
An Overview of Epigenetic Changes in the Parkinson’s Disease Brain

Abstract
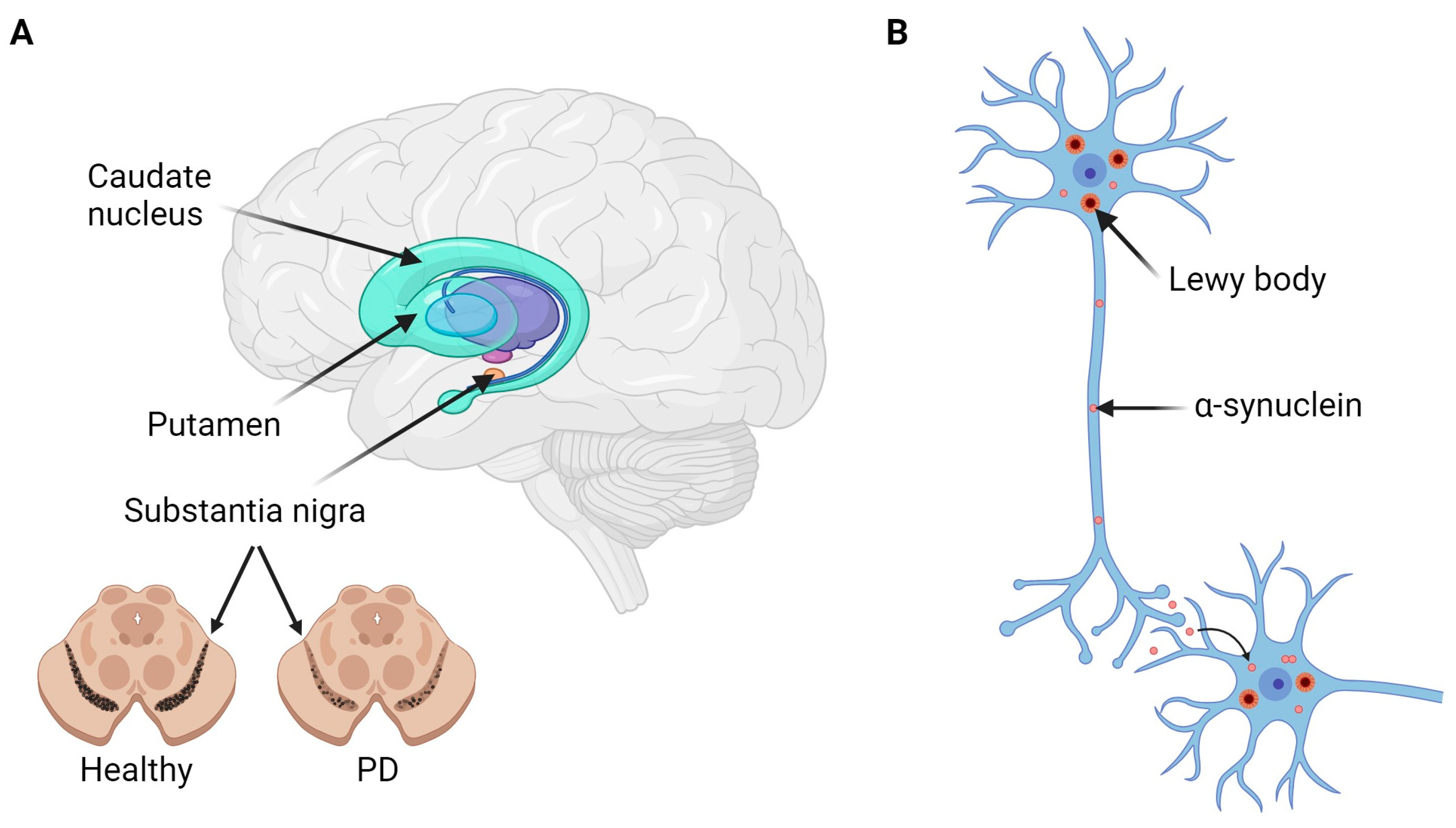
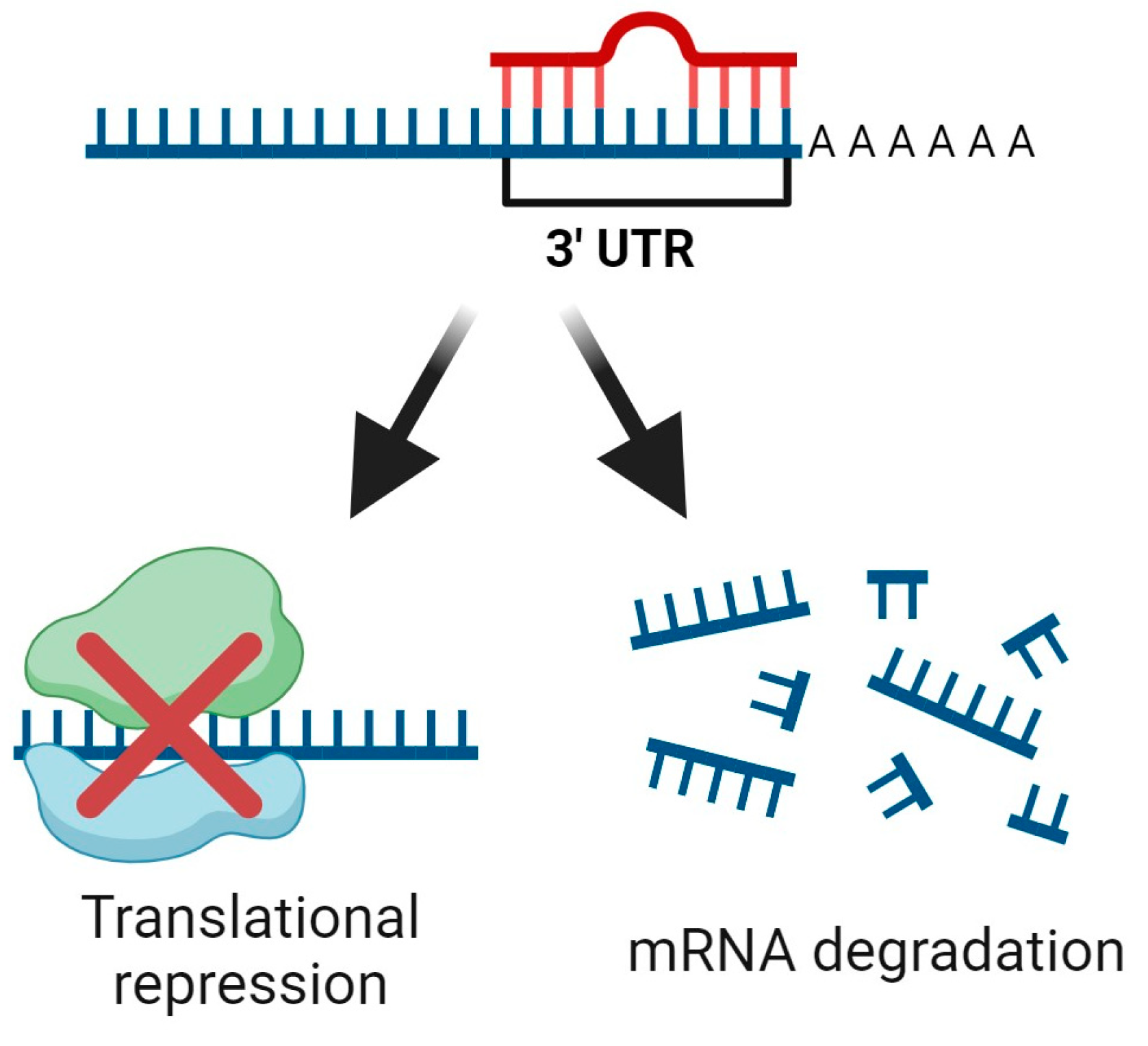
:1. Introduction
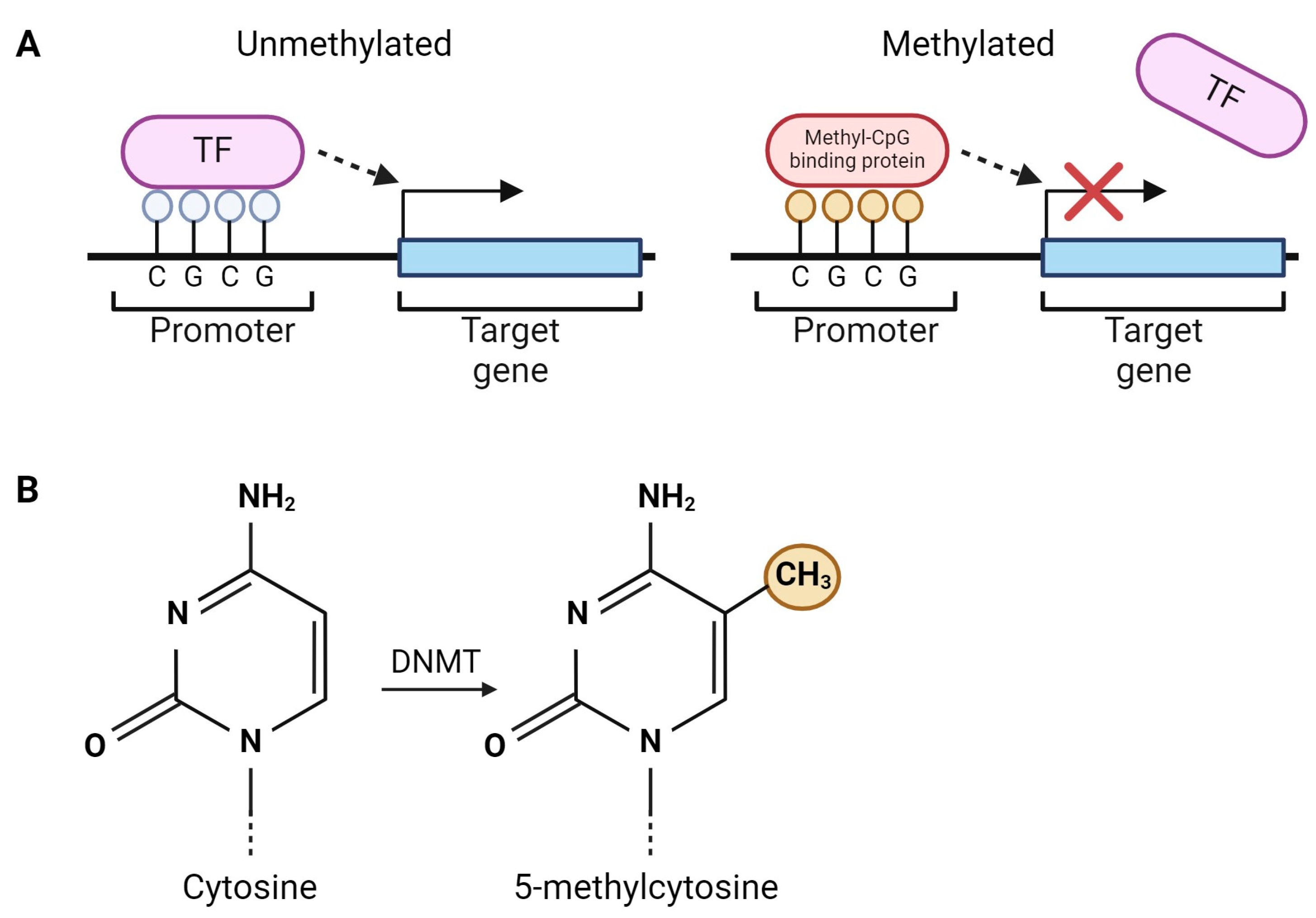
2. DNA Methylation
2.1. Single-Gene Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene(s) | Samples | Methylation Sites Studied | Key Findings | Reference/Year |
|---|---|---|---|---|
| SNCA | Substantia nigra and cortex (6 PD, 6 controls), putamen (6 PD, 8 controls) | 23 CpGs in intron 1 | Hypomethylation in all brain areas | [67]/2010 |
| SNCA | Anterior cingulate cortex (11 PD, 8 controls), putamen (6 PD, 4 controls), substantia nigra (3 PD, 3 controls) | 13 CpGs in intron 1 | Hypomethylation in substantia nigra only | [68]/2010 |
| SNCA | Brain homogenates (4 PD, 4 controls) | Gross methylation of intron 1 | Hypomethylation | [69]/2011 |
| SNCA | Cerebral cortex (12 PD, 12 controls) | Gross methylation of intron 1 | No overall differences but methylation associated with rs3756063 SNP | [38]/2015 |
| SNCA | Substantia nigra (8 PD, 8 controls) | 23 CpGs in intron 1 | No differences | [71]/2017 |
| SNCA | Bulk frontal cortex (20 PD, 20 controls), sorted neuronal and glial nuclei (12 PD, 12 controls) | 23 CpGs in intron 1 | Bulk tissue and glial nuclei: No differences across all CpGs Neuronal nuclei: Hypomethylation in PD across all CpGs (borderline significant) | [72]/2021 |
| SNCA | Frontal cortex (9 PD-GBA1, 11 idiopathic PD, 6 controls), putamen (6 PD-GBA1, 9 idiopathic PD, 6 controls), substantia nigra (8 PD-GBA1, 13 idiopathic PD, 3 controls) | 17 CpGs (8 sites in intron 1 located further from TSS, 6 CpGs in intron 1 located close to TSS and 3 CpGs within promotor) | Frontal cortex: Hypomethylation in 5 CpGs located further from TSS in intron 1 in PD-GBA1 compared to controls Hypomethylation in 2 CpGs in intron 1 in PD-GBA1 compared to idiopathic PD No significant differences in any CpGs located close to TSS in intron 1 or in the promoter Putamen: No significant differences in intron 1 or the promoter Substantia nigra: 1 CpG in the promoter hypomethylated in idiopathic PD compared to both controls and PD-GBA1 No significant differences in intron 1 | [73]/2023 |
| SNCA | Cortex (2PD, 2 controls) | 23 CpGs in intron 1 | Hypomethylation | [70]/2024 |
| PAD2 | Cortex (white matter) (2 PD, 0 controls) | Gross methylation of portion of intron 1, exon 1, and distal extension | No differences | [81]/2007 |
| TNF-α | Substantia nigra and cortex (7 PD, 8 controls), striatum (3 PD, 2 controls) | 10 CpGs in promoter | No differences | [82]/2008 |
| MAPT, PSEN1, APP and UCHL1 | Frontal cortex (8 PD, 17 controls) | MAPT: 20 CpGs near exon 0.41 + 31 CpGs in intronic regions PESN1: 26 CpGs at TSS APP: 18 + 16 CpGs in promoter regions UCHL1: 37 CpGs around TSS | No differences | [76]/2009 |
| PRKN | Substantia nigra, cerebellum, occipital cortex (5 PD, 2 controls) | Gross methylation of promoter | No differences | [74]/2013 |
| MAPT | Cerebellum, putamen, anterior cingulate cortex (28 PD, 12 controls) | 6 CpGs within promoter/intron 1 | Hypermethylation in cerebellum, hypomethylation in putamen, no significant differences in anterior cingulate cortex | [77]/2014 |
| ADORA2A | Putamen (25 PD, 26 controls) | 108 CpGs in 5′ untranslated region | Hypomethylation at 2 CpGs | [83]/2014 |
| PGC-1α | Substantia nigra (10 PD, 10 controls) | Gross methylation of promoter | Hypermethylation (of mainly non-CpG dinucleotides) and decreased expression | [78]/2015 |
| mtDNA D-loop | Substantia nigra (10 PD, 10 controls) | CpGSs and non-CpGs in D-loop | Hypomethylation in nearly all CpGs and non-CpGs | [84]/2016 |
| SNCA, LRRK2, PRKN, PINK1 and DJ-1 | Substantia nigra, occipital cortex, parietal cortex (5 PD, 5 controls) | SNCA: 6–8 CpGs in promoter, 5 CpGs in exon 1 LRRK2: 9 CpGs in promoter, 9 CpGs in exon 1 PRKN: 4 CpGs in promoter, 9 CpGs in promoter overlapping with intron 1 PINK1: 5 CpGs in promoter/exon 1 DJ-1: 6 CpGs in promoter, 8 CpGs in promoter overlapping with exon 1 | No significant differences at whole CpG islands, although differential methylation observed at some specific CpGs in SNCA, PRKN and PINK1 | [75]/2018 |
| CYP2E1, TP73, C21ORF56 and CDH13 | Cortex (14 PD, 10 controls) | CYP2E1: 10 CpGs (including promoter region) TP73: 5 CpGs C21ORF56: 2 CpGs CDH13: 1 CpG | Hypomethylation of CYP2E1, TP73 and C21ORF56 No difference in CDH13 methylation | [85]/2022 |
2.2. Epigenome-Wide Association Studies
| Samples | Method | Key Findings | Reference/Year |
|---|---|---|---|
| Frontal cortex, cerebellum (399 healthy individuals) | Illumina HumanMethylation27 BeadChip | Differential methylation of 1 or more CpGs correlated with SNPs at PARK16/1q32, GPNMB/7p15, and STX1B/16p11 loci | [103]/2011 |
| Cortex, putamen (6 PD, 6 controls) | Illumina HumanMethylation27 BeadChip | Cortex: Hypomethylation of CYP2E1 and PPP4R2 Putamen: Hypomethylation of CYP2E1 and LOC84245 Hypermethylation of DEFA1 and CHFR | [86]/2012 |
| Frontal cortex (5 PD, 6 controls) | Illumina HumanMethylation450 BeadChip | 317 hypermethylated and 2591 hypomethylated CpGs | [87]/2013 |
| Substantia nigra (39 PD, 13 controls) | Illumina HumanMethylation450 BeadChip | Hypermethylation of 1 CpG (cg10917602) associated with PD susceptibility | [104]/2016 |
| Frontal cortex (12 PD, 12 controls) | Illumina HumanMethylation450 BeadChip | 2794 differentially methylated CpGs in the frontal cortex of PD cases and 328 differentially methylated CpGs, majority hypomethylated. Clear pattern of SNCAIP hypermethylation | [96]/2017 |
| Dorsal motor nucleus of the vagus, substantia nigra, cingulate gyrus (38 PD, 41 controls) | Illumina HumanMethylation450 BeadChip and Infinium MethylationEPIC BeadChip | 234 differentially methylated regions in the dorsal motor nucleus of the vagus (including ARFGAP1 hypermethylation), 44 in the substantia nigra and 141 in the cingulate gyrus | [98]/2019 |
| Temporal lobe (13 PD with 0 years of plantation work, 4 PD with 10+ years of plantation work) (12 PD with 0–2 organochlorines, 4 PD with 4+ organochlorines) | Illumina HumanMethylation450 BeadChip | 7 differentially methylated loci between PD individuals with 10+ vs. 0 years of plantation work exposure 8 differentially methylated loci between PD individuals with 4+ vs. 0–2 organochlorines in the brain 2 different loci annotated to DNAJC15 which were differentially methylated (in both brain and blood) between the organochlorine exposure groups | [88]/2020 |
| Prefrontal cortex neuronal nuclei (discovery cohort: 57 PD, 48 controls) (replication cohort: 26 PD, 31 controls) | Bisulfite padlock probe sequencing of enhancers and promoters (633,803 modified cytosines) | 6207 CpGs in PD showing hemispheric asymmetry in DNA methylation (3894 CpGs showed a greater hemispheric asymmetry in PD compared to controls). These targeted 4691 genes, including PD risk genes. More DNA methylation and transcriptomic differences seen in hemisphere matched to symptom-dominant side Above findings validated in replication cohort 37 PD risk genes showing more hemispheric asymmetry in PD and/or greater differences in symptom-dominant hemisphere, including SNCA, ITPKB, SATB1, ANK2 and CAMK2D | [94]/2020 |
| Prefrontal cortex neuronal nuclei (57 PD, 48 controls) (22 PD Braak 3–4, 48 controls) | Bisulfite padlock probe sequencing (31,590 enhancers) | 1799 differentially methylated cytosines in enhancers (mainly hypermethylated) 2172 differentially methylated cytosines in enhancers when comparing PD Braak stage 3–4 (prior to Lewy body pathology reaching the cortex) and controls Differentially methylated enhancers targeted 2885 genes, including 15 different PD risk genes and TET2 76 of the genes with dysregulated enhancers were also transcriptionally altered | [93]/2020 |
| Olfactory bulb (9 PD, 14 controls), prefrontal cortex neuronal nuclei—discovery cohort (52 PD including 20 Braak stage 3–4, 42 controls), prefrontal cortex neuronal nuclei—replication cohort (13 PD, 15 controls) | Bisulfite padlock probe sequencing of autophagy-lysosome pathway genes (143,553 CpGs in olfactory bulb) (130,733 CpGs and 696,665 non-CpGs in prefrontal cortex discovery cohort) (110,397 CpGs in prefrontal cortex replication cohort) | Olfactory bulb: 1142 differentially methylated CpGs affecting 353 genes (mostly hypermethylated) (SNCA hypomethylation) Prefrontal cortex neuronal nuclei: 70 differentially methylated CpGs affecting 58 genes (mostly hypermethylated) in discovery cohort 110 differentially methylated CpGs affecting 87 genes in PD Braak stage 3–4 1131 differentially methylated CpGs affecting 341 genes in replication cohort | [95]/2021 |
| Prefrontal cortex (27 PD, 26 controls) | Whole-genome bisulfite sequencing | No association between mitochondrial DNA methylation and disease status | [105]/2022 |
| Cortex (14 PD, 10 controls) | Illumina HumanMethylation450 BeadChip | 35 hypomethylated and 22 hypermethylated genes (not significant after p-value adjustment). Included 5 CpGs hypomethylated in CYP2E1 and 6 CpGs hypomethylated in C21ORF56 | [85]/2022 |
| Sorted neuronal nuclei from parietal cortex (50 PD, 50 controls) | Infinium MethylationEPIC BeadChip | 3 and 87 differentially methylated CpGs in males and females, respectively, including PARK7 hypomethylation (males), ATXN1 hypermethylation (females) and SLC17A6 hypomethylation (females) 258 and 214 differentially methylated regions in males and females, respectively, including NR4A2 (males) and SLC17A6 (females) 1 differentially methylated region completely overlaps between sexes (annotated to PTPRN2)—hypermethylated in males and hypomethylated in females | [91]/2022 |
| Primary motor cortex (40 PD, 38 controls) | Infinium MethylationEPIC BeadChip | 3062 hypomethylated and 1251 hypermethylated CpGs 2.07 years of accelerated epigenetic age in PD compared to controls | [101]/2022 |
| Prefrontal cortex neurons | Targeted bisulfite sequencing | 667 differentially methylated genes, including 107 associated with stool butyrate levels | [106]/2022 |
| Prefrontal cortex (19 PDD, 18 controls) | Infinium MethylationEPIC BeadChip | 1151 differentially methylated CpGs (82% hypomethylated) 1 differentially methylated region in OTX2 gene | [107]/2023 |
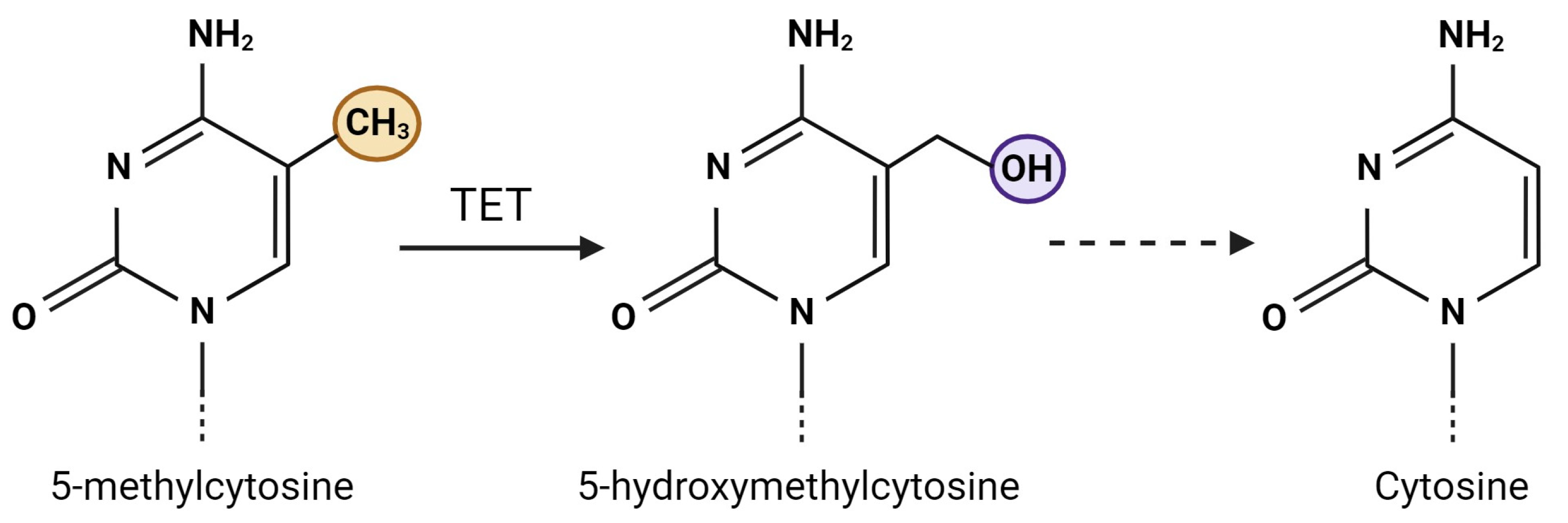
3. DNA Hydroxymethylation
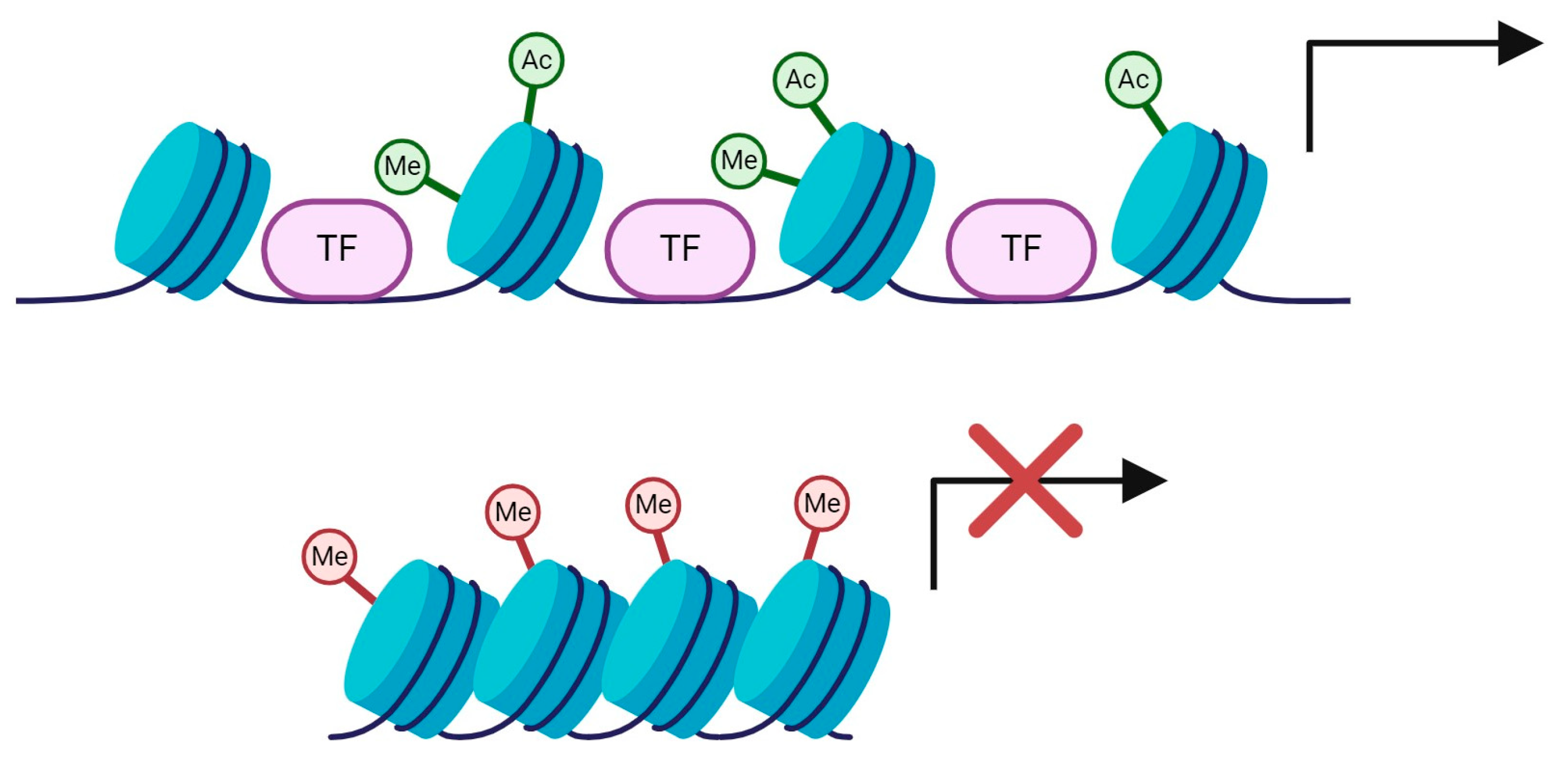
4. Histone Modifications
| Samples | Method | Key Findings | Reference/Year |
|---|---|---|---|
| Midbrain, cerebral cortex, cerebellar cortex (5 PD, 5 controls) | Western blotting and immunostaining | Midbrain: Increased acetylation of H2AK5, H2BK15, H3K9 and H4K5 (in 2–3 PD individuals) Downregulation of HDAC1, HDAC2, HDAC4, HDAC6 and SirT1 Higher proportion of acetylated midbrain dopaminergic neurons Cerebral cortex: Increased acetylation (H2AK5, H2BK15, H3K9 and H4K5) in 1 PD individual only Cerebellar cortex: Increased acetylation of H2BK15 | [121]/2016 |
| Primary motor cortex (9 PD, 8 controls) | Western botting | Increased acetylated H3–total H3 ratio Increased acetylated H3K14–total H3 ratio Increased acetylated H3K18–total H3 ratio Decreased acetylated H3K9–total H3 ratio | [117]/2016 |
| Substantia nigra (8 early PD, 12 late PD, 10 controls) | Western blotting | Increased acetylation at H3K9 in late PD Correlation between level of histone acetylation and Braak stage | [118]/2018 |
| Prefrontal cortex (global acetylation: 13 PD, 13 controls), Prefrontal cortex, striatum and cerebellar cortex (7 PD, 7 controls), Prefrontal cortex (discovery cohort: 17 PD, 11 controls), Prefrontal cortex (replication cohort: 10 PD, 11 controls) | Western blotting ChIP-seq (genome-wide H3K27 acetylation) | Prefrontal cortex: Increased global histone acetylation Increased acetylation at H3K27, H2BK15, H3K9/14, H3K56 and H4K12 No significant changes at H2AK5, H4K5 and H4K16 Striatum and cerebellar cortex: Increased acetylation at H3K27 Discovery study: 2877 H3K27-hyperacetylated regions (corresponding to 1434 genes) and 14 hypoacetylated regions (corresponding to 9 genes) Replication study: 2486 H3K27-hyperacetylated regions (corresponding to 946 genes) and 227 hypoacetylated regions (corresponding to 253 genes) 275 hyperacetylated genes (DLG2 and TNRC6B most significant) and 2 hypoacetylated genes (PTPRH, JUP) replicated across both cohorts | [119]/2021 |
| Substantia nigra (18 PD, 9 controls), substantia nigra neuronal nuclei (7 PD, 6 controls) | ChIP (H3K4me3, H3K27ac and H3K27me3) | Increased H3K4me3 at SNCA regulatory region No significant difference in H3K27ac Increased H3K4me3 at SNCA promoter/intron 1 Positive correlation between H3K4me3 and α-synuclein expression | [120]/2021 |
| Substantia nigra (9 PD, 9 controls) | ChIP-seq (H3K27ac) | Identification of 2770 downregulated and 2910 upregulated cis-regulatory elements | [39]/2023 |
5. Non-Coding RNAs
| Samples | Method | Key Findings | PD-Associated Pathophysiology | Reference/Year |
|---|---|---|---|---|
| Midbrain, cerebellum and cortex (3 PD, 5 controls) | RT-qPCR (panel of 224 miRNA precursors), RNase protection assay, qPCR and Northern blotting (including mature miR-133b) | Downregulation of miR-133b (precursor and mature) | Involved in the regulation of dopaminergic neuron maturation and function | [126]/2007 |
| Amygdala (11 PD, 6 controls) Amygdala (13 PD, 12 controls), frontal cortex (14 PD, 21 controls), cerebellum (11 PD, 17 controls) and substantia nigra (7 PD, 6 controls) | miRCURY LNA™ miRNA microarray (17 miRNAs) RT-qPCR (miR-637, miR-34b, miR-34c) | Downregulation of miR-637 and miR-34c-5p in amygdala Downregulation of miR-34b and miR-34c validated in amygdala, frontal cortex, substantia nigra and cerebellum (only miR-34c significant in cerebellum) Could not confirm change in miR-637 expression (measured in amygdala) | Depletion of miR-34b and miR-34c led to DJ-1 and PRKN downregulation and SNCA upregulation | [129]/2011 |
| Substantia nigra and amygdala (6 PD, 5 controls) | RT-qPCR (8 miRNAs) | 6 miRNAs upregulated in substantia nigra 2 miRNAs upregulated in amygdala | Target and associated with a reduction in lamp-2a and hsc70 levels, key proteins involved in chaperone-mediated autophagy | [128]/2013 |
| Frontal cortex (15 PD, 11 controls), striatum (5 PD and 4 controls) | RT-qPCR (miR-205) | Downregulation of miR-205 | Downregulation of miR-205 resulted in LRRK2 upregulation | [131]/2013 |
| Substantia nigra (8 PD, 4 controls) | TaqMan low-density arrays (733 miRNAs) and TaqMan assays (miR-198, miR-548d, miR-385-5p and miR-135b) | 10 miRNAs downregulated (including miR-135b) 1 miRNA upregulated (miR-548d) | Predicted target genes included SNCA, PRKN, LRRK2, ATXN1, SNCAIP and GBA | [137]/2014 |
| Laser capture microdissected dopaminergic neurons (8 PD, 8 controls) | Human MicroRNA TaqMan Arrays (379 miRNAs) | Dysregulation of miRNA expression profile Upregulation of miR-126 | miR-126 overexpression impaired IGF-1/PI3K/AKT signaling | [127]/2014 |
| Midbrain tissue, laser capture microdissected dopaminergic neurons (5 PD, 8 controls) | RT-qPCR (miR-133b) | Downregulation of miR-133b in midbrain tissue No differences in miR-133b levels in dopaminergic neurons | [146]/2014 | |
| Putamen (25 PD, 26 controls) | RT-qPCR (miR-34b and c) | Downregulation of miR-34b, particularly in early stages | Adenosine A2A receptor (A2AR) identified as potential target of miR-34b | [83]/2014 |
| Laser capture microdissected dopaminergic neurons (8 PD, 8 controls) | Human MicroRNA TaqMan Arrays | 109 miRNAs upregulated (miR-132 significantly upregulated) 50 miRNAs downregulated 14 significantly differentially expressed miRNAs associated with target genes Trend toward upregulation in males and downregulation in females | Targets associated with several aspects of PD pathogenesis, including cellular function and dopaminergic neuron identity | [134]/2015 |
| Prefrontal cortex (29 PD, 33 controls) | Small RNA sequencing—Illumina HiSeq 2000 (911 miRNAs) | 125 differentially expressed miRNAs (downregulation of miR-10b-5p) | Including miR-127-5p and miR-16-5p, both previously shown to regulate GBA1 expression | [132]/2016 |
| Putamen (12 PD—mostly l-DOPA treated, 12 controls) | Human v2 miRNA expression assay kit (800 miRNAs) and RT-qPCR (4 miRNAs) | 6 miRNAs upregulated 7 miRNAs downregulated Upregulation of miR-3195 and miR-204-5p Downregulation of miR-155-5p and miR-219-2-3p | miRNAs associated with inflammatory response and oxidative stress | [138]/2016 |
| Amygdala (14 PD, 7 controls) | RNA-seq | 42 differentially expressed miRNAs in premotor-stage PD compared to controls 103 differentially expressed miRNAs in motor-stage PD compared to controls | [155]/2016 | |
| Anterior cingulate gyrus (22 PD, 10 controls) | TaqMan miR array (744 miRNAs) RT-qPCR (13 miRNAs) | 43 miRNAs upregulated 5 miRNAs upregulated | 13 of these each predicted to regulate at least one of DJ-1, PRKN, PINK1, LRRK2, SNCA or HTRA2 Predicted to each regulate at least one of SNCA, PRKN or LRRK2 and additional genes involved in normal cellular function | [135]/2016 |
| Prefrontal cortex (29 PD, 36 controls) | RNA-seq (99 novel miRNAs) | Upregulation of miR-46 and miR-236 Downregulation of miR-225 | [156]/2016 | |
| Prefrontal cortex (29 PD, 33 controls) | RNA-seq | 321 differentially expressed miRNAs | [136]/2017 | |
| Substantia nigra (6 PD, 5 controls) | RT-qPCR (miR-7) | Downregulation of miR-7 | Depletion of miR-7 results in increased α-synuclein expression, dopaminergic neuron loss and reduced striatal dopamine content | [157]/2017 |
| Cingulate gyrus (8 PD, 8 controls) | RNA-seq | 44 miRNAs upregulated 55 miRNAs downregulated | [158]/2018 | |
| Substantia nigra (4 PD, 4 controls) | In situ hybridization (miR-425) | Downregulation of miR-425 | miR-425 deficiency triggers necroptosis of dopaminergic neurons | [159]/2019 |
| Prefrontal cortex (15 PD, 10 controls) | RT-qPCR (10 miRNAs) | 3 miRNAs downregulated (miR-124, miR-144 and miR-218) | Target KPNB1/A3/A4, which were all upregulated in PD brains. Inhibition of these miRNAs activates NF-κB signaling | [149]/2020 |
| Midbrain (19 PD, 12 controls) | Small and total RNA-seq, RT-qPCR (4 miRNAs) | 4 miRNAs upregulated (miR-539-3p, miR-376a-5p, miR-218-5p, miR-369-3p) | Targets of miR-369-3p (GTF2H3) and miR-218-5p (RAB6C) downregulated | [160]/2022 |
| Superior temporal gyrus (214 PD, 47 controls) | TaqMan Advanced miRNA Assays (10 miRNAs) | 3 miRNAs downregulated (miR-132-3p, miR-132-5p and miR-129-5p) | miR-132-3p/-5p significantly associated with α-synuclein Braak stage and may interact with SNCA mRNA | [161]/2022 |
| Midbrain (5 PD, 5 controls) | RT-qPCR (miR-132-3p) | Upregulation of miR-132-3p | GLRX identified as potential miR-132-3p target, GLRX mRNA and protein expression decreased in PD | [162]/2022 |
| Middle frontal gyrus (16 PD Braak stage 4, 9 PD Braak stage 5–6, 19 PDD (Braak stage 5–6), 19 controls) | RNA-seq RT-qPCR (let-7e-3p, miR-424-3p and miR-543) | 9 miRNAs downregulated 3 miRNAs upregulated (combined PD groups) Upregulation of let-7e-3p in PD with Braak 5–6 compared to PDD in both gray and white matter Upregulation of miR-424-3p in PD with Braak 5–6 in both gray and white matter compared to controls, and in PD in gray matter compared to PDD Upregulation of miR-543 in PD compared to controls in white matter only | SIRT1 identified as potential miR-543 target | [150]/2022 |
| Samples | Method | Key Findings | PD-Associated Pathophysiology | Reference/Year |
|---|---|---|---|---|
| Substantia nigra, amygdala (5 PD, 5 controls) | RT-qPCR (3 lncRNAs) | 3 lncRNAs upregulated in amygdala (RP11-462G22.1, RP11-79P5.3 and U1) RP11-462G22.1 and RP11-79P5.3 upregulated in substantia nigra | [163]/2014 | |
| Anterior cingulate gyrus neurons (20 PD, 10 controls) | RT-qPCR (90 lncRNAs) | 4 lncRNAs upregulated (lincRNA-p21, Malat1, SNHG1 and TncRNA) Downregulation of H19 lncRNA | [164]/2017 | |
| Substantia nigra (11 PD, 14 controls) | Affymetrix Human Genome U133A Array (698 lncRNAs) | 42 lncRNAs upregulated (AL049437 most significantly upregulated) 45 lncRNAs downregulated (AK021630 most significantly downregulated) | Reduction in AL049437 expression led to increases in cell viability, tyrosine hydroxylase secretion and mitochondrial transmembrane potential and mass | [153]/2017 |
| Substantia nigra and cerebellum (9 PD, 8 controls) | RT-qPCR (6 lncRNAs) | 6 lncRNAs downregulated in substantia nigra 3 lncRNAs downregulated in cerebellum (AK127687, UCHL1-AS1, MAPT-AS1) | Accompanied by increased SNCA mRNA levels in the substantia nigra and decreased LRRK2 and PINK1 mRNA levels in both brain regions | [154]/2019 |
| Substantia nigra (29 PD, 24 controls) | RT-qPCR (NEAT1 lncRNA) | Upregulation of NEAT1 | Neuroprotective agents induce NEAT1 upregulation | [165]/2019 |
| Superior frontal gyrus (23 Braak Lewy body stage 0 controls, 61 PD/PDD/incidental Lewy body disease—subdivided into 19 Braak Lewy body stage 1–4, 19 Braak Lewy body stage 5, 23 Braak Lewy body stage 6) | RNA-seq | Differential expression of 34 lncRNAs between groups | [166]/2023 | |
| Substantia nigra (57 PD, 43 controls) | Bioinformatics analysis of microarray data | 37 lncRNAs upregulated 68 lncRNAs downregulated | [167]/2023 |
6. Discussion
6.1. Limitations of Current Studies
6.2. Utility of Combining Genetics and Epigenetics Studies
6.3. Epigenetic Therapies
6.4. Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Parkinson’s Foundation Statistics|Parkinson’s Foundation. Available online: https://www.parkinson.org/Understanding-Parkinsons/Statistics (accessed on 12 March 2024).
- Parkinson’s UK. The Incidence and Prevalence of Parkinson’s in the UK; Parkinson’s UK: London, UK, 2017. [Google Scholar]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloemd, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Ray Dorsey, E.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and Projected Future Economic Burden of Parkinson’s Disease in the U.S. NPJ Park. Dis. 2020, 6, 15. [Google Scholar] [CrossRef]
- Fahn, S. Description of Parkinson’s Disease as a Clinical Syndrome. In Proceedings of the Annals of the New York Academy of Sciences. N. Y. Acad. Sci. 2003, 991, 1–14. [Google Scholar] [CrossRef]
- Pont-Sunyer, C.; Hotter, A.; Gaig, C.; Seppi, K.; Compta, Y.; Katzenschlager, R.; Mas, N.; Hofeneder, D.; Brücke, T.; Bayés, A.; et al. The Onset of Nonmotor Symptoms in Parkinson’s Disease (the Onset Pd Study). Mov. Disord. 2015, 30, 229–237. [Google Scholar] [CrossRef]
- Poewe, W. Non-Motor Symptoms in Parkinson’s Disease. Eur. J. Neurol. 2008, 15, 14–20. [Google Scholar] [CrossRef]
- Hobson, P.; Meara, J. Mortality and Quality of Death Certification in a Cohort of Patients with Parkinson’s Disease and Matched Controls in North Wales, UK at 18 Years: A Community-Based Cohort Study. BMJ Open 2018, 8, 18969. [Google Scholar] [CrossRef]
- Dickson, D.W. Parkinson’s Disease and Parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef]
- Yasuda, T.; Nakata, Y.; Mochizuki, H. α-Synuclein and Neuronal Cell Death. Mol. Neurobiol. 2013, 47, 466–483. [Google Scholar] [CrossRef]
- Luk, K.C.; Lee, V.M.Y. Modeling Lewy Pathology Propagation in Parkinson’s Disease. Park. Relat. Disord. 2014, 20, S85–S87. [Google Scholar] [CrossRef]
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-like Mechanisms in Parkinson’s Disease. Front. Neurosci. 2019, 13, 552. [Google Scholar] [CrossRef]
- Stefanoni, G.; Sala, G.; Tremolizzo, L.; Brighina, L.; Ferrarese, C. Role of Autophagy in Parkinson’s Disease. In Autophagy: Principles, Regulation and Roles in Disease; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2012; Volume 26, pp. 243–264. ISBN 9781619422667. [Google Scholar]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative Stress and Cellular Pathologies in Parkinson’s Disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial Dysfunction in Parkinson’s Disease. J. Neurochem. 2016, 139 (Suppl. 1), 216–231. [Google Scholar] [CrossRef] [PubMed]
- De Virgilio, A.; Greco, A.; Fabbrini, G.; Inghilleri, M.; Rizzo, M.I.; Gallo, A.; Conte, M.; Rosato, C.; Ciniglio Appiani, M.; de Vincentiis, M. Parkinson’s Disease: Autoimmunity and Neuroinflammation. Autoimmun. Rev. 2016, 15, 1005–1011. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental Toxins and Parkinson’s Disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s Disease: Mechanisms, Translational Models and Management Strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Nardin, A.; Schrepfer, E.; Ziviani, E. Counteracting PINK/Parkin Deficiency in the Activation of Mitophagy: A Potential Therapeutic Intervention for Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, A.; Theuns, J.; Van Broeckhoven, C. Progress in Unraveling the Genetic Etiology of Parkinson Disease in a Genomic Era. Trends Genet. 2015, 31, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Schapira, A.H.V. The Relationship between Glucocerebrosidase Mutations and Parkinson Disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef]
- Balestrino, R.; Tunesi, S.; Tesei, S.; Lopiano, L.; Zecchinelli, A.L.; Goldwurm, S. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord. 2020, 35, 2111–2114. [Google Scholar] [CrossRef]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaïti, B.; Bonaïti-Pellié, C.; Brice, A.; et al. Penetrance of PD in Glucocerebrosidase Gene Mutation Carriers. Neurology 2012, 79, 106–107. [Google Scholar] [CrossRef]
- McNeill, A.; Duran, R.; Hughes, D.A.; Mehta, A.; Schapira, A.H.V. A Clinical and Family History Study of Parkinson’s Disease in Heterozygous Glucocerebrosidase Mutation Carriers. J. Neurol. Neurosurg. Psychiatry 2012, 83, 853–854. [Google Scholar] [CrossRef]
- Menozzi, E.; Schapira, A.H.V. Exploring the Genotype–Phenotype Correlation in GBA-Parkinson Disease: Clinical Aspects, Biomarkers, and Potential Modifiers. Front. Neurol. 2021, 12, 694764. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Vitale, D.; Otani, D.V.; Lian, M.M.; Heilbron, K.; Aslibekyan, S.; Auton, A.; Babalola, E.; Bell, R.K.; Bielenberg, J.; et al. Multi-Ancestry Genome-Wide Association Meta-Analysis of Parkinson’s Disease. Nat. Genet. 2023, 56, 27–36. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M.; Marek, K.; Ottman, R.; Meng, C.; Comyns, K.; Chan, P.; Ma, J.; Marras, C.; Langston, J.W.; Ross, G.W.; et al. Concordance for Parkinson’s Disease in Twins: A 20-Year Update. Ann. Neurol. 2019, 85, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Modern Epigenetics Methods in Biological Research. Methods 2021, 187, 104. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in Epigenetics Link Genetics to the Environment and Disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. The Emerging Field of Epigenetics in Neurodegeneration and Neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Kim, S.; Kaang, B.K. Epigenetic Regulation and Chromatin Remodeling in Learning and Memory. Exp. Mol. Med. 2017, 49, e281. [Google Scholar] [CrossRef]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer Epigenetics: Moving Forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef]
- Smith, R.G.; Pishva, E.; Shireby, G.; Smith, A.R.; Roubroeks, J.A.Y.; Hannon, E.; Wheildon, G.; Mastroeni, D.; Gasparoni, G.; Riemenschneider, M.; et al. A Meta-Analysis of Epigenome-Wide Association Studies in Alzheimer’s Disease Highlights Novel Differentially Methylated Loci across Cortex. Nat. Commun. 2021, 12, 3517. [Google Scholar] [CrossRef]
- Pihlstrøm, L.; Berge, V.; Rengmark, A.; Toft, M. Parkinson’s Disease Correlates with Promoter Methylation in the α-Synuclein Gene. Mov. Disord. 2015, 30, 577–580. [Google Scholar] [CrossRef]
- Lee, A.J.; Kim, C.; Park, S.; Joo, J.; Choi, B.; Yang, D.; Jun, K.; Eom, J.; Lee, S.J.; Chung, S.J.; et al. Characterization of Altered Molecular Mechanisms in Parkinson’s Disease through Cell Type–Resolved Multiomics Analyses. Sci. Adv. 2023, 9, 15–18. [Google Scholar] [CrossRef]
- Sharma, A.; Osato, N.; Liu, H.; Asthana, S.; Dakal, T.C.; Ambrosini, G.; Bucher, P.; Schmitt, I.; Wüllner, U. Common Genetic Variants Associated with Parkinson’s Disease Display Widespread Signature of Epigenetic Plasticity. Sci. Rep. 2019, 9, 18464. [Google Scholar] [CrossRef]
- Vermunt, M.W.; Reinink, P.; Korving, J.; de Bruijn, E.; Creyghton, P.M.; Basak, O.; Geeven, G.; Toonen, P.W.; Lansu, N.; Meunier, C.; et al. Large-Scale Identification of Coregulated Enhancer Networks in the Adult Human Brain. Cell Rep. 2014, 9, 767–779. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic Regulation of Gene Expression: How the Genome Integrates Intrinsic and Environmental Signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the Environment: Emerging Patterns and Implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Babenko, O.; Golubov, A.; Ilnytskyy, Y.; Kovalchuk, I.; Metz, G.A. Genomic and Epigenomic Responses to Chronic Stress Involve MiRNA-Mediated Programming. PLoS ONE 2012, 7, e29441. [Google Scholar] [CrossRef]
- Thomas, E.A. DNA Methylation in Huntington’s Disease: Implications for Transgenerational Effects. Neurosci. Lett. 2016, 625, 34–39. [Google Scholar] [CrossRef]
- Park, C.; Rosenblat, J.D.; Brietzke, E.; Pan, Z.; Lee, Y.; Cao, B.; Zuckerman, H.; Kalantarova, A.; McIntyre, R.S. Stress, Epigenetics and Depression: A Systematic Review. Neurosci. Biobehav. Rev. 2019, 102, 139–152. [Google Scholar] [CrossRef]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P.A. Environmental Risk Factors and Parkinson’s Disease: An Umbrella Review of Meta-Analyses. Park. Relat. Disord. 2016, 23, 1–9. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic Differences Arise during the Lifetime of Monozygotic Twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Talens, R.P.; Christensen, K.; Putter, H.; Willemsen, G.; Christiansen, L.; Kremer, D.; Suchiman, H.E.D.; Slagboom, P.E.; Boomsma, D.I.; Heijmans, B.T. Epigenetic Variation during the Adult Lifespan: Cross-Sectional and Longitudinal Data on Monozygotic Twin Pairs. Aging Cell 2012, 11, 694. [Google Scholar] [CrossRef]
- Reynolds, C.A.; Tan, Q.; Munoz, E.; Jylhävä, J.; Hjelmborg, J.; Christiansen, L.; Hägg, S.; Pedersen, N.L. A Decade of Epigenetic Change in Aging Twins: Genetic and Environmental Contributions to Longitudinal DNA Methylation. Aging Cell 2020, 19, e13197. [Google Scholar] [CrossRef]
- Oertel, W.; Schulz, J.B. Current and Experimental Treatments of Parkinson Disease: A Guide for Neuroscientists. J. Neurochem. 2016, 139 (Suppl. 1), 325–337. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the Treatment of Parkinson’s Disease: An Old Drug Still Going Strong. Clin. Interv. Aging 2010, 5, 229–238. [Google Scholar]
- Laird, P.W. Principles and Challenges of Genome-Wide DNA Methylation Analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA Methylation: A Historical Perspective. Trends Genet. 2022, 38, 676–707. [Google Scholar] [CrossRef]
- Tate, P.H.; Bird, A.P. Effects of DNA Methylation on DNA-Binding Proteins and Gene Expression. Curr. Opin. Genet. Dev. 1993, 3, 226–231. [Google Scholar] [CrossRef]
- Nan, X.; Campoy, F.J.; Bird, A. MeCP2 Is a Transcriptional Repressor with Abundant Binding Sites in Genomic Chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef]
- Watt, F.; Molloy, P.L. Cytosine Methylation Prevents Binding to DNA of a HeLa Cell Transcription Factor Required for Optimal Expression of the Adenovirus Major Late Promoter. Genes. Dev. 1988, 2, 1136–1143. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; DeCarvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Wang, Q.; Xiong, F.; Wu, G.; Liu, W.; Chen, J.; Wang, B.; Chen, Y. Gene Body Methylation in Cancer: Molecular Mechanisms and Clinical Applications. Clin. Epigenetics 2022, 14, 154. [Google Scholar] [CrossRef]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; Leproust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and Genome-Scale Strategies Reveal Gene-Body Methylation Signatures in Human Cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef]
- Tost, J.; Gut, I.G. DNA Methylation Analysis by Pyrosequencing. Nat. Protoc. 2007, 2, 2265–2275. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro Mutation in the Gene Encoding α-Synuclein in Parkinson’s Disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation Regulates Alpha-Synuclein Expression and Is Decreased in Parkinson’s Disease Patients’ Brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG Demethylation Enhances Alpha-Synuclein Expression and Affects the Pathogenesis of Parkinson’s Disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. α-Synuclein Sequesters Dnmt1 from the Nucleus: A Novel Mechanism for Epigenetic Alterations in Lewy Body Diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, I.; Evert, B.O.; Sharma, A.; Khazneh, H.; Murgatroyd, C.; Wüllner, U. The Alpha-Synuclein Gene (SNCA) Is a Genomic Target of Methyl-CpG Binding Protein 2 (MeCP2)—Implications for Parkinson’s Disease and Rett Syndrome. Mol. Neurobiol. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Guhathakurta, S.; Evangelista, B.A.; Ghosh, S.; Basu, S.; Kim, Y.S. Hypomethylation of Intron1 of α-Synuclein Gene Does Not Correlate with Parkinson’s Disease. Mol. Brain 2017, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Barrera, J.; Yun, Y.; Murphy, S.K.; Beach, T.G.; Woltjer, R.L.; Serrano, G.E.; Kantor, B.; Chiba-Falek, O. Cell-Type Specific Changes in DNA Methylation of SNCA Intron 1 in Synucleinopathy Brains. Front. Neurosci. 2021, 15, 652226. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Richards, D.M.; Lunnon, K.; Schapira, A.H.V.; Migdalska-Richards, A. DNA Methylation of α-Synuclein Intron 1 Is Significantly Decreased in the Frontal Cortex of Parkinson’s Individuals with GBA1 Mutations. Int. J. Mol. Sci. 2023, 24, 2687. [Google Scholar] [CrossRef]
- De Mena, L.; Cardo, L.F.; Coto, E.; Alvarez, V. No Differential DNA Methylation of PARK2 in Brain of Parkinson’s Disease Patients and Healthy Controls. Mov. Disord. 2013, 28, 2032–2033. [Google Scholar] [CrossRef]
- Navarro-Sánchez, L.; Águeda-Gómez, B.; Aparicio, S.; Pérez-Tur, J. Epigenetic Study in Parkinson’s Disease: A Pilot Analysis of DNA Methylation in Candidate Genes in Brain. Cells 2018, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Barrachina, M.; Ferrer, I. DNA Methylation of Alzheimer Disease and Tauopathy-Related Genes in Postmortem Brain. J. Neuropathol. Exp. Neurol. 2009, 68, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Coupland, K.G.; Mellick, G.D.; Silburn, P.A.; Mather, K.; Armstrong, N.J.; Sachdev, P.S.; Brodaty, H.; Huang, Y.; Halliday, G.M.; Hallupp, M.; et al. DNA Methylation of the MAPT Gene in Parkinson’s Disease Cohorts and Modulation by Vitamin E In Vitro. Mov. Disord. 2014, 29, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Chu, Y.; Kordower, J.H.; Li, B.; Cao, H.; Huang, L.; Nishida, M.; Song, L.; Wang, D.; Federoff, H.J. PGC-1α Promoter Methylation in Parkinson’s Disease. PLoS ONE 2015, 10, e0134087. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin Functionally Interacts with PGC-1α to Preserve Mitochondria and Protect Dopaminergic Neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Mastronardi, F.G.; Noor, A.; Wood, D.D.; Paton, T.; Moscarello, M.A. Peptidyl Argininedeiminase 2 CpG Island in Multiple Sclerosis White Matter Is Hypomethylated. J. Neurosci. Res. 2007, 85, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Pieper, H.C.; Evert, B.O.; Kaut, O.; Riederer, P.F.; Waha, A.; Wüllner, U. Different Methylation of the TNF-Alpha Promoter in Cortex and Substantia Nigra: Implications for Selective Neuronal Vulnerability. Neurobiol. Dis. 2008, 32, 521–527. [Google Scholar] [CrossRef]
- Villar-Menéndez, I.; Porta, S.; Buira, S.P.; Pereira-Veiga, T.; Díaz-Sánchez, S.; Albasanz, J.L.; Ferrer, I.; Martín, M.; Barrachina, M. Increased Striatal Adenosine A2A Receptor Levels Is an Early Event in Parkinson’s Disease-Related Pathology and It Is Potentially Regulated by MiR-34b. Neurobiol. Dis. 2014, 69, 206–214. [Google Scholar] [CrossRef]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered Mitochondrial DNA Methylation Pattern in Alzheimer Disease-Related Pathology and in Parkinson Disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef]
- Kaut, O.; Schmitt, I.; Stahl, F.; Fröhlich, H.; Hoffmann, P.; Gonzalez, F.J.; Wüllner, U. Epigenome-Wide Analysis of DNA Methylation in Parkinson’s Disease Cortex. Life 2022, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Kaut, O.; Schmitt, I.; Wüllner, U. Genome-Scale Methylation Analysis of Parkinson’s Disease Patients’ Brains Reveals DNA Hypomethylation and Increased MRNA Expression of Cytochrome P450 2E1. Neurogenetics 2012, 13, 87–91. [Google Scholar] [CrossRef]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive Patterns of DNA Methylation Associated with Parkinson Disease: Identification of Concordant Epigenetic Changes in Brain and Peripheral Blood Leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Go, R.C.P.; Corley, M.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.H.; Maunakea, A.K.; He, Q.; Tiirikainen, M.I. Genome-Wide Epigenetic Analyses in Japanese Immigrant Plantation Workers with Parkinson’s Disease and Exposure to Organochlorines Reveal Possible Involvement of Glial Genes and Pathways Involved in Neurotoxicity. BMC Neurosci. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.; Cockburn, M.; Bronstein, J.; Zhang, X.; Ritz, B. Parkinson’s Disease and Residential Exposure to Maneb and Paraquat From Agricultural Applications in the Central Valley of California. Am. J. Epidemiol. 2009, 169, 919. [Google Scholar] [CrossRef]
- Sinha, D.; D’Silva, P. Chaperoning Mitochondrial Permeability Transition: Regulation of Transition Pore Complex by a J-Protein, DnaJC15. Cell Death Dis. 2014, 5, e1101. [Google Scholar] [CrossRef] [PubMed]
- Kochmanski, J.; Kuhn, N.C.; Bernstein, A.I. Parkinson’s Disease-Associated, Sex-Specific Changes in DNA Methylation at PARK7 (DJ-1), SLC17A6 (VGLUT2), PTPRN2 (IA-2β), and NR4A2 (NURR1)in Cortical Neurons. NPJ Park. Dis. 2022, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 Is a Redox-Dependent Molecular Chaperone That Inhibits α-Synuclein Aggregate Formation. PLoS Biol. 2004, 2, 308. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.L.; Killinger, B.A.; Ensink, E.; Li, P.; Li, K.X.; Cui, W.; Lubben, N.; Weiland, M.; Wang, X.; Gordevicius, J.; et al. Epigenomic Analysis of Parkinson’s Disease Neurons Identifies Tet2 Loss as Neuroprotective. Nat. Neurosci. 2020, 23, 1203–1214. [Google Scholar] [CrossRef]
- Li, P.; Ensink, E.; Lang, S.; Marshall, L.; Schilthuis, M.; Lamp, J.; Vega, I.; Labrie, V. Hemispheric Asymmetry in the Human Brain and in Parkinson’s Disease Is Linked to Divergent Epigenetic Patterns in Neurons. Genome Biol. 2020, 21, 61. [Google Scholar] [CrossRef]
- Gordevicius, J.; Li, P.; Marshall, L.L.; Killinger, B.A.; Lang, S.; Ensink, E.; Kuhn, N.C.; Cui, W.; Maroof, N.; Lauria, R.; et al. Epigenetic Inactivation of the Autophagy–Lysosomal System in Appendix in Parkinson’s Disease. Nat. Commun. 2021, 12, 5134. [Google Scholar] [CrossRef] [PubMed]
- Dashtipour, K.; Tafreshi, A.; Adler, C.; Beach, T.; Chen, X.; Serrano, G.; Tashiro, S.; Wang, C. Hypermethylation of Synphilin-1, Alpha-Synuclein-Interacting Protein (SNCAIP) Gene in the Cerebral Cortex of Patients with Sporadic Parkinson’s Disease. Brain Sci. 2017, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Castelao, B.; Castaño, J.G. Synphilin-1 Inhibits Alpha-Synuclein Degradation by the Proteasome. Cell. Mol. Life Sci. 2011, 68, 2643–2654. [Google Scholar] [CrossRef] [PubMed]
- Young, J.I.; Sivasankaran, S.K.; Wang, L.; Ali, A.; Mehta, A.; Davis, D.A.; Dykxhoorn, D.M.; Petito, C.K.; Beecham, G.W.; Martin, E.R.; et al. Genome-Wide Brain DNA Methylation Analysis Suggests Epigenetic Reprogramming in Parkinson Disease. Neurol. Genet. 2019, 5, e342. [Google Scholar] [CrossRef] [PubMed]
- Stafa, K.; Trancikova, A.; Webber, P.J.; Glauser, L.; West, A.B.; Moore, D.J. GTPase Activity and Neuronal Toxicity of Parkinson’s Disease-Associated LRRK2 Is Regulated by ArfGAP1. PLoS Genet. 2012, 8, e1002526. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yuan, C.; Chen, R.; Dawson, T.M.; Dawson, V.L. ArfGAP1 Is a GTPase Activating Protein for LRRK2: Reciprocal Regulation of ArfGAP1 by LRRK2. J. Neurosci. 2012, 32, 3877–3886. [Google Scholar] [CrossRef]
- Vishweswaraiah, S.; Akyol, S.; Yilmaz, A.; Ugur, Z.; Gordevičius, J.; Oh, K.J.; Brundin, P.; Radhakrishna, U.; Labrie, V.; Graham, S.F. Methylated Cytochrome P450 and the Solute Carrier Family of Genes Correlate With Perturbations in Bile Acid Metabolism in Parkinson’s Disease. Front. Neurosci. 2022, 16, 804261. [Google Scholar] [CrossRef] [PubMed]
- Mansell, G.; Gorrie-Stone, T.J.; Bao, Y.; Kumari, M.; Schalkwyk, L.S.; Mill, J.; Hannon, E. Guidance for DNA Methylation Studies: Statistical Insights from the Illumina EPIC Array. BMC Genom. 2019, 20, 366. [Google Scholar]
- International Parkinson’s Disease Genomics Consortium; Wellcome Trust Case Control Consortium 2. A Two-Stage Meta-Analysis Identifies Several New Loci for Parkinson’s Disease. PLoS Genet. 2011, 7, e1002142. [Google Scholar] [CrossRef]
- Rawlik, K.; Rowlatt, A.; Tenesa, A. Imputation of DNA Methylation Levels in the Brain Implicates a Risk Factor for Parkinson’s Disease. Genetics 2016, 204, 771–781. [Google Scholar] [CrossRef]
- Guitton, R.; Dölle, C.; Alves, G.; Ole-Bjørn, T.; Nido, G.S.; Tzoulis, C. Ultra-Deep Whole Genome Bisulfite Sequencing Reveals a Single Methylation Hotspot in Human Brain Mitochondrial DNA. Epigenetics 2022, 17, 906–921. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Ensink, E.; Li, P.; Gordevi, J.; Marshall, L.L.; George, S.; Pospisilik, J.A.; Aho, V.T.E.; Houser, M.C.; Pereira, P.A.B.; et al. Bacterial Butyrate in Parkinson’ s Disease Is Linked to Epigenetic Changes and Depressive Symptoms. Mov. Disord. 2022, 37, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.W.; Tulloch, J.; Yu, C.-E.; Tsuang, D. A Preliminary Comparison of the Methylome and Transcriptome from the Prefrontal Cortex Across Alzheimer’s Disease and Lewy Body Dementia. J. Alzheimers Dis. Rep. 2023, 7, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Song, C.X.; He, C.; Zhang, Y. Mechanism and Function of Oxidative Reversal of DNA and RNA Methylation. Annu. Rev. Biochem. 2014, 83, 585–614. [Google Scholar] [CrossRef] [PubMed]
- Richa, R.; Sinha, R.P. Hydroxymethylation of DNA: An Epigenetic Marker. EXCLI J. 2014, 13, 592–610. [Google Scholar] [PubMed]
- Wen, L.; Tang, F. Genomic Distribution and Possible Functions of DNA Hydroxymethylation in the Brain. Genomics 2014, 104, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Rustad, S.R.; Papale, L.A.; Alisch, R.S. DNA Methylation and Hydroxymethylation and Behavior. In Behavioral Neurogenomics. Current Topics in Behavioral Neurosciences; Binder, E.B., Klengel, T., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 51–82. ISBN 978-3-030-31265-7. [Google Scholar]
- Stöger, R.; Scaife, P.J.; Shephard, F.; Chakrabarti, L. Elevated 5hmC Levels Characterize DNA of the Cerebellum in Parkinson’s Disease. NPJ Park. Dis. 2017, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Kaut, O.; Kuchelmeister, K.; Moehl, C.; Wüllner, U. 5-Methylcytosine and 5-Hydroxymethylcytosine in Brains of Patients with Multiple System Atrophy and Patients with Parkinson’s Disease. J. Chem. Neuroanat. 2019, 96, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Xu, Q.; Qin, L.; Li, Y.; Li, Z.; Chen, C.; Wu, H.; Han, J.; Zhu, X.; Jin, P.; et al. Altered Hydroxymethylome in the Substantia Nigra of Parkinson’s Disease. Hum. Mol. Genet. 2022, 31, 3494–3503. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone Post-Translational Modifications—Cause and Consequence of Genome Function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Gebremedhin, K.G.; Rademacher, D.J. Histone H3 Acetylation in the Postmortem Parkinson’s Disease Primary Motor Cortex. Neurosci. Lett. 2016, 627, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Smith, A.D.; Dexter, D.T. Pathological Histone Acetylation in Parkinson’s Disease: Neuroprotection and Inhibition of Microglial Activation through SIRT 2 Inhibition. Neurosci. Lett. 2018, 666, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Toker, L.; Tran, G.T.; Sundaresan, J.; Tysnes, O.B.; Alves, G.; Haugarvoll, K.; Nido, G.S.; Dölle, C.; Tzoulis, C. Genome-Wide Histone Acetylation Analysis Reveals Altered Transcriptional Regulation in the Parkinson’s Disease Brain. Mol. Neurodegener. 2021, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Guhathakurta, S.; Kim, J.; Adams, L.; Basu, S.; Song, M.K.; Adler, E.; Je, G.; Fiadeiro, M.B.; Kim, Y. Targeted Attenuation of Elevated Histone Marks at SNCA Alleviates A-synuclein in Parkinson’s Disease. EMBO Mol. Med. 2021, 13, e12188. [Google Scholar] [CrossRef]
- Park, G.; Tan, J.; Garcia, G.; Kang, Y.; Salvesen, G.; Zhang, Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J. Biol. Chem. 2016, 291, 3531–3540. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-Coding RNAs in Human Disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and Their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.; Cairns, M.J. Identifying MiRNAs, Targets and Functions. Brief. Bioinform. 2014, 15, 1–19. [Google Scholar] [CrossRef]
- Shang, R.; Lee, S.; Senavirathne, G.; Lai, E.C. MicroRNAs in Action: Biogenesis, Function and Regulation. Nat. Rev. Genet. 2023, 24, 816–833. [Google Scholar] [CrossRef]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA Feedback Circuit in Midbrain Dopamine Neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, Y.; McKenna, N.D.; Yi, M.; Simunovic, F.; Wang, Y.; Kong, B.; Rooney, R.J.; Seo, H.; Stephens, R.M.; et al. MiR-126 Contributes to Parkinson’s Disease by Dysregulating the Insulin-like Growth Factor/Phosphoinositide 3-Kinase Signaling. Neurobiol. Aging 2014, 35, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.V.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of MicroRNA Deregulation on Chaperone-Mediated Autophagy and α-Synuclein Pathology in Parkinson’s Disease. Cell Death Dis. 2013, 4, e545. [Google Scholar] [CrossRef] [PubMed]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA Profiling of Parkinson’s Disease Brains Identifies Early Downregulation of MiR-34b/c Which Modulate Mitochondrial Function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef] [PubMed]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of MiR-34b and MiR-34c Enhances α-Synuclein Expression in Parkinson’s Disease. FEBS Lett. 2015, 589, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Jin Cho, H.; Liu, G.; Min Jin, S.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 Regulates the Expression of Parkinson’s Disease-Related Leucine-Rich Repeat Kinase 2 Protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Labadorf, A.; Beach, T.G.; Latourelle, J.C.; Myers, R.H. MicroRNA Profiles in Parkinson’s Disease Prefrontal Cortex. Front. Aging Neurosci. 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Siebert, M.; Westbroek, W.; Chen, Y.C.; Moaven, N.; Li, Y.; Velayati, A.; Luiza Saraiva-Pereira, M.; Martin, S.E.; Sidransky, E. Identification of MiRNAs That Modulate Glucocerebrosidase Activity in Gaucher Disease Cells. RNA Biol. 2014, 11, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Briggs, C.E.; Wang, Y.; Kong, B.; Woo, T.U.W.; Iyer, L.K.; Sonntag, K.C. Midbrain Dopamine Neurons in Parkinson’s Disease Exhibit a Dysregulated MiRNA and Target-Gene Network. Brain Res. 2015, 1618, 111–121. [Google Scholar] [CrossRef]
- Tatura, R.; Kraus, T.; Giese, A.; Arzberger, T.; Buchholz, M.; Höglinger, G.; Müller, U. Parkinson’s Disease: SNCA-, PARK2-, and LRRK2- Targeting MicroRNAs Elevated in Cingulate Gyrus. Park. Relat. Disord. 2016, 33, 115–121. [Google Scholar] [CrossRef]
- Chatterjee, P.; Roy, D. Comparative Analysis of RNA-Seq Data from Brain and Blood Samples of Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2017, 484, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Cardo, L.F.; Coto, E.; Ribacoba, R.; Menéndez, M.; Moris, G.; Suárez, E.; Alvarez, V. MiRNA Profile in the Substantia Nigra of Parkinson’s Disease and Healthy Subjects. J. Mol. Neurosci. 2014, 54, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.D.; Ge, Y. Alterations of MiRNAs Reveal a Dysregulated Molecular Regulatory Network in Parkinson’s Disease Striatum. Neurosci. Lett. 2016, 629, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.X.; Liu, H.; Zhang, L.S.; Lv, W.; Hu, X.Y. Altered MicroRNA Profiles in Cerebrospinal Fluid Exosome in Parkinson Disease and Alzheimer Disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [PubMed]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of Extracellular MiRNA in Cerebrospinal Fluid and Serum from Patients with Alzheimer’s and Parkinson’s Diseases Correlate with Disease Status and Features of Pathology. PLoS ONE 2014, 9, e94839. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; Rosa, A.; Guedes, L.C.; Fonseca, B.V.; Gotovac, K.; Violante, S.; Mestre, T.; Coelho, M.; Rosa, M.M.; Martin, E.R.; et al. Convergence of MiRNA Expression Profiling, α-Synuclein Interacton and GWAS in Parkinson’s Disease. PLoS ONE 2011, 6, e25443. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Lü, J.; Cao, S.; Zhao, Q.; Yu, Z. Identification of Aberrant Circulating MiRNAs in Parkinson’s Disease Plasma Samples. Brain Behav. 2018, 8, e00941. [Google Scholar] [CrossRef] [PubMed]
- Botta-Orfila, T.; Morató, X.; Compta, Y.; Lozano, J.J.; Falgàs, N.; Valldeoriola, F.; Pont-Sunyer, C.; Vilas, D.; Mengual, L.; Fernández, M.; et al. Identification of Blood Serum Micro-RNAs Associated with Idiopathic and LRRK2 Parkinson’s Disease. J. Neurosci. Res. 2014, 92, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Serafin, A.; Foco, L.; Zanigni, S.; Blankenburg, H.; Picard, A.; Zanon, A.; Giannini, G.; Pichler, I.; Facheris, M.F.; Cortelli, P.; et al. Overexpression of Blood MicroRNAs 103a, 30b, and 29a in L-Dopa-Treated Patients with PD. Neurology 2015, 84, 645–653. [Google Scholar] [CrossRef]
- Cao, X.Y.; Lu, J.M.; Zhao, Z.Q.; Li, M.C.; Lu, T.; An, X.S.; Xue, L.J. MicroRNA Biomarkers of Parkinson’s Disease in Serum Exosome-like Microvesicles. Neurosci. Lett. 2017, 644, 94–99. [Google Scholar] [CrossRef]
- Schlaudraff, F.; Gründemann, J.; Fauler, M.; Dragicevic, E.; Hardy, J.; Liss, B. Orchestrated Increase of Dopamine and PARK MRNAs but Not MiR-133b in Dopamine Neurons in Parkinson’s Disease. Neurobiol. Aging 2014, 35, 2302–2315. [Google Scholar] [CrossRef]
- Zhao, N.; Jin, L.; Fei, G.; Zheng, Z.; Zhong, C. Serum MicroRNA-133b Is Associated with Low Ceruloplasmin Levels in Parkinson’s Disease. Park. Relat. Disord. 2014, 20, 1177–1180. [Google Scholar] [CrossRef]
- Schulz, J.; Takousis, P.; Wohlers, I.; Itua, I.O.G.; Dobricic, V.; Rücker, G.; Binder, H.; Middleton, L.; Ioannidis, J.P.A.; Perneczky, R.; et al. Meta-analyses Identify Differentially Expressed MicroRNAs in Parkinson’s Disease. Ann. Neurol. 2019, 85, 835–851. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Li, L.; Liu, X.; Tian, B.; Cheng, Y. Down Regulation of MiR-218, MiR-124, and MiR-144 Relates to Parkinson’s Disease via Activating NF-κB Signaling. Kaohsiung J. Med. Sci. 2020, 36, 786–792. [Google Scholar] [CrossRef]
- Scheper, M.; Iyer, A.; Anink, J.J.; Mesarosova, L.; Mills, J.D.; Aronica, E. Dysregulation of MiR-543 in Parkinson’s Disease: Impact on the Neuroprotective Gene SIRT1. Neuropathol. Appl. Neurobiol. 2022, 19, e12864. [Google Scholar] [CrossRef] [PubMed]
- Ravanidis, S.; Bougea, A.; Papagiannakis, N.; Maniati, M.; Koros, C.; Simitsi, A.; Bozi, M.; Pachi, I.; Stamelou, M.; Paraskevas, G.P.; et al. Circulating Brain-Enriched MicroRNAs for Detection and Discrimination of Idiopathic and Genetic Parkinson’s Disease. Mov. Disord. 2020, 35, 457–467. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique Features of Long Non-Coding RNA Biogenesis and Function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Ni, Y.; Huang, H.; Chen, Y.; Cao, M.; Zhou, H.; Zhang, Y. Investigation of Long Non-Coding RNA Expression Profiles in the Substantia Nigra of Parkinson’s Disease. Cell Mol. Neurobiol. 2017, 37, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Elkouris, M.; Kouroupi, G.; Vourvoukelis, A.; Papagiannakis, N.; Kaltezioti, V.; Matsas, R.; Stefanis, L.; Xilouri, M.; Politis, P.K. Long Non-Coding RNAs Associated with Neurodegeneration-Linked Genes Are Reduced in Parkinson’s Disease Patients. Front. Cell Neurosci. 2019, 13, 58. [Google Scholar] [CrossRef]
- Pantano, L.; Friedländer, M.R.; Escaramís, G.; Lizano, E.; Pallarès-Albanell, J.; Ferrer, I.; Estivill, X.; Martí, E. Specific Small-RNA Signatures in the Amygdala at Premotor and Motor Stages of Parkinson’s Disease Revealed by Deep Sequencing Analysis. Bioinformatics 2016, 32, 673–681. [Google Scholar] [CrossRef]
- Wake, C.; Labadorf, A.; Dumitriu, A.; Hoss, A.G.; Bregu, J.; Albrecht, K.H.; DeStefano, A.L.; Myers, R.H. Novel MicroRNA Discovery Using Small RNA Sequencing in Post-Mortem Human Brain. BMC Genom. 2016, 17, 776. [Google Scholar] [CrossRef] [PubMed]
- McMillan, K.J.; Murray, T.K.; Bengoa-Vergniory, N.; Cordero-Llana, O.; Cooper, J.; Buckley, A.; Wade-Martins, R.; Uney, J.B.; O’Neill, M.J.; Wong, L.F.; et al. Loss of MicroRNA-7 Regulation Leads to α-Synuclein Accumulation and Dopaminergic Neuronal Loss In Vivo. Mol. Ther. 2017, 25, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.; Sommer, A.; Plötz, S.; Farrell, M.; Winner, B.; Grosch, J.; Winkler, J.; Riemenschneider, M.J. Sporadic Parkinson’s Disease Derived Neuronal Cells Show Disease-Specific MRNA and Small RNA Signatures with Abundant Deregulation of PiRNAs. Acta Neuropathol. Commun. 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-B.; Zhang, Y.-F.; Wang, H.; Ren, R.-J.; Cui, H.-L.; Huang, W.-Y.; Cheng, Q.; Chen, H.-Z.; Wang, G. MiR-425 Deficiency Promotes Necroptosis and Dopaminergic Neurodegeneration in Parkinson’s Disease. Cell Death Dis. 2019, 10, 589. [Google Scholar] [CrossRef] [PubMed]
- Caldi Gomes, L.; Galhoz, A.; Jain, G.; Roser, A.E.; Maass, F.; Carboni, E.; Barski, E.; Lenz, C.; Lohmann, K.; Klein, C.; et al. Multi-Omic Landscaping of Human Midbrains Identifies Disease-Relevant Molecular Targets and Pathways in Advanced-Stage Parkinson’s Disease. Clin. Transl. Med. 2022, 12, e692. [Google Scholar] [CrossRef] [PubMed]
- Dobricic, V.; Schilling, M.; Farkas, I.; Gveric, D.O.; Ohlei, O.; Schulz, J.; Middleton, L.; Gentleman, S.M.; Parkkinen, L.; Bertram, L.; et al. Common Signatures of Differential MicroRNA Expression in Parkinson’s and Alzheimer’s Disease Brains. Brain Commun. 2022, 4, fcac274. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Huang, M.; Chen, L. Mechanism of MiR-132-3p Promoting Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson’s Disease. eNeuro 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Soreq, L.; Guffanti, A.; Salomonis, N.; Simchovitz, A.; Israel, Z.; Bergman, H.; Soreq, H. Long Non-Coding RNA and Alternative Splicing Modulations in Parkinson’s Leukocytes Identified by RNA Sequencing. PLoS Comput. Biol. 2014, 10, 1003517. [Google Scholar] [CrossRef] [PubMed]
- Kraus, T.F.J.; Haider, M.; Spanner, J.; Steinmaurer, M.; Dietinger, V.; Kretzschmar, H.A. Altered Long Noncoding RNA Expression Precedes the Course of Parkinson’s Disease—A Preliminary Report. Mol. Neurobiol. 2017, 54, 2869–2877. [Google Scholar] [CrossRef]
- Simchovitz, A.; Hanan, M.; Niederhoffer, N.; Madrer, N.; Yayon, N.; Bennett, E.R.; Greenberg, D.S.; Kadener, S.; Soreq, H. NEAT1 Is Overexpressed in Parkinson’s Disease Substantia Nigra and Confers Drug-Inducible Neuroprotection from Oxidative Stress. FASEB J. 2019, 33, 11223–11234. [Google Scholar] [CrossRef]
- Cappelletti, C.; Henriksen, S.P.; Geut, H.; Rozemuller, A.J.M.; van de Berg, W.D.J.; Pihlstrøm, L.; Toft, M. Transcriptomic Profiling of Parkinson’s Disease Brains Reveals Disease Stage Specific Gene Expression Changes. Acta Neuropathol. 2023, 146, 227–244. [Google Scholar] [CrossRef]
- Asad Samani, L.; Ghaedi, K.; Majd, A.; Peymani, M.; Etemadifar, M. Coordinated Modification in Expression Levels of HSPA1A/B, DGKH, and NOTCH2 in Parkinson’s Patients’ Blood and Substantia Nigra as a Diagnostic Sign: The Transcriptomes’ Relationship. Neurol. Sci. 2023, 44, 2753–2761. [Google Scholar] [CrossRef]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific IPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A Single-Cell Atlas of the Human Substantia Nigra Reveals Cell-Specific Pathways Associated with Neurological Disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef] [PubMed]
- Bryois, J.; Skene, N.G.; Hansen, T.F.; Kogelman, L.J.A.; Watson, H.J.; Liu, Z.; Adan, R.; Alfredsson, L.; Ando, T.; Andreassen, O.; et al. Genetic Identification of Cell Types Underlying Brain Complex Traits Yields Insights into the Etiology of Parkinson’s Disease. Nat. Genet. 2020, 52, 482–493. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of Major Histocompatibility Complex Class II-Positive Microglia and Cytokine Profile of Parkinson’s Disease Brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Microglial Inflammation in the Parkinsonian Substantia Nigra: Relationship to Alpha-Synuclein Deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Nolz, J.; Sekar, S.; Delvaux, E.; Serrano, G.; Cuyugan, L.; Liang, W.S.; Beach, T.G.; Rogers, J.; Coleman, P.D. Laser-Captured Microglia in the Alzheimer’s and Parkinson’s Brain Reveal Unique Regional Expression Profiles and Suggest a Potential Role for Hepatitis B in the Alzheimer’s Brain. Neurobiol. Aging 2018, 63, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-Cell Sequencing of Human Midbrain Reveals Glial Activation and a Parkinson-Specific Neuronal State. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-Synuclein Immunoreactive Astrocytes in the Forebrain Parallels Stages of Intraneuronal Pathology in Sporadic Parkinson’s Disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Rostami, J.; Fotaki, G.; Sirois, J.; Mzezewa, R.; Bergström, J.; Essand, M.; Healy, L.; Erlandsson, A. Astrocytes Have the Capacity to Act as Antigen-Presenting Cells in the Parkinson’s Disease Brain. J. Neuroinflamm. 2020, 17, 119. [Google Scholar] [CrossRef]
- Iwamoto, K.; Bundo, M.; Ueda, J.; Oldham, M.C.; Ukai, W.; Hashimoto, E.; Saito, T.; Geschwind, D.H.; Kato, T. Neurons Show Distinctive DNA Methylation Profile and Higher Interindividual Variations Compared with Non-Neurons. Genome Res. 2011, 21, 688–696. [Google Scholar] [CrossRef]
- Jeffries, A.R.; Mill, J. Profiling Regulatory Variation in the Brain: Methods for Exploring the Neuronal Epigenome. Biol. Psychiatry 2017, 81, 90–91. [Google Scholar] [CrossRef]
- Nott, A.; Schlachetzki, J.C.M.; Fixsen, B.R.; Glass, C.K. Nuclei Isolation of Multiple Brain Cell Types for Omics Interrogation. Nat. Protoc. 2021, 16, 1629–1646. [Google Scholar] [CrossRef]
- Sakib, M.S.; Sokpor, G.; Nguyen, H.P.; Fischer, A.; Tuoc, T. Intranuclear Immunostaining-Based FACS Protocol from Embryonic Cortical Tissue. STAR Protoc. 2021, 2, 100318. [Google Scholar] [CrossRef]
- Policicchio, S.S.; Davies, J.P.; Chioza, B.; Burrage, J.; Mill, J.; Dempster, E.L.; Policicchio, S. Fluorescence-Activated Nuclei Sorting (FANS) on Human Post-Mortem Cortex Tissue Enabling the Isolation of Distinct Neural Cell Populations for Multiple Omic Profiling; Neurodegeneration Method Development Community Complex Disease Epigenetics Group: Exeter, UK, 2020. [Google Scholar] [CrossRef]
- Hannon, E.; Gorrie-Stone, T.J.; Smart, M.C.; Burrage, J.; Hughes, A.; Bao, Y.; Kumari, M.; Schalkwyk, L.C.; Mill, J. Leveraging DNA-Methylation Quantitative-Trait Loci to Characterize the Relationship between Methylomic Variation, Gene Expression, and Complex Traits. Am. J. Hum. Genet. 2018, 103, 654–665. [Google Scholar] [CrossRef]
- Scherzer, C.R.; Grass, J.A.; Liao, Z.; Pepivani, I.; Zheng, B.; Eklund, A.C.; Ney, P.A.; Ng, J.; McGoldrick, M.; Mollenhauer, B.; et al. GATA Transcription Factors Directly Regulate the Parkinson’s Disease-Linked Gene α-Synuclein. Proc. Natl. Acad. Sci. USA 2008, 105, 10907–10912. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of Summary Data from GWAS and EQTL Studies Predicts Complex Trait Gene Targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Jacobs, B.M.; Taylor, T.; Awad, A.; Baker, D.; Giovanonni, G.; Noyce, A.J.; Dobson, R. Summary-Data-Based Mendelian Randomization Prioritizes Potential Druggable Targets for Multiple Sclerosis. Brain Commun. 2020, 2, fcaa119. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The Emerging Role of Epigenetic Therapeutics in Immuno-Oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef]
- Peng, G.S.; Li, G.; Tzeng, N.S.; Chen, P.S.; Chuang, D.M.; Hsu, Y.D.; Yang, S.; Hong, J.S. Valproate Pretreatment Protects Dopaminergic Neurons from LPS-Induced Neurotoxicity in Rat Primary Midbrain Cultures: Role of Microglia. Mol. Brain Res. 2005, 134, 162–169. [Google Scholar] [CrossRef]
- Kidd, S.K.; Schneider, J.S. Protection of Dopaminergic Cells from MPP+-Mediated Toxicity by Histone Deacetylase Inhibition. Brain Res. 2010, 1354, 172–178. [Google Scholar] [CrossRef]
- Kidd, S.K.; Schneider, J.S. Protective Effects of Valproic Acid on the Nigrostriatal Dopamine System in a 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson’s Disease. Neuroscience 2011, 194, 189–194. [Google Scholar] [CrossRef]
- St. Laurent, R.; O’Brien, L.M.; Ahmad, S.T. Sodium Butyrate Improves Locomotor Impairment and Early Mortality in a Rotenone-Induced Drosophila Model of Parkinson’s Disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef]
- Sharma, S.; Taliyan, R.; Singh, S. Beneficial Effects of Sodium Butyrate in 6-OHDA Induced Neurotoxicity and Behavioral Abnormalities: Modulation of Histone Deacetylase Activity. Behav. Brain Res. 2015, 291, 306–314. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F.; Liu, S.; Du, J.; Hu, X.; Xiong, J.; Fang, R.; Chen, W.; Sun, J. Sodium Butyrate Exerts Protective Effect against Parkinson’s Disease in Mice via Stimulation of Glucagon like Peptide-1. J. Neurol. Sci. 2017, 381, 176–181. [Google Scholar] [CrossRef]
- Li, B.; Yang, Y.; Wang, Y.; Zhang, J.; Ding, J.; Liu, X.; Jin, Y.; Lian, B.; Ling, Y.; Sun, C. Acetylation of NDUFV1 Induced by a Newly Synthesized HDAC6 Inhibitor HGC Rescues Dopaminergic Neuron Loss in Parkinson Models. iScience 2021, 24, 102302. [Google Scholar] [CrossRef]
- Mazzocchi, M.; Goulding, S.R.; Wyatt, S.L.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. LMK235, a Small Molecule Inhibitor of HDAC4/5, Protects Dopaminergic Neurons against Neurotoxin- and α-Synuclein-Induced Degeneration in Cellular Models of Parkinson’s Disease. Mol. Cell. Neurosci. 2021, 115, 103642. [Google Scholar] [CrossRef]
- Mazzocchi, M.; Goulding, S.R.; Morales-Prieto, N.; Foley, T.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. Peripheral Administration of the Class-IIa HDAC Inhibitor MC1568 Partially Protects against Nigrostriatal Neurodegeneration in the Striatal 6-OHDA Rat Model of Parkinson’s Disease. Brain Behav. Immun. 2022, 102, 151–160. [Google Scholar] [CrossRef]
- El-Saiy, K.A.; Sayed, R.H.; El-Sahar, A.E.; Kandil, E.A. Modulation of Histone Deacetylase, the Ubiquitin Proteasome System, and Autophagy Underlies the Neuroprotective Effects of Venlafaxine in a Rotenone-Induced Parkinson’s Disease Model in Rats. Chem. Biol. Interact. 2022, 354, 109841. [Google Scholar] [CrossRef]
- Wang, L.; Liu, L.; Han, C.; Jiang, H.; Ma, K.; Guo, S.; Xia, Y.; Wan, F.; Huang, J.; Xiong, N.; et al. Histone Deacetylase 4 Inhibition Reduces Rotenone-Induced Alpha-Synuclein Accumulation via Autophagy in SH-SY5Y Cells. Brain Sci. 2023, 13, 670. [Google Scholar] [CrossRef]
- Meka, S.T.; Bojja, S.L.; Kumar, G.; Birangal, S.R.; Rao, C.M. Novel HDAC Inhibitors Provide Neuroprotection in MPTP-Induced Parkinson’s Disease Model of Rats. Eur. J. Pharmacol. 2023, 959, 176067. [Google Scholar] [CrossRef]
- Izco, M.; Blesa, J.; Schleef, M.; Schmeer, M.; Porcari, R.; Al-Shawi, R.; Ellmerich, S.; de Toro, M.; Gardiner, C.; Seow, Y.; et al. Systemic Exosomal Delivery of ShRNA Minicircles Prevents Parkinsonian Pathology. Mol. Ther. 2019, 27, 2111–2122. [Google Scholar] [CrossRef]
- Sun, Z.; Kantor, B.; Chiba-Falek, O. Neuronal-Type-Specific Epigenome Editing to Decrease SNCA Expression: Implications for Precision Medicine in Synucleinopathies. Mol. Ther. Nucleic Acids 2024, 35, 102084. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, Y.; Li, Z.; Zhu, M.; Wang, Z.; Li, Y.; Xu, T.; Feng, D.; Zhang, S.; Tang, F.; et al. MiR-103a-3p Regulates Mitophagy in Parkinson’s Disease through Parkin/Ambra1 Signaling. Pharmacol. Res. 2020, 160, 105197. [Google Scholar] [CrossRef]
- Leggio, L.; Vivarelli, S.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Marchetti, B.; Iraci, N. MicroRNAs in Parkinson’s Disease: From Pathogenesis to Novel Diagnostic and Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 2698. [Google Scholar] [CrossRef] [PubMed]

| Gene Name | Abbreviation | Mode of Inheritance |
|---|---|---|
| α-synuclein | SNCA | Autosomal dominant |
| leucine-rich repeat kinase 2 | LRRK2 | Autosomal dominant |
| PTEN-induced kinase 1 | PINK1 | Autosomal recessive |
| parkin | PRKN | Autosomal recessive |
| Parkinsonism-associated deglycase | DJ-1 | Autosomal recessive |
| ATPase cation transporting 13A2 | ATP13A2 | Autosomal recessive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klokkaris, A.; Migdalska-Richards, A. An Overview of Epigenetic Changes in the Parkinson’s Disease Brain. Int. J. Mol. Sci. 2024, 25, 6168. https://doi.org/10.3390/ijms25116168
Klokkaris A, Migdalska-Richards A. An Overview of Epigenetic Changes in the Parkinson’s Disease Brain. International Journal of Molecular Sciences. 2024; 25(11):6168. https://doi.org/10.3390/ijms25116168
Chicago/Turabian StyleKlokkaris, Anthony, and Anna Migdalska-Richards. 2024. "An Overview of Epigenetic Changes in the Parkinson’s Disease Brain" International Journal of Molecular Sciences 25, no. 11: 6168. https://doi.org/10.3390/ijms25116168