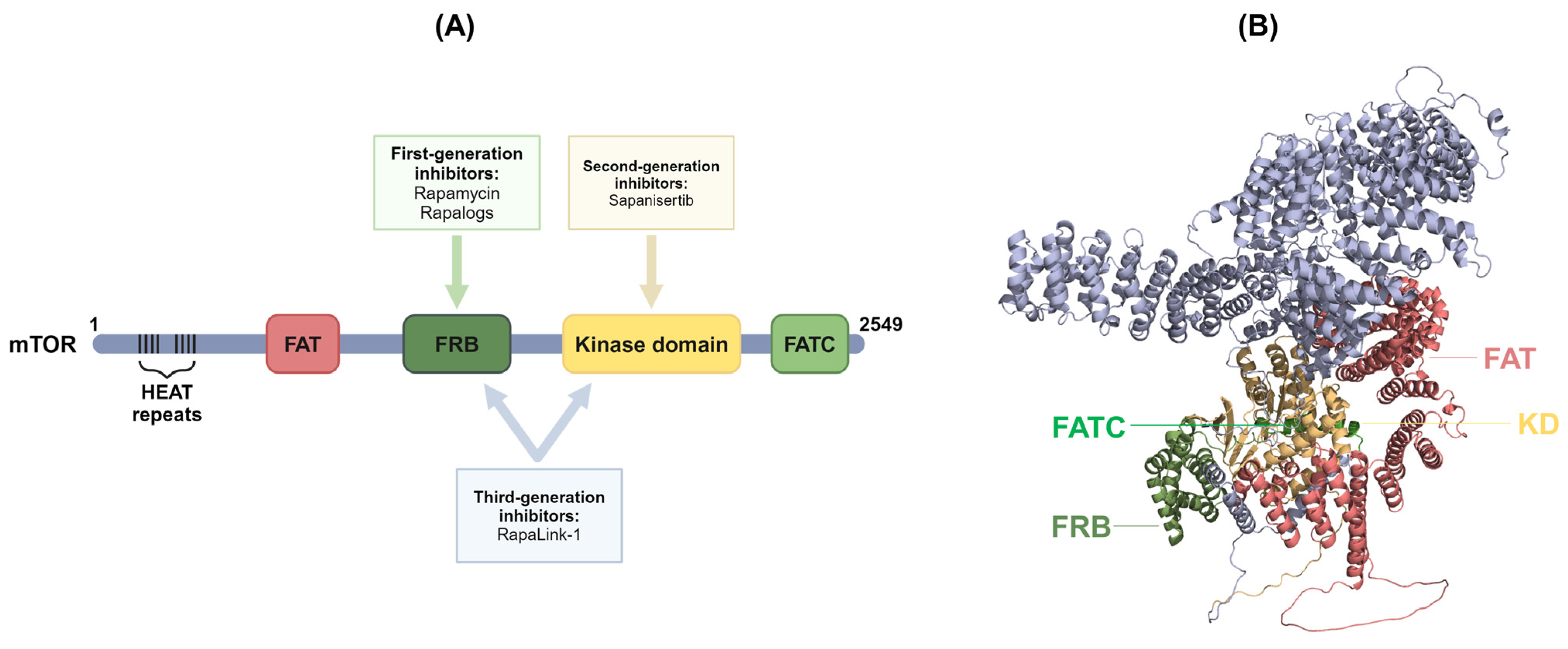
4.4. Other Inhibitors of the mTOR Signaling Pathway
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leung, A.; Rangamani, P. Computational modeling of AMPK and mTOR crosstalk in glutamatergic synapse calcium signaling. NPJ Syst. Biol. Appl. 2023, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Blenis, J.; Proud, C.G. Analysis of mTOR signaling by the small G-proteins, Rheb and RhebL1. FEBS Lett. 2005, 579, 4763–4768. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, Z.; Peng, J.; Loor, J.J. Methionine and valine activate the mammalian target of rapamycin complex 1 pathway through heterodimeric amino acid taste receptor (TAS1R1/TAS1R3) and intracellular Ca(2+) in bovine mammary epithelial cells. J. Dairy Sci. 2018, 101, 11354–11363. [Google Scholar] [CrossRef]
- Pende, M.; Kozma, S.C.; Jaquet, M.; Oorschot, V.; Burcelin, R.; Le Marchand-Brustel, Y.; Klumperman, J.; Thorens, B.; Thomas, G. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 2000, 408, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Xia, Z.-F.; Ben, D.-F.; Duo, W. mTOR partly mediates insulin resistance by phosphorylation of insulin receptor substrate-1 on serine(307) residues after burn. Burns 2011, 37, 86–93. [Google Scholar]
- Yan, X.; Huang, S.; Li, H.; Feng, Z.; Kong, J.; Liu, J. The causal effect of mTORC1-dependent circulating protein levels on nonalcoholic fatty liver disease: A Mendelian randomization study. Dig. Liver Dis. 2024, 56, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Tosta, B.R.; de Almeida, I.M.; da Cruz Pena, L.; Dos Santos Silva, H.; Reis-Goes, F.S.; Silva, N.N.; Cruz, J.V.A.; Dos Anjos Silva, M.; de Araujo, J.F.; Rodrigues, J.L.; et al. MTOR gene variants are associated with severe COVID-19 outcomes: A multicenter study. Int. Immunopharmacol. 2023, 125 Pt B, 111155. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.M.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR interacts with Raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.; Natsume, T.; Mizushima, N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.J.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.; Ali, S.; Sengupta, S.; Sheen, J.; Hsu, P.; Bagley, A.; Markhard, A.; Sabatini, D. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Lara, P.N. Belzutifan versus Everolimus in Advanced Kidney Cancer: A Commentary on LITESPARK-005 Trial from ESMO 2023. Kidney Cancer 2024, 8, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Salaun, H.; Djerroudi, L.; Haik, L.; Schnitzler, A.; Bataillon, G.; Deniziaut, G.; Bieche, I.; Vincent-Salomon, A.; Debled, M.; Cottu, P. The prognosis of patients treated with everolimus for advanced ER-positive, HER2-negative breast cancer is driven by molecular features. J. Pathol. Clin. Res. 2024, 10, e12372. [Google Scholar] [CrossRef] [PubMed]
- Nghiemphu, P.L.; Vitte, J.; Dombi, E.; Nguyen, T.; Wagle, N.; Ishiyama, A.; Sepahdari, A.R.; Cachia, D.; Widemann, B.C.; Brackmann, D.E.; et al. Imaging as an early biomarker to predict sensitivity to everolimus for progressive NF2-related vestibular schwannoma. J. Neuro-Oncol. 2024, 167, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Man, S.M.; Malireddi, R.K.S.; Kesavardhana, S.; Zhu, Q.; Burton, A.R.; Sharma, B.R.; Qi, X.; Pelletier, S.; Vogel, P.; et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature 2016, 540, 583–587. [Google Scholar] [CrossRef]
- Oh, W.; Wu, C.; Kim, S.; Facchinetti, V.; Julien, L.; Finlan, M.; Roux, P.; Su, B.; Jacinto, E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010, 29, 3939–3951. [Google Scholar] [CrossRef]
- Deng, Y.F.; Wu, S.T.; Peng, H.Y.; Tian, L.; Li, Y.N.; Yang, Y.; Meng, M.; Huang, L.L.; Xiong, P.W.; Li, S.Y.; et al. mTORC2 acts as a gatekeeper for mTORC1 deficiency-mediated impairments in ILC3 development. Acta Pharmacol. Sin. 2023, 44, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor Phosphorylation Sites Reveals Direct Regulation of mTOR Complex 2 by S6K1. Mol. Cell. Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 2009, 32 (Suppl. 1), S62–S67. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martin, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Tzatsos, A.; Kandror, K.V. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol. Cell Biol. 2006, 26, 63–76. [Google Scholar] [CrossRef]
- Marafie, S.K.; Al-Shawaf, E.M.; Abubaker, J.; Arefanian, H. Palmitic acid-induced lipotoxicity promotes a novel interplay between Akt-mTOR, IRS-1, and FFAR1 signaling in pancreatic β-cells. Biol. Res. 2019, 52, 44. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, B.; Coppola, I.; Vints, K.; Dislich, B.; Jouvet, N.; Van Lommel, L.; Segers, C.; Gounko, N.V.; Thorrez, L.; Schuit, F.; et al. Loss of Furin in β-Cells Induces an mTORC1-ATF4 Anabolic Pathway That Leads to β-Cell Dysfunction. Diabetes 2021, 70, 492–503. [Google Scholar] [CrossRef]
- Seidah, N.G. The proprotein convertases, 20 years later. Methods Mol. Biol. 2011, 768, 23–57. [Google Scholar]
- Smeekens, S.P.; Montag, A.G.; Thomas, G.; Albiges-Rizo, C.; Carroll, R.; Benig, M.; Phillips, L.A.; Martin, S.; Ohagi, S.; Gardner, P.; et al. Proinsulin processing by the subtilisin-related proprotein convertases furin, PC2, and PC3. Proc. Natl. Acad. Sci. USA 1992, 89, 8822–8826. [Google Scholar] [CrossRef] [PubMed]
- Marafie, S.K.; Al-Mulla, F. An Overview of the Role of Furin in Type 2 Diabetes. Cells 2023, 12, 2407. [Google Scholar] [CrossRef] [PubMed]
- Ursino, G.; Ramadori, G.; Hofler, A.; Odouard, S.; Teixeira, P.D.S.; Visentin, F.; Veyrat-Durebex, C.; Lucibello, G.; Firnkes, R.; Ricci, S.; et al. Hepatic non-parenchymal S100A9-TLR4-mTORC1 axis normalizes diabetic ketogenesis. Nat. Commun. 2022, 13, 4107. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E. Ketone bodies as a fuel for the brain during starvation. Biochem. Mol. Biol. Educ. 2005, 33, 246–251. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 4, e127737. [Google Scholar] [CrossRef] [PubMed]
- Gayatri, M.B.; Gajula, N.N.; Chava, S.; Reddy, A.B.M. High glutamine suppresses osteogenesis through mTORC1-mediated inhibition of the mTORC2/AKT-473/RUNX2 axis. Cell Death Discov. 2022, 8, 277. [Google Scholar] [CrossRef] [PubMed]
- Uno, K.; Yamada, T.; Ishigaki, Y.; Imai, J.; Hasegawa, Y.; Sawada, S.; Kaneko, K.; Ono, H.; Asano, T.; Oka, Y.; et al. A hepatic amino acid/mTOR/S6K-dependent signalling pathway modulates systemic lipid metabolism via neuronal signals. Nat. Commun. 2015, 6, 7940. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Populo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhong, W.; Cao, R. Phosphorylation of the mRNA cap-binding protein eIF4E and cancer. Cell Signal 2020, 73, 109689. [Google Scholar] [CrossRef] [PubMed]
- Gentilella, A.; Kozma, S.C.; Thomas, G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim. Biophys. Acta 2015, 1849, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Mafi, S.; Ahmadi, E.; Meehan, E.; Chiari, C.; Mansoori, B.; Sadeghi, H.; Milani, S.; Jafarinia, M.; Taeb, S.; Mafakheri Bashmagh, B.; et al. The mTOR Signaling Pathway Interacts with the ER Stress Response and the Unfolded Protein Response in Cancer. Cancer Res. 2023, 83, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Seo, M.; Otto, N.M.; Kim, D.H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 2011, 7, 1212–1221. [Google Scholar] [CrossRef]
- Lopez-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef]
- Zhang, H.P.; Jiang, R.Y.; Zhu, J.Y.; Sun, K.N.; Huang, Y.; Zhou, H.H.; Zheng, Y.B.; Wang, X.J. PI3K/AKT/mTOR signaling pathway: An important driver and therapeutic target in triple-negative breast cancer. Breast Cancer 2024. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, C.; Xu, X.; Li, A.; Cai, Q.; Long, X. Androgen receptor, EGFR, and BRCA1 as biomarkers in triple-negative breast cancer: A meta-analysis. BioMed Res. Int. 2015, 2015, 357485. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.-H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR Complex 2 Is Required for the Development of Prostate Cancer Induced by Pten Loss in Mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Maiti, S.; Mandal, C. PTEN negatively regulates mTORC2 formation and signaling in grade IV glioma via Rictor hyperphosphorylation at Thr1135 and direct the mode of action of an mTORC1/2 inhibitor. Oncogenesis 2016, 5, e227. [Google Scholar] [CrossRef]
- Ting, J.P.; Willingham, S.B.; Bergstralh, D.T. NLRs at the intersection of cell death and immunity. Nat. Rev. Immunol. 2008, 8, 372–379. [Google Scholar] [CrossRef]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef]
- Liu, R.; Truax, A.D.; Chen, L.; Hu, P.; Li, Z.; Chen, J.; Song, C.; Chen, L.; Ting, J.P. Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: Correlation with cancer stages and inflammasome components. Oncotarget 2015, 6, 33456–33469. [Google Scholar] [CrossRef]
- Lau, H.C.H.; Yu, J. Gut microbiome alters functions of mutant p53 to promote tumorigenesis. Signal Transduct. Target. Ther. 2020, 5, 232. [Google Scholar] [CrossRef] [PubMed]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.M.; Li, G.F.; Ren, Y.L.; Xu, X.Y.; Xu, Z.H.; Geng, Y.; Mao, Y. A Free Amino Acid Diet Alleviates Colorectal Tumorigenesis through Modulating Gut Microbiota and Metabolites. Nutrients 2024, 16, 1040. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Muller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Chen, D.; Riddle, D.L. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 2004, 131, 3897–3906. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Powers, R.W.; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013, 4, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Powers, R.W., 3rd; Kaeberlein, M.; Caldwell, S.D.; Kennedy, B.K.; Fields, S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006, 20, 174–184. [Google Scholar] [CrossRef]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012, 15, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. Regulation of the aging process by autophagy. Trends Mol. Med. 2009, 15, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Carosi, J.M.; Fourrier, C.; Bensalem, J.; Sargeant, T.J. The mTOR-lysosome axis at the centre of ageing. FEBS Open Bio 2022, 12, 739–757. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A. The role of the immunosuppressive PD-1/PD-L1 checkpoint pathway in the aging process and age-related diseases. J. Mol. Med. 2024, 102, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Tian, X.; Liu, Q.; Jin, J.; Shi, J.; Hou, Y. Role of autophagy on cancer immune escape. Cell Commun. Signal 2021, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Chen, Y. Autophagy controls programmed death-ligand 1 expression on cancer cells (Review). Biomed. Rep. 2021, 15, 84. [Google Scholar] [CrossRef]
- Hudson, K.; Cross, N.; Jordan-Mahy, N.; Leyland, R. The Extrinsic and Intrinsic Roles of PD-L1 and Its Receptor PD-1: Implications for Immunotherapy Treatment. Front. Immunol. 2020, 11, 568931. [Google Scholar] [CrossRef]
- Garcia-Perez, B.E.; Perez-Torres, C.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Castrejon-Jimenez, N.S. Autophagy as a Target for Non-Immune Intrinsic Functions of Programmed Cell Death-Ligand 1 in Cancer. Int. J. Mol. Sci. 2023, 24, 15016. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, J.; Ren, X. PD-L1 regulates tumorigenesis and autophagy of ovarian cancer by activating mTORC signaling. Biosci. Rep. 2019, 39, BSR20191041. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef]
- Riera, C.E.; Dillin, A. Tipping the metabolic scales towards increased longevity in mammals. Nat. Cell Biol. 2015, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Dillin, A.; Hsu, A.L.; Arantes-Oliveira, N.; Lehrer-Graiwer, J.; Hsin, H.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Rates of behavior and aging specified by mitochondrial function during development. Science 2002, 298, 2398–2401. [Google Scholar] [CrossRef]
- Monaghan, R.M.; Barnes, R.G.; Fisher, K.; Andreou, T.; Rooney, N.; Poulin, G.B.; Whitmarsh, A.J. A nuclear role for the respiratory enzyme CLK-1 in regulating mitochondrial stress responses and longevity. Nat. Cell Biol. 2015, 17, 782–792. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, H.; Zhang, W.; Liang, Z.; Huang, Q.; Xu, G.; Zhen, X.; Zheng, L.T. Clk1-regulated aerobic glycolysis is involved in glioma chemoresistance. J. Neurochem. 2017, 142, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Albert, L.; Karsy, M.; Murali, R.; Jhanwar-Uniyal, M. Inhibition of mTOR Activates the MAPK Pathway in Glioblastoma Multiforme. Cancer Genom. Proteom. 2009, 6, 255–261. [Google Scholar]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.C.; Debled, M.; Spaeth, D.; et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.; Karsy, M.; Albert, L.; Murali, R.; Jhanwar-Uniyal, M. Involvement of mTORC1 and mTORC2 in regulation of glioblastoma multiforme growth and motility. Int. J. Oncol. 2009, 35, 731–740. [Google Scholar] [PubMed]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; Jaeckle, K.A.; Buckner, J.C. North Central Cancer Treatment Group Phase I trial N057K of everolimus (RAD001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol. 2015, 17, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Dietz, A.B.; Kaufmann, T.J.; Gustafson, M.P.; Brown, P.D.; Uhm, J.H.; Rao, R.D.; Doyle, L.; et al. Combination of temsirolimus (CCI-779) with chemoradiation in newly diagnosed glioblastoma multiforme (GBM) (NCCTG trial N027D) is associated with increased infectious risks. Clin. Cancer Res. 2010, 16, 5573–5580. [Google Scholar] [CrossRef]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.S.; Zhang, J.M.; Gao, Y.; Reichling, L.J.; Sim, T.B.; Sabatini, D.M.; Gray, N.S. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Zheng, B.; Mao, J.H.; Qian, L.; Zhu, H.; Gu, D.H.; Pan, X.D.; Yi, F.; Ji, D.M. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015, 357, 468–475. [Google Scholar] [CrossRef]
- Janes, M.R.; Vu, C.; Mallya, S.; Shieh, M.P.; Limon, J.J.; Li, L.S.; Jessen, K.A.; Martin, M.B.; Ren, P.; Lilly, M.B.; et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia 2013, 27, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Gokhale, P.C.; Crew, A.P.; Cooke, A.; Yao, Y.; Mantis, C.; Kahler, J.; Workman, J.; Bittner, M.; Dudkin, L.; et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: Distinct from rapamycin. Mol. Cancer Ther. 2011, 10, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Korets, S.B.; Musa, F.; Curtin, J.; Blank, S.V.; Schneider, R.J. Dual mTORC1/2 inhibition in a preclinical xenograft tumor model of endometrial cancer. Gynecol. Oncol. 2014, 132, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Gordon, M.S.; Mita, M.; Rini, B.; Makker, V.; Macarulla, T.; Smith, D.C.; Cervantes, A.; Puzanov, I.; Pili, R.; et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br. J. Cancer 2020, 123, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Stephen, B.; Fu, S.; Karp, D.; Subbiah, V.; Ahnert, J.R.; Piha-Paul, S.A.; Wright, J.; Fessahaye, S.N.; Ouyang, F.; et al. Phase I study of sapanisertib (CB-228/TAK-228/MLN0128) in combination with ziv-aflibercept in patients with advanced solid tumors. Cancer Med. 2024, 13, e6877. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, Y.; Xiong, Z.Y.; Deng, Z.Y.; Song, H.L.; An, Z.M. Changes of plasma fibroblast growth factor-21 (FGF-21) in oral glucose tolerance test and effects of metformin on FGF-21 levels in type 2 diabetes mellitus. Endokrynol. Pol. 2013, 64, 220–224. [Google Scholar] [PubMed]
- Subbiah, V.; Coleman, N.; Piha-Paul, S.A.; Tsimberidou, A.M.; Janku, F.; Rodon, J.; Pant, S.; Dumbrava, E.E.I.; Fu, S.; Hong, D.S.; et al. Phase I Study of mTORC1/2 Inhibitor Sapanisertib (CB-228/TAK-228) in Combination with Metformin in Patients with mTOR/AKT/PI3K Pathway Alterations and Advanced Solid Malignancies. Cancer Res. Commun. 2024, 4, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.K.; Hong, S.E.; Lee, D.H.; Kim, J.Y.; Kim, J.Y.; Ye, S.K.; Hong, J.; Park, I.C.; Jin, H.O. Inhibition of mTORC1 through ATF4-induced REDD1 and Sestrin2 expression by Metformin. BMC Cancer 2021, 21, 803. [Google Scholar]
- Melnik, B.C.; Schmitz, G. Metformin: An Inhibitor of mTORC1 Signaling. J. Endocrinol. Diabetes Obes. 2014, 2, 1029. [Google Scholar]
- Jhanwar-Uniyal, M.; Gillick, J.L.; Neil, J.; Tobias, M.; Thwing, Z.E.; Murali, R. Distinct signaling mechanisms of mTORC1 and mTORC2 in glioblastoma multiforme: A tale of two complexes. Adv. Biol. Regul. 2015, 57, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Kandoussi, I.; Abbou, H.; Haddoumi, G.E.; Mansouri, M.; Belyamani, L.; Ibrahimi, A. Virtual docking screening and quantitative structure-activity relationship studies to explore AKT and PI3K inhibitors acting on mTOR in cancers by theoretical biology and medical modeling. Contemp. Oncol. 2023, 27, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhou, K.; Liang, Z.; Sun, R.; Tang, H.; Yang, Z.; Zhao, W.; Peng, Y.; Song, P.; Zheng, S.; et al. RapaLink-1 outperforms rapamycin in alleviating allogeneic graft rejection by inhibiting the mTORC1-4E-BP1 pathway in mice. Int. Immunopharmacol. 2023, 125 Pt B, 111172. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Huang, Y.; Zheng, Y.; Chen, W.; Zhang, Y.; Yang, Y.; Huang, W. Taurine Inhibits Lung Metastasis in Triple-Negative Breast Cancer by Modulating Macrophage Polarization through PTEN-PI3K/Akt/mTOR Pathway. J. Immunother. 2024. [Google Scholar] [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major pathways involved in macrophage polarization in cancer. Front. Immunol. 2022, 13, 1026954. [Google Scholar] [CrossRef]
- Gao, J.; Liang, Y.; Wang, L. Shaping Polarization Of Tumor-Associated Macrophages In Cancer Immunotherapy. Front. Immunol. 2022, 13, 888713. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marafie, S.K.; Al-Mulla, F.; Abubaker, J. mTOR: Its Critical Role in Metabolic Diseases, Cancer, and the Aging Process. Int. J. Mol. Sci. 2024, 25, 6141. https://doi.org/10.3390/ijms25116141
Marafie SK, Al-Mulla F, Abubaker J. mTOR: Its Critical Role in Metabolic Diseases, Cancer, and the Aging Process. International Journal of Molecular Sciences. 2024; 25(11):6141. https://doi.org/10.3390/ijms25116141
Chicago/Turabian StyleMarafie, Sulaiman K., Fahd Al-Mulla, and Jehad Abubaker. 2024. "mTOR: Its Critical Role in Metabolic Diseases, Cancer, and the Aging Process" International Journal of Molecular Sciences 25, no. 11: 6141. https://doi.org/10.3390/ijms25116141
APA StyleMarafie, S. K., Al-Mulla, F., & Abubaker, J. (2024). mTOR: Its Critical Role in Metabolic Diseases, Cancer, and the Aging Process. International Journal of Molecular Sciences, 25(11), 6141. https://doi.org/10.3390/ijms25116141