
CaMKIV-Mediated Phosphorylation Inactivates Freud-1/CC2D1A Repression for Calcium-Dependent 5-HT1A Receptor Gene Induction


Abstract
1. Introduction
2. Results
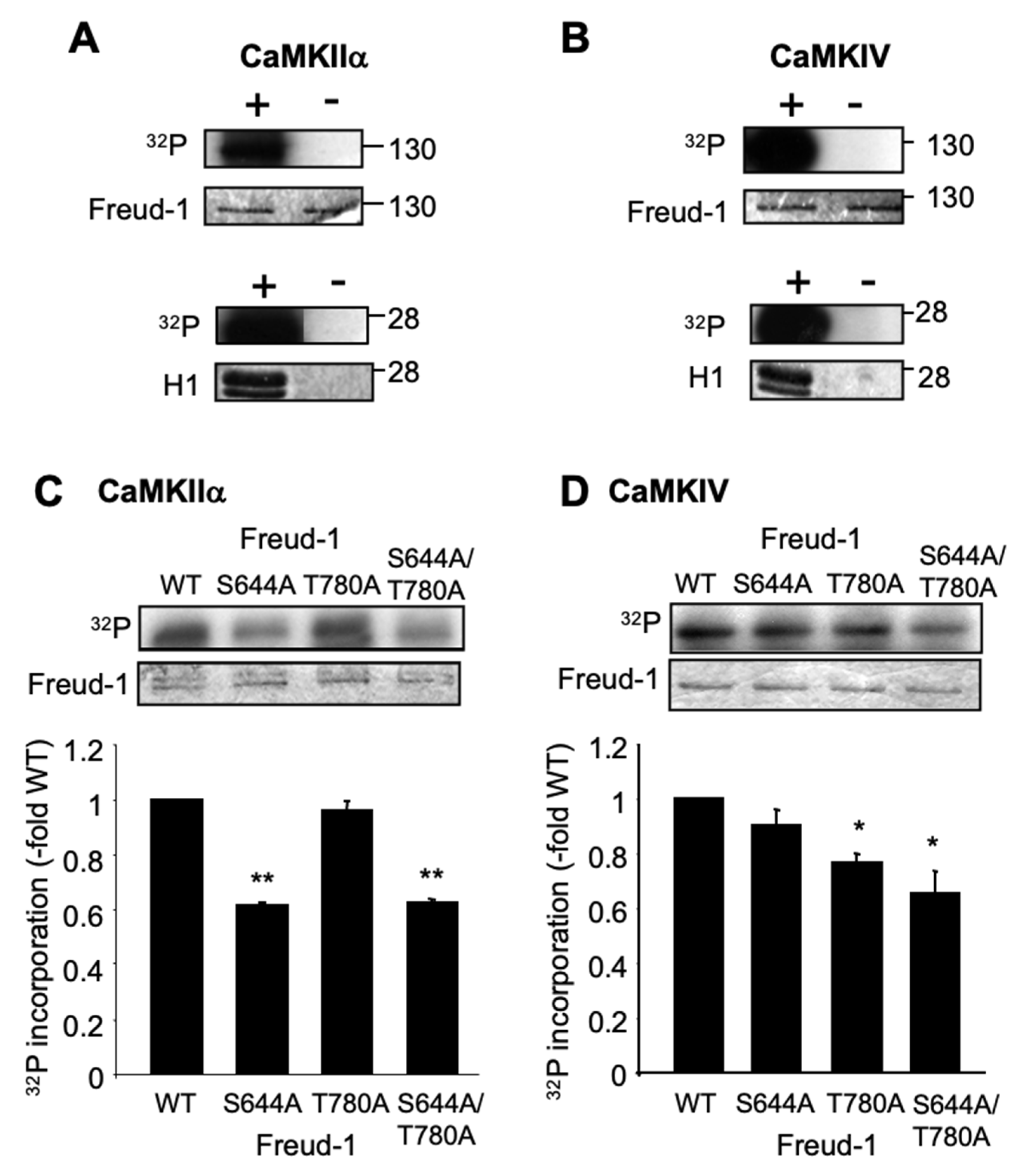
2.1. Human Freud-1 Phosphorylation by CaMKIIα and CaMKIV In Vitro
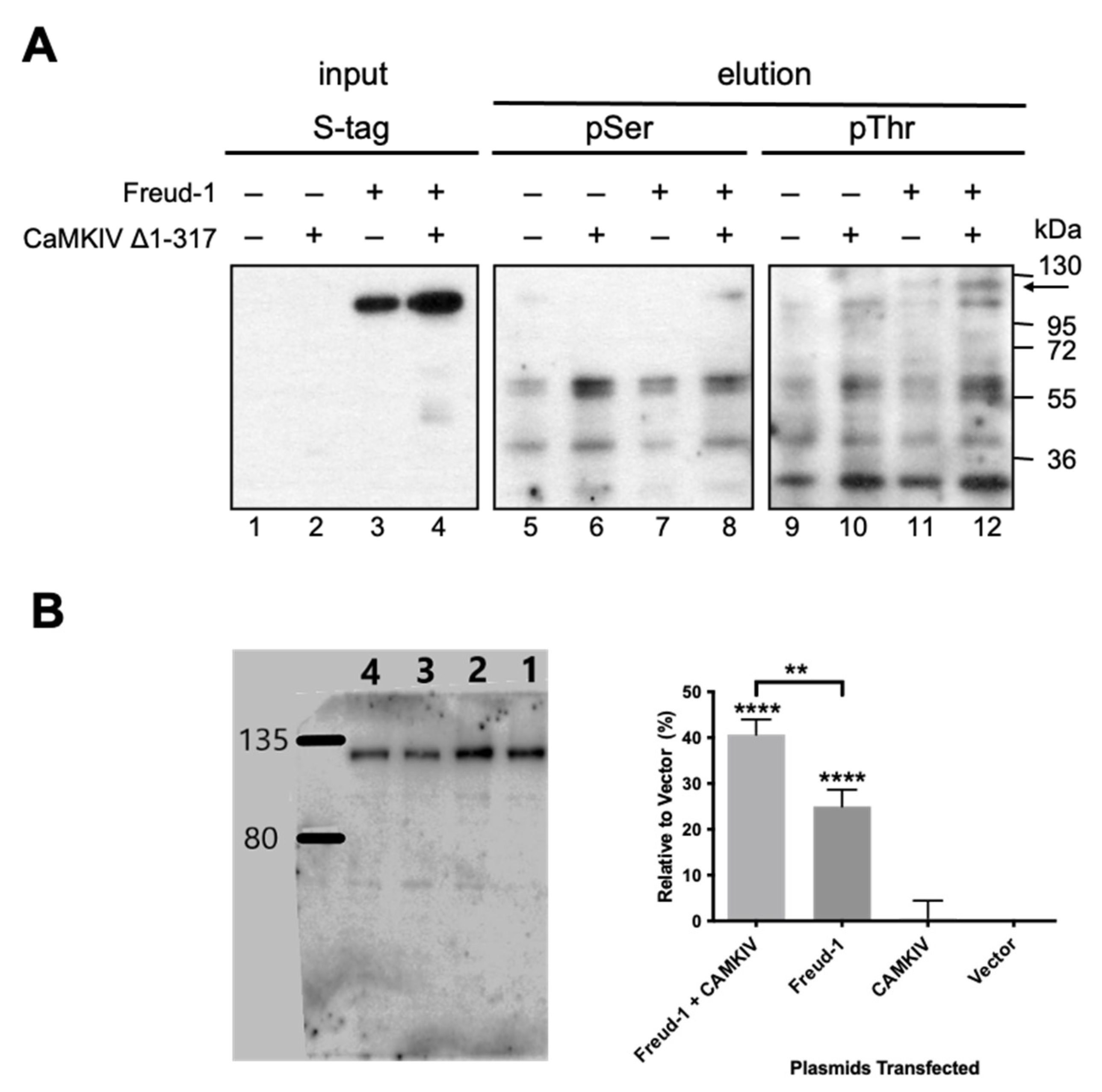
2.2. CaMKIV-Mediated Phosphorylation of Freud-1 in Cells
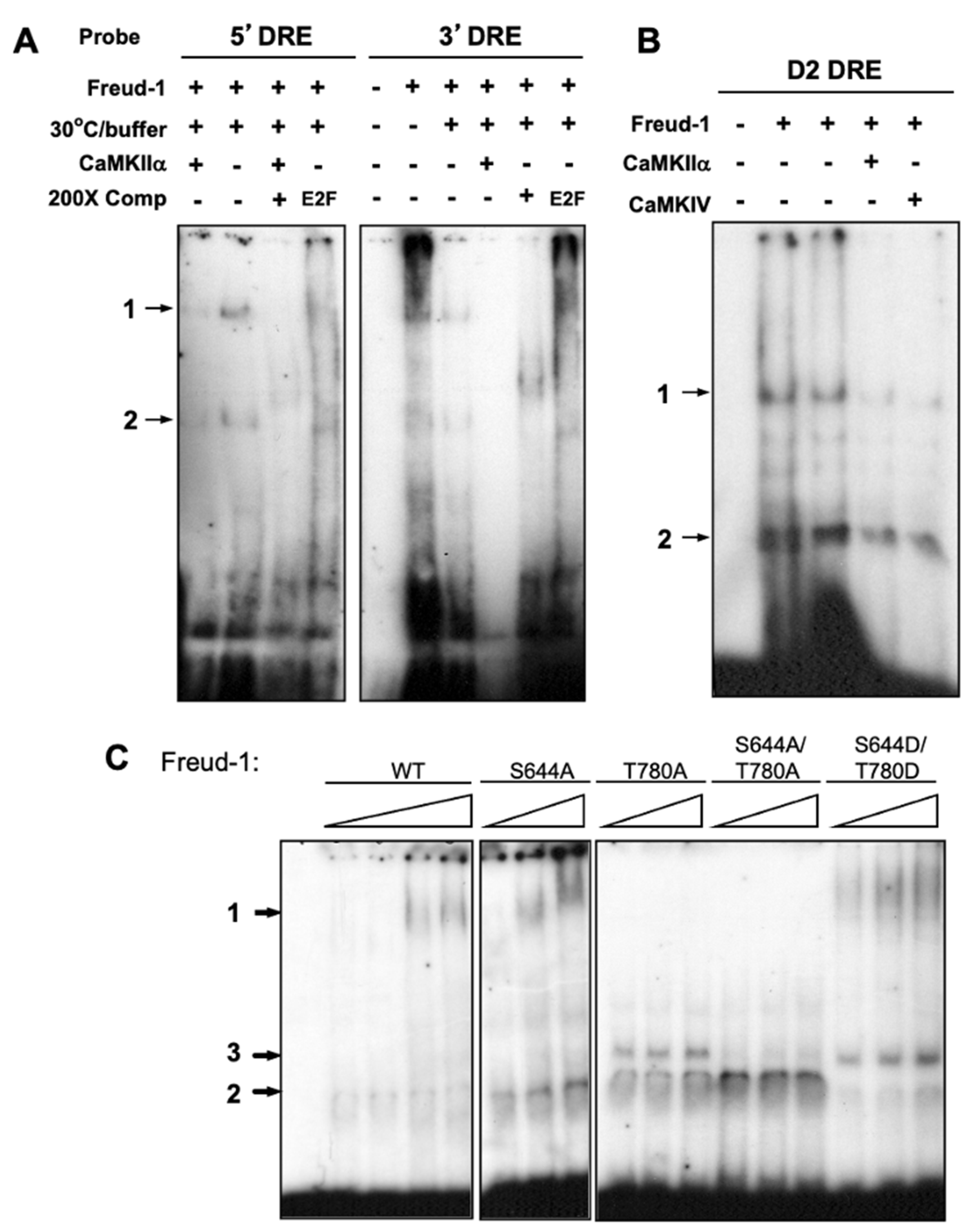
2.3. CaMK Activation Decreases the Freud-1 DNA Binding Activity In Vitro
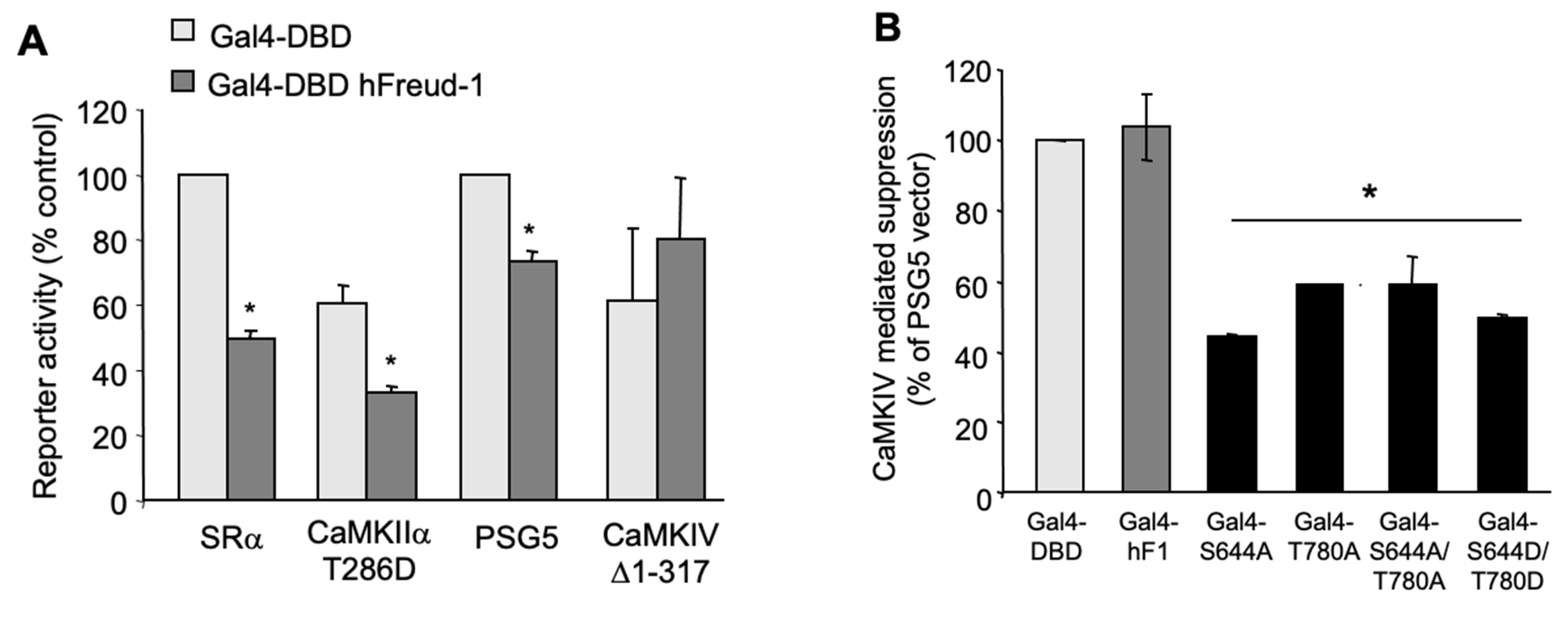
2.4. Activated CaMKIV Attenuates the Freud-1-Induced Repressor Activity
2.5. CaMKIV Inactivates the Freud-1-Induced Repression of the Human 5-HT1A Promoter
3. Discussion
3.1. CaMKIV Inhibits the Freud-1 Repression of the 5-HT1A Receptor Gene
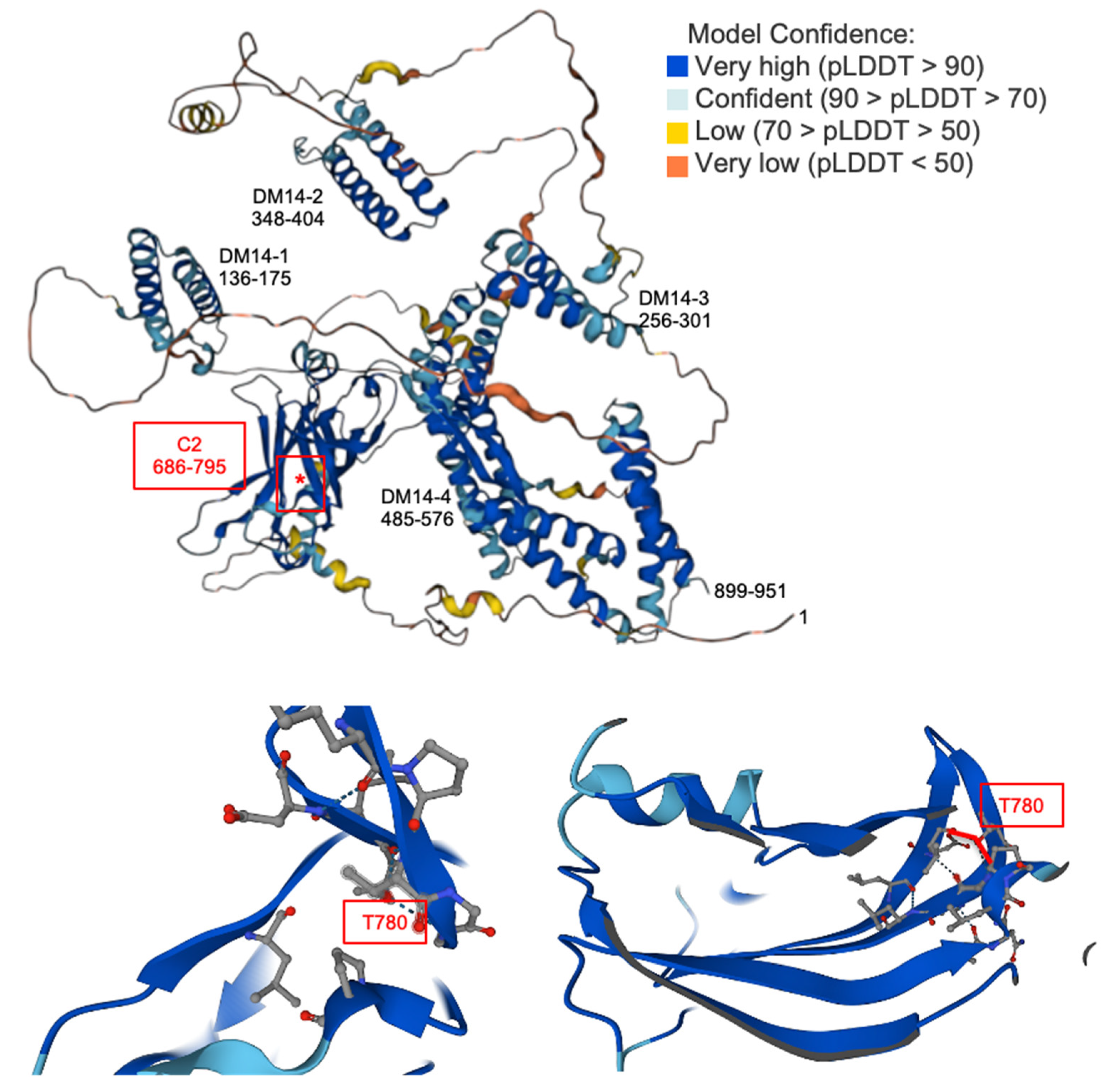
3.2. Sites of the CaMKIV-Induced Phosphorylation and Inhibition of Freud-1
3.3. Freud-1-Containing DNA Complexes
3.4. Potential Biological Roles for the CaMKIV-Dependent Inactivation of Freud-1
4. Materials and Methods
4.1. Plasmid Construction and Mutagenesis
4.2. Cell Culture and Transient Transfection
4.3. Freud-1 Protein Expression and Purification
4.4. CaMK Kinase Assay
4.5. Pulldown Assay
4.6. Immunoprecipitation (CREB)
4.7. Phospho-T780-Freud-1 Antibody
4.8. Electrophoretic Shift Mobility Assay (EMSA)
4.9. Western Blot Analysis
4.10. Luciferase and β-Galactosidase Assays
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matsuda, A.; Suzuki, Y.; Honda, G.; Muramatsu, S.; Matsuzaki, O.; Nagano, Y.; Doi, T.; Shimotohno, K.; Harada, T.; Nishida, E.; et al. Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene 2003, 22, 3307–3318. [Google Scholar] [CrossRef]
- Rogaeva, A.; Galaraga, K.; Albert, P.R. The Freud-1/CC2D1A family: Transcriptional regulators implicated in mental retardation. J. Neurosci. Res. 2007, 85, 2833–2838. [Google Scholar] [CrossRef]
- Millar, A.M.; Souslova, T.; Albert, P.R. The Freud-1/CC2D1A family: Multifunctional regulators implicated in mental retardation. In Latest Findings in Intellectual and Developmental Disabilities Research; Tan, P.U., Ed.; InTech: Rijeka, Croatia, 2012; pp. 279–302. [Google Scholar] [CrossRef]
- Basel-Vanagaite, L.; Attia, R.; Yahav, M.; Ferland, R.J.; Anteki, L.; Walsh, C.A.; Olender, T.; Straussberg, R.; Magal, N.; Taub, E.; et al. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J. Med. Genet. 2006, 43, 203–210. [Google Scholar] [CrossRef]
- Manzini, M.C.; Xiong, L.; Shaheen, R.; Tambunan, D.E.; Di Costanzo, S.; Mitisalis, V.; Tischfield, D.J.; Cinquino, A.; Ghaziuddin, M.; Christian, M.; et al. CC2D1A regulates human intellectual and social function as well as NF-kappaB signaling homeostasis. Cell Rep. 2014, 8, 647–655. [Google Scholar] [CrossRef]
- Shi, Z.Y.; Li, Y.J.; Zhang, K.J.; Gao, X.C.; Zheng, Z.J.; Han, N.; Zhang, F.C. Positive association of CC2D1A and CC2D2A gene haplotypes with mental retardation in a Han Chinese population. DNA Cell Biol. 2012, 31, 80–87. [Google Scholar] [CrossRef]
- Oaks, A.W.; Zamarbide, M.; Tambunan, D.E.; Santini, E.; Di Costanzo, S.; Pond, H.L.; Johnson, M.W.; Lin, J.; Gonzalez, D.M.; Boehler, J.F.; et al. Cc2d1a Loss of Function Disrupts Functional and Morphological Development in Forebrain Neurons Leading to Cognitive and Social Deficits. Cereb. Cortex 2017, 27, 1670–1685. [Google Scholar] [CrossRef]
- Yang, C.Y.; Yu, T.H.; Wen, W.L.; Ling, P.; Hsu, K.S. Conditional Deletion of CC2D1A Reduces Hippocampal Synaptic Plasticity and Impairs Cognitive Function through Rac1 Hyperactivation. J. Neurosci. 2019, 39, 4959–4975. [Google Scholar] [CrossRef]
- Albert, P.R.; Le Francois, B.; Millar, A.M. Transcriptional dysregulation of 5-HT1A autoreceptors in mental illness. Mol. Brain 2011, 4, 21. [Google Scholar] [CrossRef]
- Vahid-Ansari, F.; Daigle, M.; Manzini, M.C.; Tanaka, K.F.; Hen, R.; Geddes, S.D.; Beique, J.C.; James, J.; Merali, Z.; Albert, P.R. Abrogated Freud-1/Cc2d1a Repression of 5-HT1A Autoreceptors Induces Fluoxetine-Resistant Anxiety/Depression-Like Behavior. J. Neurosci. 2017, 37, 11967–11978. [Google Scholar] [CrossRef]
- Vahid-Ansari, F.; Zahrai, A.; Daigle, M.; Albert, P.R. Chronic Desipramine Reverses Deficits in Cell Activity, Norepinephrine Innervation, and Anxiety-Depression Phenotypes in Fluoxetine-Resistant cF1ko Mice. J. Neurosci. 2024, 44, e1147232023. [Google Scholar] [CrossRef]
- Le Francois, B.; Czesak, M.; Steubl, D.; Albert, P.R. Transcriptional regulation at a HTR1A polymorphism associated with mental illness. Neuropharmacology 2008, 55, 977–985. [Google Scholar] [CrossRef]
- Savitz, J.; Lucki, I.; Drevets, W.C. 5-HT(1A) receptor function in major depressive disorder. Prog. Neurobiol. 2009, 88, 17–31. [Google Scholar] [CrossRef]
- Hesselgrave, N.; Parsey, R.V. Imaging the serotonin 1A receptor using [11C]WAY100635 in healthy controls and major depression. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120004. [Google Scholar] [CrossRef]
- Richardson-Jones, J.W.; Craige, C.P.; Guiard, B.P.; Stephen, A.; Metzger, K.L.; Kung, H.F.; Gardier, A.M.; Dranovsky, A.; David, D.J.; Beck, S.G.; et al. 5-HT1A autoreceptor levels determine vulnerability to stress and response to antidepressants. Neuron 2010, 65, 40–52. [Google Scholar] [CrossRef]
- Bortolozzi, A.; Castane, A.; Semakova, J.; Santana, N.; Alvarado, G.; Cortes, R.; Ferres-Coy, A.; Fernandez, G.; Carmona, M.C.; Toth, M.; et al. Selective siRNA-mediated suppression of 5-HT1A autoreceptors evokes strong anti-depressant-like effects. Mol. Psychiatry 2012, 17, 612–623. [Google Scholar] [CrossRef]
- Ou, X.M.; Lemonde, S.; Jafar-Nejad, H.; Bown, C.D.; Goto, A.; Rogaeva, A.; Albert, P.R. Freud-1: A novel calcium-regulated repressor of the 5-HT1A receptor gene. J. Neurosci. 2003, 23, 7415–7425. [Google Scholar] [CrossRef]
- Rogaeva, A.; Albert, P.R. The mental retardation gene CC2D1A/Freud-1 encodes a long isoform that binds conserved DNA elements to repress gene transcription. Eur. J. Neurosci. 2007, 26, 965–974. [Google Scholar] [CrossRef]
- Rogaeva, A.; Ou, X.M.; Jafar-Nejad, H.; Lemonde, S.; Albert, P.R. Differential repression by freud-1/CC2D1A at a polymorphic site in the dopamine-D2 receptor gene. J. Biol. Chem. 2007, 282, 20897–20905. [Google Scholar] [CrossRef]
- Cohen, S.; Greenberg, M.E. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu. Rev. Cell Dev. Biol. 2008, 24, 183–209. [Google Scholar] [CrossRef]
- Robison, A.J. Emerging role of CaMKII in neuropsychiatric disease. Trends Neurosci. 2014, 37, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Gaudilliere, B.; Konishi, Y.; de la Iglesia, N.; Yao, G.; Bonni, A. A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron 2004, 41, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Kashani, A.H.; Qiu, Z.; Jurata, L.; Lee, S.K.; Pfaff, S.; Goebbels, S.; Nave, K.A.; Ghosh, A. Calcium activation of the LMO4 transcription complex and its role in the patterning of thalamocortical connections. J. Neurosci. 2006, 26, 8398–8408. [Google Scholar] [CrossRef] [PubMed]
- Bilbao, A.; Parkitna, J.R.; Engblom, D.; Perreau-Lenz, S.; Sanchis-Segura, C.; Schneider, M.; Konopka, W.; Westphal, M.; Breen, G.; Desrivieres, S.; et al. Loss of the Ca2+/calmodulin-dependent protein kinase type IV in dopaminoceptive neurons enhances behavioral effects of cocaine. Proc. Natl. Acad. Sci. USA 2008, 105, 17549–17554. [Google Scholar] [CrossRef] [PubMed]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef] [PubMed]
- Czesak, M.; Lemonde, S.; Peterson, E.A.; Rogaeva, A.; Albert, P.R. Cell-specific repressor or enhancer activities of Deaf-1 at a serotonin 1A receptor gene polymorphism. J. Neurosci. 2006, 26, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Rich, R.C.; Schulman, H. Substrate-directed function of calmodulin in autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1998, 273, 28424–28429. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, R.; Hanson, P.I.; Schulman, H. Multifunctional Ca2+/calmodulin-dependent protein kinase made Ca2+ independent for functional studies. Biochemistry 1990, 29, 1679–1684. [Google Scholar] [CrossRef]
- Chatila, T.; Anderson, K.A.; Ho, N.; Means, A.R. A unique phosphorylation-dependent mechanism for the activation of Ca2+/calmodulin-dependent protein kinase type IV/GR. J. Biol. Chem. 1996, 271, 21542–21548. [Google Scholar] [CrossRef] [PubMed]
- Souslova, T. Transcriptional Regulatory Mechanisms of Freud-1, a Novel Mental Retrardation Gene. Ph.D. Thesis, University of Ottawa, Ottawa, ON, Canada, 2011. [Google Scholar] [CrossRef]
- Lemonde, S.; Rogaeva, A.; Albert, P.R. Cell type-dependent recruitment of trichostatin A-sensitive repression of the human 5-HT1A receptor gene. J. Neurochem. 2004, 88, 857–868. [Google Scholar] [CrossRef]
- Impey, S.; Fong, A.L.; Wang, Y.; Cardinaux, J.R.; Fass, D.M.; Obrietan, K.; Wayman, G.A.; Storm, D.R.; Soderling, T.R.; Goodman, R.H. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron 2002, 34, 235–244. [Google Scholar] [CrossRef]
- Kasahara, J.; Fukunaga, K.; Miyamoto, E. Activation of calcium/calmodulin-dependent protein kinase IV in long term potentiation in the rat hippocampal CA1 region. J. Biol. Chem. 2001, 276, 24044–24050. [Google Scholar] [CrossRef] [PubMed]
- Wayman, G.A.; Lee, Y.S.; Tokumitsu, H.; Silva, A.J.; Soderling, T.R. Calmodulin-kinases: Modulators of neuronal development and plasticity. Neuron 2008, 59, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Maeda, R.; Suzuki, R.; Suzuki, A.; Nomoto, M.; Toyoda, H.; Wu, L.J.; Xu, H.; Zhao, M.G.; Ueda, K.; et al. Upregulation of calcium/calmodulin-dependent protein kinase IV improves memory formation and rescues memory loss with aging. J. Neurosci. 2008, 28, 9910–9919. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, E.E.; Joseph, J.D.; MacDonald, J.A.; Kane, C.D.; Haystead, T.A.; Means, A.R. Proteomic analysis of calcium/calmodulin-dependent protein kinase I and IV in vitro substrates reveals distinct catalytic preferences. J. Biol. Chem. 2003, 278, 10516–10522. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ito, A.; Kane, C.D.; Liao, T.S.; Bolger, T.A.; Lemrow, S.M.; Means, A.R.; Yao, T.P. The modular nature of histone deacetylase HDAC4 confers phosphorylation-dependent intracellular trafficking. J. Biol. Chem. 2001, 276, 35042–35048. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Kwon, Y.G.; Taing, M.; Lawrence, D.S.; Edelman, A.M. Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J. Biol. Chem. 1998, 273, 3166–3172. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T. Neuronal Ca2+/calmodulin-dependent protein kinase II—Discovery, progress in a quarter of a century, and perspective: Implication for learning and memory. Biol. Pharm. Bull. 2005, 28, 1342–1354. [Google Scholar] [CrossRef]
- Nakamura, A.; Naito, M.; Arai, H.; Fujita, N. Mitotic phosphorylation of Aki1 at Ser208 by cyclin B1-Cdk1 complex. Biochem. Biophys. Res. Commun. 2010, 393, 872–876. [Google Scholar] [CrossRef]
- Hadjighassem, M.R.; Austin, M.C.; Szewczyk, B.; Daigle, M.; Stockmeier, C.A.; Albert, P.R. Human Freud-2/CC2D1B: A novel repressor of postsynaptic serotonin-1A receptor expression. Biol. Psychiatry 2009, 66, 214–222. [Google Scholar] [CrossRef]
- Hadjighassem, M.R.; Galaraga, K.; Albert, P.R. Freud-2/CC2D1B mediates dual repression of the serotonin-1A receptor gene. Eur. J. Neurosci. 2011, 33, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Souslova, T.; Miredin, K.; Millar, A.M.; Albert, P.R. Recruitment by the Repressor Freud-1 of Histone Deacetylase-Brg1 Chromatin Remodeling Complexes to Strengthen HTR1A Gene Repression. Mol. Neurobiol. 2017, 54, 8263–8277. [Google Scholar] [CrossRef] [PubMed]
- Ronan, J.L.; Wu, W.; Crabtree, G.R. From neural development to cognition: Unexpected roles for chromatin. Nat. Rev. Genet. 2013, 14, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Raingo, J.; Chen, Z.J.; Kavalali, E.T. Cc2d1a, a C2 domain containing protein linked to nonsyndromic mental retardation, controls functional maturation of central synapses. J. Neurophysiol. 2011, 105, 1506–1515. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B.; Albert, P.R.; Rogaeva, A.; Fitzgibbon, H.; May, W.L.; Rajkowska, G.; Miguel-Hidalgo, J.J.; Stockmeier, C.A.; Woolverton, W.L.; Kyle, P.B.; et al. Decreased expression of Freud-1/CC2D1A, a transcriptional repressor of the 5-HT1A receptor, in the prefrontal cortex of subjects with major depression. Int. J. Neuropsychopharmacol. 2010, 13, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Goswami, D.B.; May, W.L.; Stockmeier, C.A.; Austin, M.C. Transcriptional expression of serotonergic regulators in laser-captured microdissected dorsal raphe neurons of subjects with major depressive disorder: Sex-specific differences. J. Neurochem. 2010, 112, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Iyo, A.H.; Kieran, N.; Chandran, A.; Albert, P.R.; Wicks, I.; Bissette, G.; Austin, M.C. Differential regulation of the serotonin 1 A transcriptional modulators five prime repressor element under dual repression-1 and nuclear-deformed epidermal autoregulatory factor by chronic stress. Neuroscience 2009, 163, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Miyagishi, H.; Tsuji, M.; Miyagawa, K.; Kurokawa, K.; Mochida-Saito, A.; Takahashi, K.; Kosuge, Y.; Ishige, K.; Takeda, H. Possible role of transcriptional regulation of 5-HT(1A) receptor in the midbrain on unadaptation to stress in mice. Brain Res. 2022, 1783, 147859. [Google Scholar] [CrossRef]
- Szewczyk, B.; Kotarska, K.; Daigle, M.; Misztak, P.; Sowa-Kucma, M.; Rafalo, A.; Curzytek, K.; Kubera, M.; Basta-Kaim, A.; Nowak, G.; et al. Stress-induced alterations in 5-HT1A receptor transcriptional modulators NUDR and Freud-1. Int. J. Neuropsychopharmacol. 2014, 17, 1763–1775. [Google Scholar] [CrossRef]
- Albert, P.R. Transcriptional regulation of the 5-HT1A receptor: Implications for mental illness. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2402–2415. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.R.; Lemonde, S. 5-HT1A receptors, gene repression, and depression: Guilt by association. Neuroscientist 2004, 10, 575–593. [Google Scholar] [CrossRef]
- Welner, S.A.; De Montigny, C.; Desroches, J.; Desjardins, P.; Suranyi-Cadotte, B.E. Autoradiographic quantification of serotonin1A receptors in rat brain following antidepressant drug treatment. Synapse 1989, 4, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, R.J.; McMonagle-Strucko, K. Alteration of 5-HT1A receptor binding sites following chronic treatment with ipsapirone measured by quantitative autoradiography. Synapse 1992, 12, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Afkhami-Dastjerdian, S.; Reader, T.A. Effects of a chronic lithium treatment on cortical serotonin uptake sites and 5-HT1A receptors. Neurochem. Res. 1997, 22, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Morley-Fletcher, S.; Darnaudery, M.; Mocaer, E.; Froger, N.; Lanfumey, L.; Laviola, G.; Casolini, P.; Zuena, A.R.; Marzano, L.; Hamon, M.; et al. Chronic treatment with imipramine reverses immobility behaviour, hippocampal corticosteroid receptors and cortical 5-HT(1A) receptor mRNA in prenatally stressed rats. Neuropharmacology 2004, 47, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.A.; Milak, M.S.; Delorenzo, C.; Ogden, R.T.; Huang, Y.Y.; Mann, J.J.; Parsey, R.V. Antidepressant treatment reduces serotonin-1A autoreceptor binding in major depressive disorder. Biol. Psychiatry 2013, 74, 26–31. [Google Scholar] [CrossRef]
- Pillai, R.L.I.; Zhang, M.; Yang, J.; Boldrini, M.; Mann, J.J.; Oquendo, M.A.; Parsey, R.V.; DeLorenzo, C. Will imaging individual raphe nuclei in males with major depressive disorder enhance diagnostic sensitivity and specificity? Depress. Anxiety 2018, 35, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Milak, M.S.; Pantazatos, S.; Rashid, R.; Zanderigo, F.; DeLorenzo, C.; Hesselgrave, N.; Ogden, R.T.; Oquendo, M.A.; Mulhern, S.T.; Miller, J.M.; et al. Higher 5-HT1A autoreceptor binding as an endophenotype for major depressive disorder identified in high risk offspring—A pilot study. Psychiatry Res. Neuroimaging 2018, 276, 15–23. [Google Scholar] [CrossRef]
- Parsey, R.V.; Olvet, D.M.; Oquendo, M.A.; Huang, Y.Y.; Ogden, R.T.; Mann, J.J. Higher 5-HT1A receptor binding potential during a major depressive episode predicts poor treatment response: Preliminary data from a naturalistic study. Neuropsychopharmacology 2006, 31, 1745–1749. [Google Scholar] [CrossRef]
- Celada, P.; Puig, M.V.; Casanovas, J.M.; Guillazo, G.; Artigas, F. Control of dorsal raphe serotonergic neurons by the medial prefrontal cortex: Involvement of serotonin-1A, GABA(A), and glutamate receptors. J. Neurosci. 2001, 21, 9917–9929. [Google Scholar] [CrossRef] [PubMed]
- Warden, M.R.; Selimbeyoglu, A.; Mirzabekov, J.J.; Lo, M.; Thompson, K.R.; Kim, S.Y.; Adhikari, A.; Tye, K.M.; Frank, L.M.; Deisseroth, K. A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 2012, 492, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.R.; Vahid-Ansari, F. The 5-HT1A receptor: Signaling to behavior. Biochimie 2019, 161, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Takebe, Y.; Seiki, M.; Fujisawa, J.; Hoy, P.; Yokota, K.; Arai, K.; Yoshida, M.; Arai, N. SR alpha promoter: An efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol. Cell. Biol. 1988, 8, 466–472. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| 5′-DRE | 5′-AGATGGCACTCTAAAACATTTGCCACA |
| 3′-DRE | 5′-AGGTGGCGACATAAAACCTCATTGCTTAGAACT |
| 5′+3′-DRE | 5′-AGATGGCACTCTAAAACATTTGCCACAAGGTGGCGACATAAAACCTCATTGCTTAGAACT |
| hD2DR DRE | 5′-CGCGTGGGATAAGCAAGCCCTTCTTGTAAAAGTTTTAAGAACAATACA |
| E2F | 5′-TATAGTGTACTCTACTATTCTGCTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galaraga, K.; Rogaeva, A.; Biniam, N.; Daigle, M.; Albert, P.R. CaMKIV-Mediated Phosphorylation Inactivates Freud-1/CC2D1A Repression for Calcium-Dependent 5-HT1A Receptor Gene Induction. Int. J. Mol. Sci. 2024, 25, 6194. https://doi.org/10.3390/ijms25116194
Galaraga K, Rogaeva A, Biniam N, Daigle M, Albert PR. CaMKIV-Mediated Phosphorylation Inactivates Freud-1/CC2D1A Repression for Calcium-Dependent 5-HT1A Receptor Gene Induction. International Journal of Molecular Sciences. 2024; 25(11):6194. https://doi.org/10.3390/ijms25116194
Chicago/Turabian StyleGalaraga, Kimberly, Anastasia Rogaeva, Nathan Biniam, Mireille Daigle, and Paul R. Albert. 2024. "CaMKIV-Mediated Phosphorylation Inactivates Freud-1/CC2D1A Repression for Calcium-Dependent 5-HT1A Receptor Gene Induction" International Journal of Molecular Sciences 25, no. 11: 6194. https://doi.org/10.3390/ijms25116194
APA StyleGalaraga, K., Rogaeva, A., Biniam, N., Daigle, M., & Albert, P. R. (2024). CaMKIV-Mediated Phosphorylation Inactivates Freud-1/CC2D1A Repression for Calcium-Dependent 5-HT1A Receptor Gene Induction. International Journal of Molecular Sciences, 25(11), 6194. https://doi.org/10.3390/ijms25116194