
The Skin–Brain Axis: From UV and Pigmentation to Behaviour Modulation
Abstract
1. Introduction
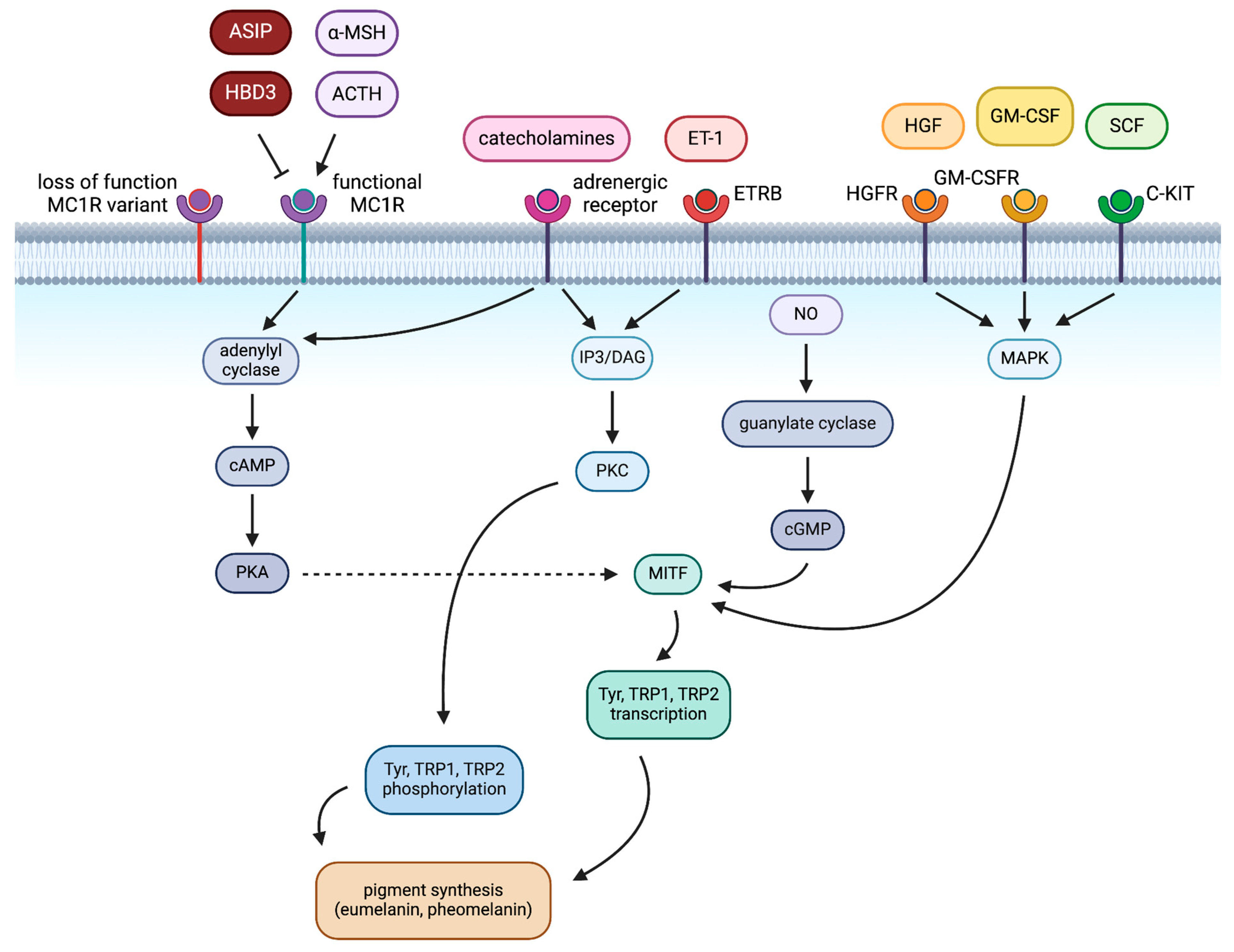
2. Pigmentation in Melanocytes
2.1. Extrinsic Regulation of Pigmentation in Melanocytes
2.2. Intrinsic Regulation of Pigmentation in Melanocytes
3. Pigmentation in the CNS—Role of Neuromelanin
3.1. Neuromelanin in Dopaminergic Neurons
3.2. Neuromelanin in Parkinson’s Disease
3.3. MC1R in Relation to Parkinson’s Disease
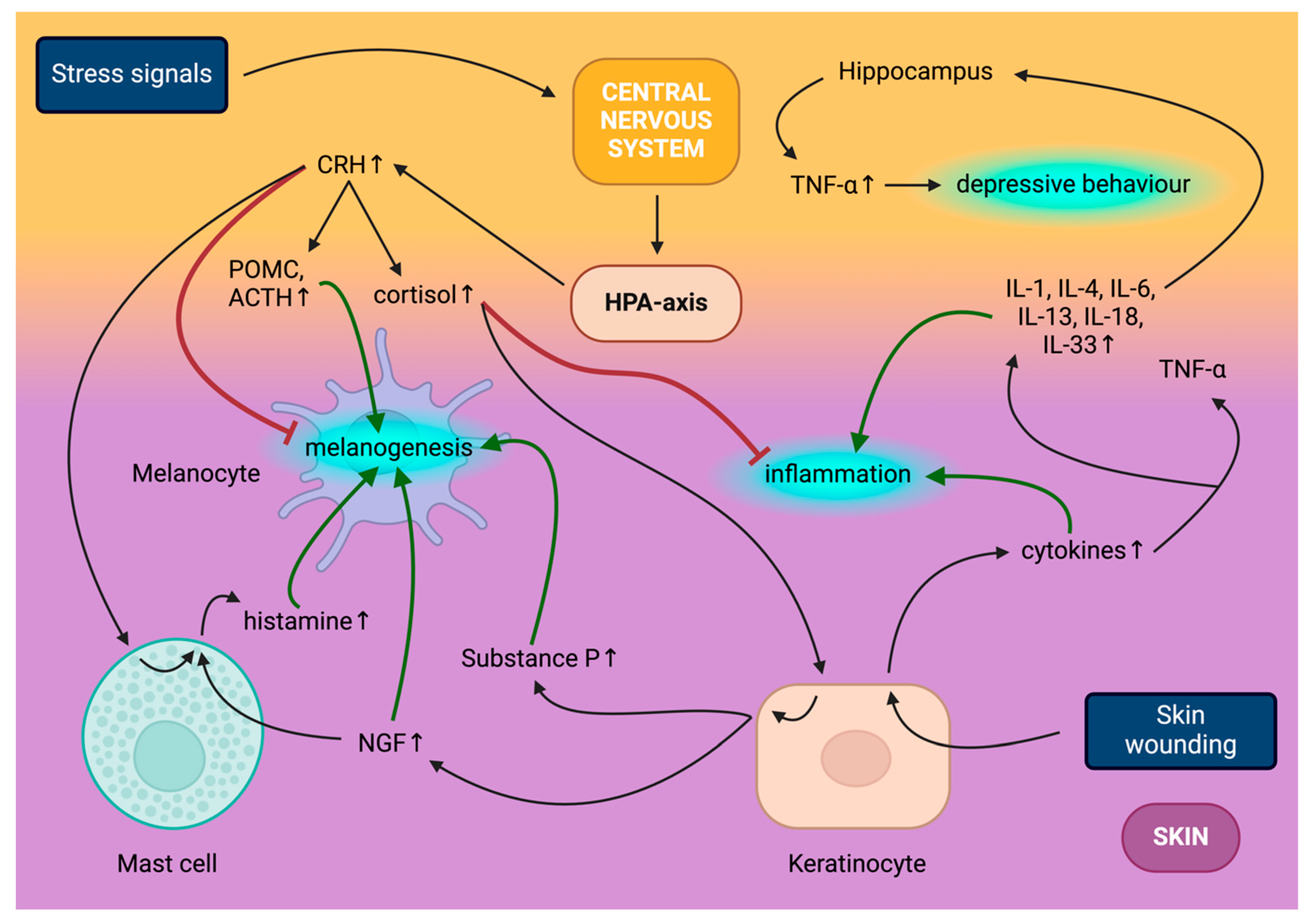
4. Skin–Brain Axis Basics—Involvement of the Skin in the HPA Axis
4.1. Central HPA Axis
4.2. Peripheral HPA Axis
5. Skin Effects on the CNS—Impact of Hair Colour on Pain
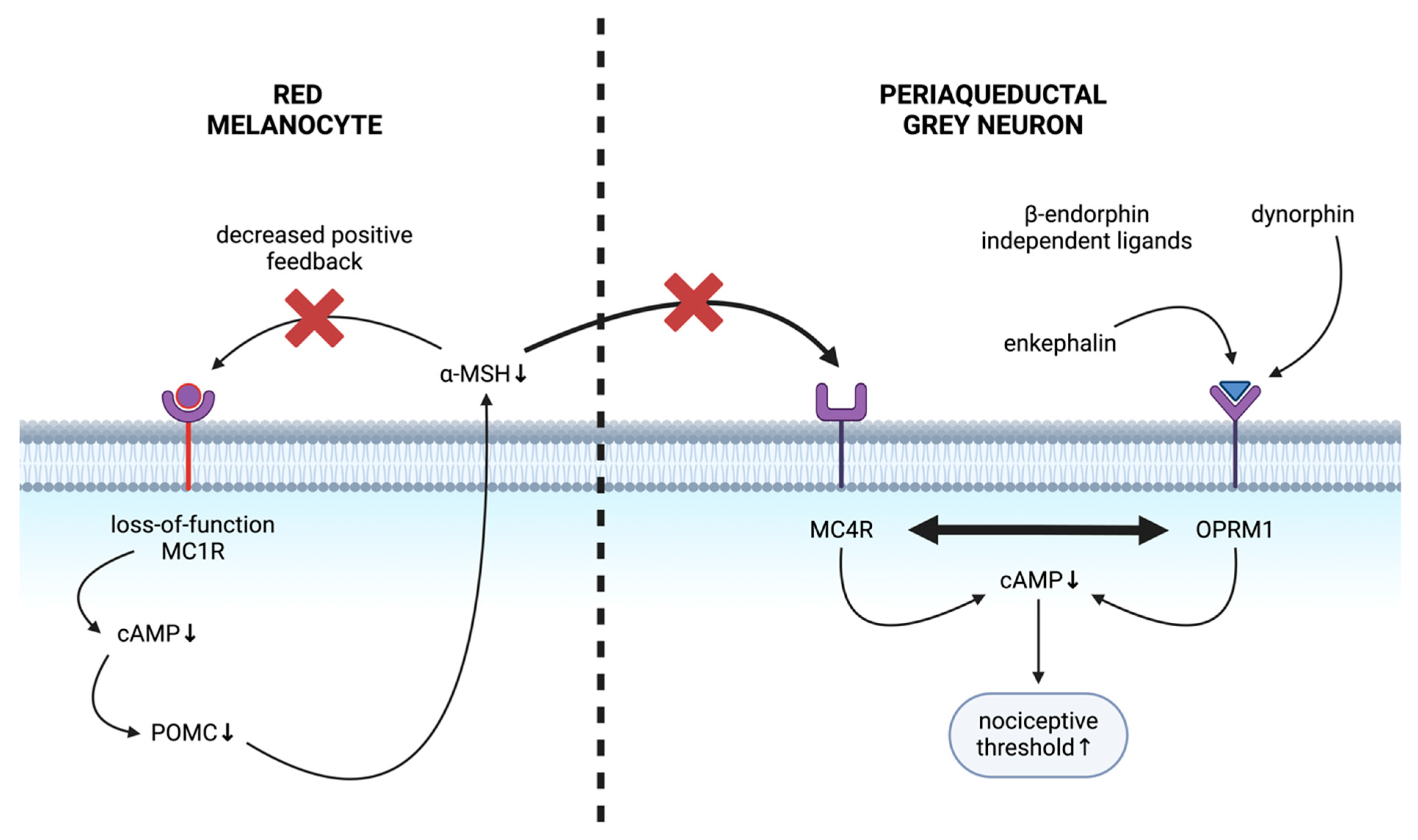
5.1. MC1R: Skin–Brain Link
5.2. MC1R and Pain
5.3. Mechanism of Altered Nociception in Red-Haired Background
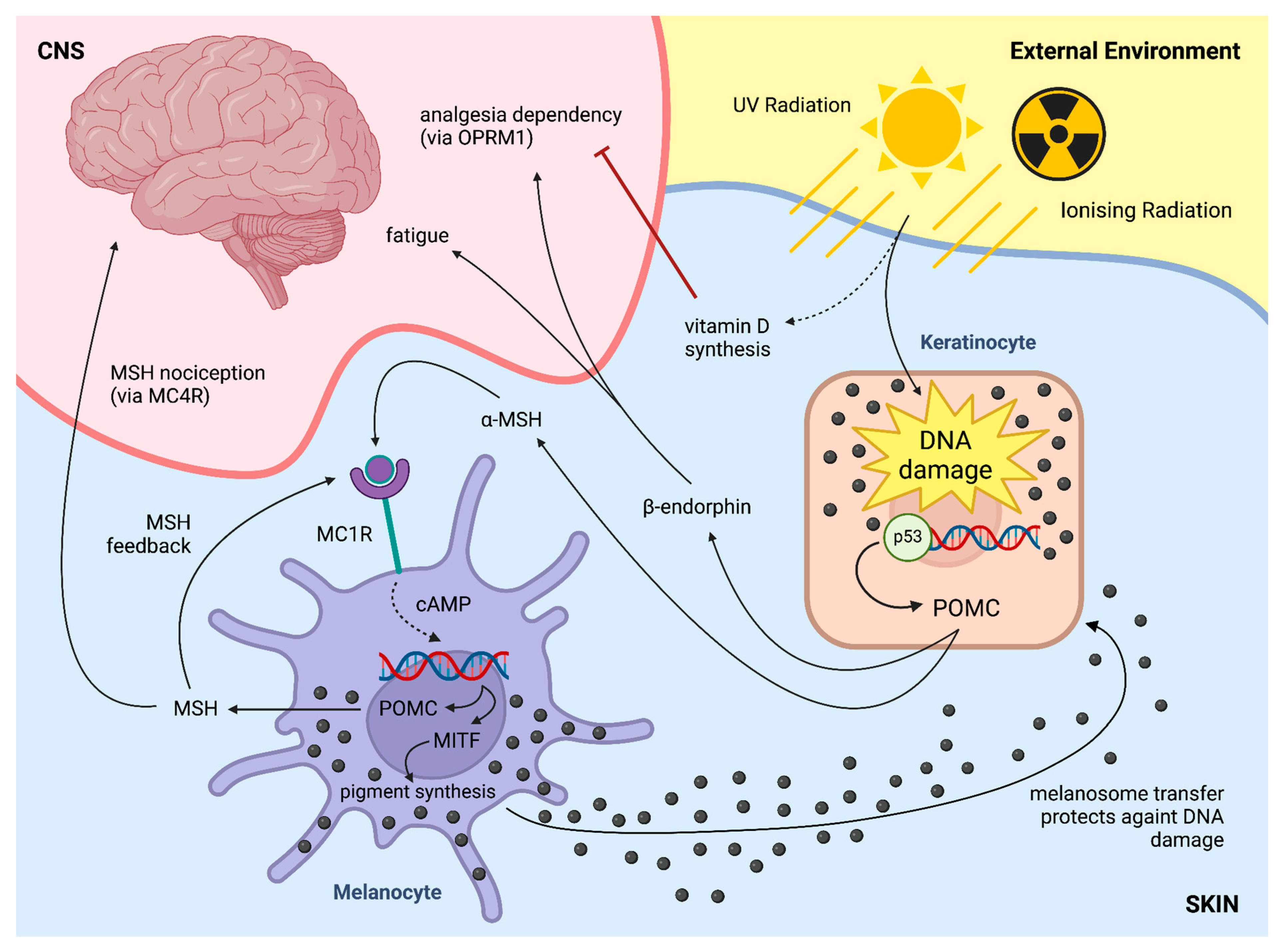
6. Skin Effects on the CNS—Skin-Derived Endogenous Opioids Driving Addiction
7. Future Directions
Funding
Conflicts of Interest
References
- Lopez-Ojeda, W.; Pandey, A.; Alhajj, M.; Oakley, A.M. Anatomy, Skin (Integument). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Slominski, A.; Wortsman, J.; Tuckey, R.C.; Paus, R. Differential expression of hpa axis homolog in the skin. Mol. Cell. Endocrinol. 2007, 265–266, 143–149. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV Light Touches the Brain and Endocrine System Through Skin, and Why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef]
- Videira, I.F.d.S.; Moura, D.F.L.; Magina, S. Mechanisms Regulating Melanogenesis. An. Bras. Dermatol. 2013, 88, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, N.G.; Chaplin, G. Human Skin Pigmentation, Migration and Disease Susceptibility. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Maynard, R.L.; Downes, N. (Eds.) Chapter 24—The Skin or the Integument. In Anatomy and Histology of the Laboratory Rat in Toxicology and Biomedical Research; Academic Press: Cambridge, MA, USA, 2019; pp. 303–315. ISBN 978-0-12-811837-5. [Google Scholar]
- Yamaguchi, Y.; Hearing, V.J. Melanocytes and Their Diseases. Cold Spring Harb. Perspect. Med. 2014, 4, a017046. [Google Scholar] [CrossRef] [PubMed]
- Serre, C.; Busuttil, V.; Botto, J.-M. Intrinsic and extrinsic regulation of human skin melanogenesis and pigmentation. Int. J. Cosmet. Sci. 2018, 40, 328–347. [Google Scholar] [CrossRef] [PubMed]
- Domingues, L.; Hurbain, I.; Gilles-Marsens, F.; Sirés-Campos, J.; André, N.; Dewulf, M.; Romao, M.; Viaris de Lesegno, C.; Macé, A.-S.; Blouin, C.; et al. Coupling of Melanocyte Signaling and Mechanics by Caveolae Is Required for Human Skin Pigmentation. Nat. Commun. 2020, 11, 2988. [Google Scholar] [CrossRef]
- Raposo, G.; Marks, M.S. Melanosomes—Dark Organelles Enlighten Endosomal Membrane Transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786. [Google Scholar] [CrossRef]
- Hermann, A.L.; Fell, G.L.; Kemény, L.V.; Fung, C.Y.; Held, K.D.; Biggs, P.J.; Rivera, P.D.; Bilbo, S.D.; Igras, V.; Willers, H.; et al. β-Endorphin Mediates Radiation Therapy Fatigue. Sci. Adv. 2022, 8, eabn6025. [Google Scholar] [CrossRef]
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central Role of P53 in the Suntan Response and Pathologic Hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar] [CrossRef]
- Fell, G.L.; Robinson, K.C.; Mao, J.; Woolf, C.J.; Fisher, D.E. Skin β-Endorphin Mediates Addiction to Ultraviolet Light. Cell 2014, 157, 1527–1534. [Google Scholar] [CrossRef]
- Swope, V.B.; Abdel-Malek, Z.A. MC1R: Front and Center in the Bright Side of Dark Eumelanin and DNA Repair. Int. J. Mol. Sci. 2018, 19, 2667. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.M.; Lo, J.; Fisher, D.E. How Does Pheomelanin Synthesis Contribute to Melanomagenesis? BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 672–676. [Google Scholar] [CrossRef]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An Ultraviolet-Radiation-Independent Pathway to Melanoma Carcinogenesis in the Red Hair/Fair Skin Background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef]
- Logesh, R.; Prasad, S.R.; Chipurupalli, S.; Robinson, N.; Mohankumar, S.K. Natural Tyrosinase Enzyme Inhibitors: A Path from Melanin to Melanoma and Its Reported Pharmacological Activities. Biochim. Biophys. Acta BBA—Rev. Cancer 2023, 1878, 188968. [Google Scholar] [CrossRef]
- Maranduca, M.A.; Branisteanu, D.; Serban, D.N.; Branisteanu, D.C.; Stoleriu, G.; Manolache, N.; Serban, I.L. Synthesis and Physiological Implications of Melanic Pigments. Oncol. Lett. 2019, 17, 4183–4187. [Google Scholar] [CrossRef]
- Dall’Olmo, L.; Papa, N.; Surdo, N.C.; Marigo, I.; Mocellin, S. Alpha-Melanocyte Stimulating Hormone (α-MSH): Biology, Clinical Relevance and Implication in Melanoma. J. Transl. Med. 2023, 21, 562. [Google Scholar] [CrossRef]
- Chelakkot, V.S.; Thomas, K.; Romigh, T.; Fong, A.; Li, L.; Ronen, S.; Chen, S.; Funchain, P.; Ni, Y.; Arbesman, J. MC1R Signaling through the cAMP-CREB/ATF-1 and ERK-NFκB Pathways Accelerates G1/S Transition Promoting Breast Cancer Progression. Npj Precis. Oncol. 2023, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The Cyclic AMP Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Chen, J.; Lu, J.; Yi, L.; Tong, X.; Kang, L.; Pei, S.; Ouyang, Y.; Jiang, L.; Ding, Y.; et al. Roles of Inflammation Factors in Melanogenesis. Mol. Med. Rep. 2020, 21, 1421–1430. [Google Scholar] [CrossRef]
- Guo, H.; Xing, Y.; Liu, Y.; Luo, Y.; Deng, F.; Yang, T.; Yang, K.; Li, Y. Wnt/β-Catenin Signaling Pathway Activates Melanocyte Stem Cells In Vitro and In Vivo. J. Dermatol. Sci. 2016, 83, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Nasti, T.H.; Timares, L. Invited Review MC1R, Eumelanin and Pheomelanin: Their Role in Determining the Susceptibility to Skin Cancer. Photochem. Photobiol. 2015, 91, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Krainc, T.; Monje, M.H.G.; Kinsinger, M.; Bustos, B.I.; Lubbe, S.J. Melanin and Neuromelanin: Linking Skin Pigmentation and Parkinson’s Disease. Mov. Disord. 2023, 38, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Kim, M.; Lee, S.H.; Kim, K.D. The Function of Autophagy as a Regulator of Melanin Homeostasis. Cells 2022, 11, 2085. [Google Scholar] [CrossRef] [PubMed]
- Murase, D.; Hachiya, A.; Takano, K.; Hicks, R.; Visscher, M.O.; Kitahara, T.; Hase, T.; Takema, Y.; Yoshimori, T. Autophagy Has a Significant Role in Determining Skin Color by Regulating Melanosome Degradation in Keratinocytes. J. Investig. Dermatol. 2013, 133, 2416–2424. [Google Scholar] [CrossRef] [PubMed]
- Białczyk, A.; Czajkowski, R. Autophagy in Psoriasis and Vitiligo. Forum Dermatol. 2022, 8, 153–157. [Google Scholar] [CrossRef]
- Qiao, Z.; Wang, X.; Xiang, L.; Zhang, C. Dysfunction of Autophagy: A Possible Mechanism Involved in the Pathogenesis of Vitiligo by Breaking the Redox Balance of Melanocytes. Oxid. Med. Cell. Longev. 2016, 2016, e3401570. [Google Scholar] [CrossRef]
- Casalou, C.; Moreiras, H.; Mayatra, J.M.; Fabre, A.; Tobin, D.J. Loss of ‘Epidermal Melanin Unit’ Integrity in Human Skin During Melanoma-Genesis. Front. Oncol. 2022, 12, 878336. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-H.; Liu, L.-H.; Chang, C.-C.; Gao, R.; Leung, C.-H.; Ma, D.-L.; David Wang, H.-M. Silencing Stem Cell Factor Gene in Fibroblasts to Regulate Paracrine Factor Productions and Enhance C-Kit Expression in Melanocytes on Melanogenesis. Int. J. Mol. Sci. 2018, 19, 1475. [Google Scholar] [CrossRef]
- Yuan, X.H.; Jin, Z.H. Paracrine Regulation of Melanogenesis. Br. J. Dermatol. 2018, 178, 632–639. [Google Scholar] [CrossRef]
- Haining, R.L.; Achat-Mendes, C. Neuromelanin, One of the Most Overlooked Molecules in Modern Medicine, Is Not a Spectator. Neural Regen. Res. 2017, 12, 372–375. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Riederer, P. Melanins. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 978-0-12-809324-5. [Google Scholar]
- Elstner, M.; Müller, S.K.; Leidolt, L.; Laub, C.; Krieg, L.; Schlaudraff, F.; Liss, B.; Morris, C.; Turnbull, D.M.; Masliah, E.; et al. Neuromelanin, Neurotransmitter Status and Brainstem Location Determine the Differential Vulnerability of Catecholaminergic Neurons to Mitochondrial DNA Deletions. Mol. Brain 2011, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Diaz-Corrales, F.J.; Ogawa, N. Quinone Formation as Dopaminergic Neuron-Specific Oxidative Stress in the Pathogenesis of Sporadic Parkinson’s Disease and Neurotoxin-Induced Parkinsonism. Acta Med. Okayama 2004, 58, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Umek, N.; Geršak, B.; Vintar, N.; Šoštarič, M.; Mavri, J. Dopamine Autoxidation Is Controlled by Acidic pH. Front. Mol. Neurosci. 2018, 11, 467. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ding, Y.; Cagniard, B.; Van Laar, A.D.; Mortimer, A.; Chi, W.; Hastings, T.G.; Kang, U.J.; Zhuang, X. Unregulated Cytosolic Dopamine Causes Neurodegeneration Associated with Oxidative Stress in Mice. J. Neurosci. 2008, 28, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Maruyama, W.; Riederer, P. Melanin. In Encyclopedia of Movement Disorders; Kompoliti, K., Metman, L.V., Eds.; Academic Press: Oxford, UK, 2010; pp. 169–171. ISBN 978-0-12-374105-9. [Google Scholar]
- Ito, S.; Wakamatsu, K.; Zecca, L. Structure and Function of Neuromelanin. In Catecholamine Research: From Molecular Insights to Clinical Medicine; Nagatsu, T., Nabeshima, T., McCarty, R., Goldstein, D.S., Eds.; Advances in Behavioral Biology; Springer: Boston, MA, USA, 2002; pp. 269–272. ISBN 978-1-4757-3538-3. [Google Scholar]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the Human Substantia Nigra: An Update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. Chapter 7—Dopamine Oxidation to Neuromelanin and Neurotoxic Metabolites. In Clinical Studies and Therapies in Parkinson’s Disease; Segura-Aguilar, J., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 213–227. ISBN 978-0-12-822120-4. [Google Scholar]
- Tondo, G.; Comi, C.; Naldi, A.; de Natale, E.R.; Politis, M. Chapter 12—Neuroimaging in Multiple System Atrophy. In Neuroimaging in Parkinson’s Disease and Related Disorders; Politis, M., Wilson, H., De Natale, E.R., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 311–354. ISBN 978-0-12-821651-4. [Google Scholar]
- Volpicelli-Daley, L. Neuromelanin as a Nidus for Neurodegeneration. Brain 2023, 146, 4794–4795. [Google Scholar] [CrossRef]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain Tyrosinase Overexpression Implicates Age-Dependent Neuromelanin Production in Parkinson’s Disease Pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [PubMed]
- Capucciati, A.; Zucca, F.A.; Monzani, E.; Zecca, L.; Casella, L.; Hofer, T. Interaction of Neuromelanin with Xenobiotics and Consequences for Neurodegeneration; Promising Experimental Models. Antioxidants 2021, 10, 824. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, M.; Calero, O. The Neuromelanin Paradox and Its Dual Role in Oxidative Stress and Neurodegeneration. Antioxidants 2021, 10, 124. [Google Scholar] [CrossRef]
- Lees, A.J.; Selikhova, M.; Andrade, L.A.; Duyckaerts, C. The Black Stuff and Konstantin Nikolaevich Tretiakoff. Mov. Disord. Off. J. Mov. Disord. Soc. 2008, 23, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Vila, M. Neuromelanin, Aging, and Neuronal Vulnerability in Parkinson’s Disease. Mov. Disord. 2019, 34, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Azcorra, M.; Gaertner, Z.; Davidson, C.; He, Q.; Kim, H.; Nagappan, S.; Hayes, C.K.; Ramakrishnan, C.; Fenno, L.; Kim, Y.S.; et al. Unique Functional Responses Differentially Map onto Genetic Subtypes of Dopamine Neurons. Nat. Neurosci. 2023, 26, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Rodriguez, E.; Vegas-Suarez, S.; Morera-Herreras, T.; De Deurwaerdere, P.; Miguelez, C. The Noradrenergic System in Parkinson’s Disease. Front. Pharmacol. 2020, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Faits, M.C.; Zhang, C.; Soto, F.; Kerschensteiner, D. Dendritic Mitochondria Reach Stable Positions during Circuit Development. eLife 2016, 5, e11583. [Google Scholar] [CrossRef] [PubMed]
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and Cellular Pathways Contributing to Brain Aging. Behav. Brain Funct. 2021, 17, 6. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Yi, H.; Yamaoka, Y.; Shamoto-Nagai, M.; Akao, Y.; Gerlach, M.; Tanaka, M.; Riederer, P. Neuromelanin Selectively Induces Apoptosis in Dopaminergic SH-SY5Y Cells by Deglutathionylation in Mitochondria: Involvement of the Protein and Melanin Component. J. Neurochem. 2008, 105, 2489–2500. [Google Scholar] [CrossRef]
- Korzhevskii, D.E.; Kirik, O.V.; Guselnikova, V.V.; Tsyba, D.L.; Fedorova, E.A.; Grigorev, I.P. Changes in Cytoplasmic and Extracellular Neuromelanin in Human Substantia Nigra with Normal Aging. Eur. J. Histochem. EJH 2021, 65, 3283. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wen, Y.; Al-Kuwari, N.; Chen, X. Association Between Parkinson’s Disease and Melanoma: Putting the Pieces Together. Front. Aging Neurosci. 2020, 12, 60. [Google Scholar] [CrossRef]
- Flores-Torres, M.H.; Bjornevik, K.; Zhang, X.; Gao, X.; Hung, A.Y.; Schwarzschild, M.A.; Chen, X.; Ascherio, A. Hair Color, Family History of Melanoma, and the Risk of Parkinson’s Disease: An Analysis Update. Park. Relat. Disord. 2024, 119, 105965. [Google Scholar] [CrossRef]
- Chen, X.; Chen, H.; Cai, W.; Maguire, M.; Ya, B.; Zuo, F.; Logan, R.; Li, H.; Robinson, K.; Vanderburg, C.R.; et al. The Melanoma-Linked “Redhead” MC1R Influences Dopaminergic Neuron Survival. Ann. Neurol. 2017, 81, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ortiz, M.E.; Seo, Y.; Posavi, M.; Carceles Cordon, M.; Clark, E.; Jain, N.; Charan, R.; Gallagher, M.D.; Unger, T.L.; Amari, N.; et al. GPNMB Confers Risk for Parkinson’s Disease through Interaction with Alpha-Synuclein. Science 2022, 377, eabk0637. [Google Scholar] [CrossRef] [PubMed]
- Mencacci, N.E.; Isaias, I.U.; Reich, M.M.; Ganos, C.; Plagnol, V.; Polke, J.M.; Bras, J.; Hersheson, J.; Stamelou, M.; Pittman, A.M.; et al. Parkinson’s Disease in GTP Cyclohydrolase 1 Mutation Carriers. Brain 2014, 137, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Saw, W.T.; Ho, P.G.H.; Zhang, Z.W.; Zeng, L.; Chang, Y.Y.; Sun, A.X.Y.; Ma, D.R.; Wang, H.Y.; Zhou, L.; et al. The Role of Tyrosine Hydroxylase–Dopamine Pathway in Parkinson’s Disease Pathogenesis. Cell. Mol. Life Sci. 2022, 79, 599. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Nishiyama, S.; Zhou, F.; Lin, S.H.; Srivastava, A.; Su, C.; Xu, Y.; Peng, W.; Levy, M.; Schwarzschild, M.; et al. Peripheral MC1R Activation Modulates Immune Responses and Is Neuroprotective in a Mouse Model of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2023, 18, 704–717. [Google Scholar] [CrossRef]
- Mykicki, N.; Herrmann, A.M.; Schwab, N.; Deenen, R.; Sparwasser, T.; Limmer, A.; Wachsmuth, L.; Klotz, L.; Köhrer, K.; Faber, C.; et al. Melanocortin-1 Receptor Activation Is Neuroprotective in Mouse Models of Neuroinflammatory Disease. Sci. Transl. Med. 2016, 8, 362ra146. [Google Scholar] [CrossRef] [PubMed]
- Galiano-Landeira, J.; Torra, A.; Vila, M.; Bové, J. CD8 T Cell Nigral Infiltration Precedes Synucleinopathy in Early Stages of Parkinson’s Disease. Brain J. Neurol. 2020, 143, 3717–3733. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Luo, X.; Wu, X.; Zhang, T.; Gu, L.; Wang, Y.; Gao, M.; Cheng, Y.; Xie, Z. Activation of the Melanocortin-1 Receptor by NDP-MSH Attenuates Oxidative Stress and Neuronal Apoptosis through PI3K/Akt/Nrf2 Pathway after Intracerebral Hemorrhage in Mice. Oxid. Med. Cell. Longev. 2020, 2020, 8864100. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fu, S.; Liu, Y.; Luo, H.; Li, F.; Wang, Y.; Gao, M.; Cheng, Y.; Xie, Z. NDP-MSH Binding Melanocortin-1 Receptor Ameliorates Neuroinflammation and BBB Disruption through CREB/Nr4a1/NF-κB Pathway after Intracerebral Hemorrhage in Mice. J. Neuroinflamm. 2019, 16, 192. [Google Scholar] [CrossRef]
- Chen, X.; Feng, D.; Schwarzschild, M.A.; Gao, X. Red Hair, MC1R Variants, and Risk for Parkinson’s Disease—A Meta-analysis. Ann. Clin. Transl. Neurol. 2017, 4, 212–216. [Google Scholar] [CrossRef]
- Dean, D.N.; Lee, J.C. Linking Parkinson’s Disease and Melanoma: Interplay between α-Synuclein and Pmel17 Amyloid Formation. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Chocarro, J.; Rico, A.J.; Ariznabarreta, G.; Roda, E.; Honrubia, A.; Collantes, M.; Peñuelas, I.; Vázquez, A.; Rodríguez-Pérez, A.I.; Labandeira-García, J.L.; et al. Neuromelanin Accumulation Drives Endogenous Synucleinopathy in Non-Human Primates. Brain J. Neurol. 2023, 146, 5000–5014. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease Gene SNCA: Evolutionary and Structural Insights with Pathological Implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Schirinzi, T.; Di Lazzaro, G.; D’Amico, J.; Colona, V.L.; Bertini, E.; Pierantozzi, M.; Mari, L.; Mercuri, N.B.; Piemonte, F.; et al. Systemic Activation of Nrf2 Pathway in Parkinson’s Disease. Mov. Disord. 2020, 35, 180–184. [Google Scholar] [CrossRef]
- Cai, W.; Srivastava, P.; Feng, D.; Lin, Y.; Vanderburg, C.R.; Xu, Y.; Mclean, P.; Frosch, M.P.; Fisher, D.E.; Schwarzschild, M.A.; et al. Melanocortin 1 Receptor Activation Protects against Alpha-Synuclein Pathologies in Models of Parkinson’s Disease. Mol. Neurodegener. 2022, 17, 16. [Google Scholar] [CrossRef]
- Frost, P.; Kleisner, K.; Flegr, J. Health Status by Gender, Hair Color, and Eye Color: Red-Haired Women Are the Most Divergent. PLoS ONE 2017, 12, e0190238. [Google Scholar] [CrossRef]
- Wong, H.Y.; Lee, R.C.; Chong, S.; Kapadia, S.; Freeman, M.; Murigneux, V.; Brown, S.; Soyer, H.P.; Roy, E.; Khosrotehrani, K. Epidermal Mutation Accumulation in Photodamaged Skin Is Associated with Skin Cancer Burden and Can Be Targeted through Ablative Therapy. Sci. Adv. 2023, 9, eadf2384. [Google Scholar] [CrossRef]
- Paus, R.; Theoharides, T.C.; Arck, P.C. Neuroimmunoendocrine Circuitry of the ‘Brain-Skin Connection’. Trends Immunol. 2006, 27, 32–39. [Google Scholar] [CrossRef]
- Hall, J.M.F.; Cruser, d.; Podawiltz, A.; Mummert, D.I.; Jones, H.; Mummert, M.E. Psychological Stress and the Cutaneous Immune Response: Roles of the HPA Axis and the Sympathetic Nervous System in Atopic Dermatitis and Psoriasis. Dermatol. Res. Pract. 2012, 2012, 403908. [Google Scholar] [CrossRef]
- Sansone, R.A.; Sansone, L.A. Sunshine, Serotonin, and Skin: A Partial Explanation for Seasonal Patterns in Psychopathology? Innov. Clin. Neurosci. 2013, 10, 20–24. [Google Scholar]
- Skobowiat, C.; Slominski, A.T. UVB Activates Hypothalamic–Pituitary–Adrenal Axis in C57BL/6 Mice. J. Investig. Dermatol. 2015, 135, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Roosterman, D.; Goerge, T.; Schneider, S.W.; Bunnett, N.W.; Steinhoff, M. Neuronal Control of Skin Function: The Skin as a Neuroimmunoendocrine Organ. Physiol. Rev. 2006, 86, 1309–1379. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lyga, J. Brain-Skin Connection: Stress, Inflammation and Skin Aging. Inflamm. Allergy Drug Targets 2014, 13, 177–190. [Google Scholar] [CrossRef]
- Gillbro, J.M.; Marles, L.K.; Hibberts, N.A.; Schallreuter, K.U. Autocrine Catecholamine Biosynthesis and the Β2-Adrenoceptor Signal Promote Pigmentation in Human Epidermal Melanocytes. J. Investig. Dermatol. 2004, 123, 346–353. [Google Scholar] [CrossRef]
- Stephens, M.A.C.; Wand, G. Stress and the HPA Axis. Alcohol Res. Curr. Rev. 2012, 34, 468–483. [Google Scholar]
- Karin, O.; Raz, M.; Tendler, A.; Bar, A.; Korem Kohanim, Y.; Milo, T.; Alon, U. A New Model for the HPA Axis Explains Dysregulation of Stress Hormones on the Timescale of Weeks. Mol. Syst. Biol. 2020, 16, e9510. [Google Scholar] [CrossRef]
- Smith, S.M.; Vale, W.W. The Role of the Hypothalamic-Pituitary-Adrenal Axis in Neuroendocrine Responses to Stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [CrossRef]
- Soto-Rivera, C.L.; Majzoub, J.A. Chapter 3—Adrenocorticotrophin. In The Pituitary, 4th ed.; Melmed, S., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 47–83. ISBN 978-0-12-804169-7. [Google Scholar]
- Irwin, M.; Hauger, R.; Brown, M. Central Corticotropin-Releasing Hormone Activates the Sympathetic Nervous System and Reduces Immune Function: Increased Responsivity of the Aged Rat. Endocrinology 1992, 131, 1047–1053. [Google Scholar] [CrossRef]
- Kim, J.E.; Cho, B.K.; Cho, D.H.; Park, H.J. Expression of Hypothalamic–Pituitary–Adrenal Axis in Common Skin Diseases: Evidence of Its Association with Stress-Related Disease Activity. Acta Derm. Venereol. 2013, 93, 387–393. [Google Scholar] [CrossRef]
- Katsu, Y.; Iguchi, T. Subchapter 95D—Cortisol. In Handbook of Hormones; Takei, Y., Ando, H., Tsutsui, K., Eds.; Academic Press: San Diego, CA, USA, 2016; p. 533-e95D-2. ISBN 978-0-12-801028-0. [Google Scholar]
- Chan, S.; Debono, M. Replication of Cortisol Circadian Rhythm: New Advances in Hydrocortisone Replacement Therapy. Ther. Adv. Endocrinol. Metab. 2010, 1, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.A.; Bales, N.J.; Myers, S.A.; Bautista, A.I.; Roueinfar, M.; Hale, T.M.; Handa, R.J. The Hypothalamic-Pituitary-Adrenal Axis: Development, Programming Actions of Hormones, and Maternal-Fetal Interactions. Front. Behav. Neurosci. 2021, 14, 601939. [Google Scholar] [CrossRef] [PubMed]
- Grigore, O.; Mihailescu, A.I.; Solomon, I.; Boda, D.; Caruntu, C. Role of Stress in Modulation of Skin Neurogenic Inflammation. Exp. Ther. Med. 2019, 17, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Jozic, I.; Stojadinovic, O.; Kirsner, R.S.F.; Tomic-Canic, M. Skin under the (Spot)-Light: Cross-Talk with the Central Hypothalamic–Pituitary–Adrenal (HPA) Axis. J. Investig. Dermatol. 2015, 135, 1469–1471. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-K.; Zhong, L.; Santiago, J.L. Association between Stress and the HPA Axis in the Atopic Dermatitis. Int. J. Mol. Sci. 2017, 18, 2131. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A. On the Role of the Corticotropin-Releasing Hormone Signalling System in the Aetiology of Inflammatory Skin Disorders. Br. J. Dermatol. 2009, 160, 229–232. [Google Scholar] [CrossRef]
- Slominski, A.; Zbytek, B.; Szczesniewski, A.; Semak, I.; Kaminski, J.; Sweatman, T.; Wortsman, J. CRH Stimulation of Corticosteroids Production in Melanocytes Is Mediated by ACTH. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E701–E706. [Google Scholar] [CrossRef] [PubMed]
- Millington, G.W.M. Proopiomelanocortin (POMC): The Cutaneous Roles of Its Melanocortin Products and Receptors. Clin. Exp. Dermatol. 2006, 31, 407–412. [Google Scholar] [CrossRef]
- Møller, C.L.; Pedersen, S.B.; Richelsen, B.; Conde-Frieboes, K.W.; Raun, K.; Grove, K.L.; Wulff, B.S. Melanocortin Agonists Stimulate Lipolysis in Human Adipose Tissue Explants but Not in Adipocytes. BMC Res. Notes 2015, 8, 559. [Google Scholar] [CrossRef]
- Salazar-Onfray, F.; López, M.; Lundqvist, A.; Aguirre, A.; Escobar, A.; Serrano, A.; Korenblit, C.; Petersson, M.; Chhajlani, V.; Larsson, O.; et al. Tissue Distribution and Differential Expression of Melanocortin 1 Receptor, a Malignant Melanoma Marker. Br. J. Cancer 2002, 87, 414–422. [Google Scholar] [CrossRef]
- Fregoso, D.R.; Hadian, Y.; Gallegos, A.C.; Degovics, D.; Maaga, J.; Keogh, C.E.; Kletenik, I.; Gareau, M.G.; Isseroff, R.R. Skin-Brain Axis Signaling Mediates Behavioral Changes after Skin Wounding. Brain Behav. Immun.—Health 2021, 15, 100279. [Google Scholar] [CrossRef] [PubMed]
- Guida, S.; Guida, G.; Goding, C.R. MC1R Functions, Expression, and Implications for Targeted Therapy. J. Investig. Dermatol. 2022, 142, 293–302.e1. [Google Scholar] [CrossRef] [PubMed]
- Savos, A.V.; Gee, J.M.; Zierath, D.; Becker, K.J. α-MSH: A Potential Neuroprotective and Immunomodulatory Agent for the Treatment of Stroke. J. Cereb. Blood Flow Metab. 2011, 31, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Feng, J.; Zhang, S.; Tong, Y.; Zhang, Q.; Yang, X.; Zhang, H. Altered Levels of α-Melanocyte Stimulating Hormone in Cerebrospinal Fluid and Plasma of Patients with Traumatic Brain Injury. Brain Res. 2018, 1696, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Schaible, E.-V.; Steinsträßer, A.; Jahn-Eimermacher, A.; Luh, C.; Sebastiani, A.; Kornes, F.; Pieter, D.; Schäfer, M.K.; Engelhard, K.; Thal, S.C. Single Administration of Tripeptide α-MSH(11–13) Attenuates Brain Damage by Reduced Inflammation and Apoptosis after Experimental Traumatic Brain Injury in Mice. PLoS ONE 2013, 8, e71056. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; McLaurin, J. α-Melanocyte Stimulating Hormone as a Potential Therapy for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Wang, J.; Lu, J.; Lu, H.; Teng, J.; Jia, Y. Neuroprotective Effects of α-Melanocyte-Stimulating Hormone against the Neurotoxicity of 1-Methyl-4-Phenylpyridinium. IUBMB Life 2017, 69, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Schiöth, H.B.; Phillips, S.R.; Rudzish, R.; Birch-Machin, M.A.; Wikberg, J.E.; Rees, J.L. Loss of Function Mutations of the Human Melanocortin 1 Receptor Are Common and Are Associated with Red Hair. Biochem. Biophys. Res. Commun. 1999, 260, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, A.H.; Couto-Souza, P.H.; Berti-Couto, S.A. Surgeons Beware: Some Red Heads Have Increased Requirements for Inhalation, Infiltration, and Topically Administered Anesthetic Agents. J. Oral Maxillofac. Surg. 2021, 79, 958–959. [Google Scholar] [CrossRef]
- Más, J.S.; Sánchez, C.O.; Ghanem, G.; Haycock, J.; Teruel, J.A.L.; García-Borrón, J.C.; Jiménez-Cervantes, C. Loss-of-Function Variants of the Human Melanocortin-1 Receptor Gene in Melanoma Cells Define Structural Determinants of Receptor Function. Eur. J. Biochem. 2002, 269, 6133–6141. [Google Scholar] [CrossRef]
- Liem, E.B.; Joiner, T.V.; Tsueda, K.; Sessler, D.I. Increased Sensitivity to Thermal Pain and Reduced Subcutaneous Lidocaine Efficacy in Redheads. Anesthesiology 2005, 102, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Delaney, A.; Keighren, M.; Fleetwood-Walker, S.M.; Jackson, I.J. Involvement of the Melanocortin-1 Receptor in Acute Pain and Pain of Inflammatory but Not Neuropathic Origin. PLoS ONE 2010, 5, e12498. [Google Scholar] [CrossRef] [PubMed]
- Binkley, C.J.; Beacham, A.; Neace, W.; Gregg, R.G.; Liem, E.B.; Sessler, D.I. Genetic Variations Associated with Red Hair Color and Fear of Dental Pain, Anxiety Regarding Dental Care and Avoidance of Dental Care. J. Am. Dent. Assoc. 2009, 140, 896–905. [Google Scholar] [CrossRef]
- Liem, E.B.; Lin, C.; Suleman, M.; Doufas, A.G.; Gregg, R.G.; Veauthier, J.M.; Loyd, G.; Sessler, D.I. Anesthetic Requirement Is Increased in Redheads. Anesthesiology 2004, 101, 279–283. [Google Scholar] [CrossRef]
- Choi, J.; Zamary, K.; Barreto, N.B.; Tennakoon, L.; Davis, K.M.; Trickey, A.W.; Spain, D.A. Intravenous Lidocaine as a Non-Opioid Adjunct Analgesic for Traumatic Rib Fractures. PLoS ONE 2020, 15, e0239896. [Google Scholar] [CrossRef]
- Malhotra, P.; Mychaskiw, G.; Rai, A. Desflurane Versus Opioid Anesthesia for Cardiac Shunt Procedures in Infants with Cyantoic Congential Heart Disease. Anesthesiol. Pain Med. 2013, 3, 191–197. [Google Scholar] [CrossRef]
- Robinson, K.C.; Kemény, L.V.; Fell, G.L.; Hermann, A.L.; Allouche, J.; Ding, W.; Yekkirala, A.; Hsiao, J.J.; Su, M.Y.; Theodosakis, N.; et al. Reduced MC4R Signaling Alters Nociceptive Thresholds Associated with Red Hair. Sci. Adv. 2021, 7, eabd1310. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.D.; Pairo-Castineira, E.; Rawlik, K.; Canela-Xandri, O.; Rees, J.; Sims, D.; Tenesa, A.; Jackson, I.J. Genome-Wide Study of Hair Colour in UK Biobank Explains Most of the SNP Heritability. Nat. Commun. 2018, 9, 5271. [Google Scholar] [CrossRef]
- Harno, E.; Gali Ramamoorthy, T.; Coll, A.P.; White, A. POMC: The Physiological Power of Hormone Processing. Physiol. Rev. 2018, 98, 2381–2430. [Google Scholar] [CrossRef]
- Pritchard, L.E.; White, A. Neuropeptide Processing and Its Impact on Melanocortin Pathways. Endocrinology 2007, 148, 4201–4207. [Google Scholar] [CrossRef]
- Rousseau, K.; Kauser, S.; Pritchard, L.E.; Warhurst, A.; Oliver, R.L.; Slominski, A.; Wei, E.T.; Thody, A.J.; Tobin, D.J.; White, A. Proopiomelanocortin (POMC), the ACTH/Melanocortin Precursor, Is Secreted by Human Epidermal Keratinocytes and Melanocytes and Stimulates Melanogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 1844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, D.; Li, H.; Sun, Y.; Jiang, X.; Gong, S.; Qian, Z.; Tao, J. Melanocortin Type 4 Receptor–Mediated Inhibition of A-Type K+ Current Enhances Sensory Neuronal Excitability and Mechanical Pain Sensitivity in Rats. J. Biol. Chem. 2019, 294, 5496–5507. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Sun, J.; Xu, H.; Niu, Z.; Xu, M. Effect of Periaqueductal Gray Melanocortin 4 Receptor in Pain Facilitation and Glial Activation in Rat Model of Chronic Constriction Injury. Neurol. Res. 2012, 34, 871–888. [Google Scholar] [CrossRef]
- Li, Z.-X.; Liu, B.-W.; He, Z.-G.; Xiang, H.-B. Melanocortin-4 Receptor Regulation of Pain. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2017, 1863, 2515–2522. [Google Scholar] [CrossRef]
- Sharfman, N.M.; Kelley, L.K.; Secci, M.E.; Gilpin, N.W. Melanocortin-4 Receptor Signaling in the Central Amygdala Mediates Chronic Inflammatory Pain Effects on Nociception. Neuropharmacology 2022, 210, 109032. [Google Scholar] [CrossRef]
- Crist, R.C.; Berrettini, W.H. Pharmacogenetics of OPRM1. Pharmacol. Biochem. Behav. 2014, 123, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Taqi, M.M.; Faisal, M.; Zaman, H. OPRM1 A118G Polymorphisms and Its Role in Opioid Addiction: Implication on Severity and Treatment Approaches. Pharmacogenom. Pers. Med. 2019, 12, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Hand, W.; Alexov, E. Opioid Addiction and Opioid Receptor Dimerization: Structural Modeling of the OPRD1 and OPRM1 Heterodimer and Its Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 10290. [Google Scholar] [CrossRef]
- Mogil, J.S.; Ritchie, J.; Smith, S.B.; Strasburg, K.; Kaplan, L.; Wallace, M.R.; Romberg, R.R.; Bijl, H.; Sarton, E.Y.; Fillingim, R.B.; et al. Melanocortin-1 Receptor Gene Variants Affect Pain and Mu-Opioid Analgesia in Mice and Humans. J. Med. Genet. 2005, 42, 583–587. [Google Scholar] [CrossRef]
- Kemény, L.V.; Robinson, K.C.; Hermann, A.L.; Walker, D.M.; Regan, S.; Yew, Y.W.; Lai, Y.C.; Theodosakis, N.; Rivera, P.D.; Ding, W.; et al. Vitamin D Deficiency Exacerbates UV/Endorphin and Opioid Addiction. Sci. Adv. 2021, 7, eabe4577. [Google Scholar] [CrossRef]
- Feldman, S.R.; Liguori, A.; Kucenic, M.; Rapp, S.R.; Fleischer, A.B.; Lang, W.; Kaur, M. Ultraviolet Exposure Is a Reinforcing Stimulus in Frequent Indoor Tanners. J. Am. Acad. Dermatol. 2004, 51, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.; Engelhardt, M.; Schaupp, P.; Lappe, C.; Ivanov, I.V. The Inner Clock—Blue Light Sets the Human Rhythm. J. Biophotonics 2019, 12, e201900102. [Google Scholar] [CrossRef] [PubMed]
- Sahay, M.; Sahay, R. Rickets–Vitamin D Deficiency and Dependency. Indian J. Endocrinol. Metab. 2012, 16, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, W.; Li, D.; Yin, X.; Zhang, X.; Olsen, N.; Zheng, S.G. Vitamin D and Chronic Diseases. Aging Dis. 2017, 8, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, N.G.; Chaplin, G. The Roles of Vitamin D and Cutaneous Vitamin D Production in Human Evolution and Health. Int. J. Paleopathol. 2018, 23, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, C.; Ness, A.R.; Wills, A.K.; Lawlor, D.A.; Lewis, S.J.; Davey Smith, G. Skin Pigmentation, Sun Exposure and Vitamin D Levels in Children of the Avon Longitudinal Study of Parents and Children. BMC Public Health 2014, 14, 597. [Google Scholar] [CrossRef] [PubMed]
- Bemanian, M.; Chowdhury, R.; Stokke, K.; Aas, C.F.; Johansson, K.A.; Vold, J.H.; Fadnes, L.T. Vitamin D Status and Associations with Substance Use Patterns among People with Severe Substance Use Disorders in Western Norway. Sci. Rep. 2022, 12, 13695. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Zhang, F.; Su, K.; LaRochelle, M.; Callahan, M.; Fisher, D.; Wharam, J.F.; Asgari, M.M. Perioperative Serum 25-Hydroxyvitamin D Levels as a Predictor of Postoperative Opioid Use and Opioid Use Disorder: A Cohort Study. J. Gen. Intern. Med. 2020, 35, 2545–2552. [Google Scholar] [CrossRef]
- De Sanctis, V.; Agolli, L.; Visco, V.; Monaco, F.; Muni, R.; Spagnoli, A.; Campanella, B.; Valeriani, M.; Minniti, G.; Osti, M.F.; et al. Cytokines, Fatigue, and Cutaneous Erythema in Early Stage Breast Cancer Patients Receiving Adjuvant Radiation Therapy. BioMed Res. Int. 2014, 2014, 523568. [Google Scholar] [CrossRef]
- Xiao, C.; Beitler, J.J.; Higgins, K.A.; Conneely, K.; Dwivedi, B.; Felger, J.; Wommack, E.C.; Shin, D.M.; Saba, N.F.; Ong, L.Y.; et al. Fatigue Is Associated with Inflammation in Patients with Head and Neck Cancer before and after Intensity-Modulated Radiation Therapy. Brain Behav. Immun. 2016, 52, 145–152. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Secretory Cell | Paracrine Factor | Corresponding Receptor | Signalling Pathway |
|---|---|---|---|
| melanocyte, keratinocyte | β-endorphin | OPRM1 | PKC |
| α-MSH (alpha-melanocyte-stimulating hormone) | MC1R | PKA | |
| β-MSH (beta-melanocyte stimulating hormone) | MC4R | ||
| BMP6 (bone morphogenetic protein 6) | BMPR1/2 | MAPK | |
| BMP4 (bone morphogenetic protein 4) | |||
| keratinocyte | Glu (glutamate) | mGLUR6 | PKC |
| EDN1 (endothelin 1) | EDNRB | ||
| norepinephrine | ADRB2 | ||
| keratinocyte, fibroblast | FGF (fibroblast growth factors) | FGFR | MAPK |
| HGF (hepatocyte growth factor) | MET | ||
| SCF (stem cell factor) | KIT | ||
| melanocyte, keratinocyte, fibroblast | CRF (corticotropin-releasing factor) | CRF-1R | PKA, PKC |
| fibroblast | NRG1 (neuregulin 1) | ErbB | MAPK |
| WNT (Wingless-type protein) | Fzd-3 | WNT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ascsillán, A.A.; Kemény, L.V. The Skin–Brain Axis: From UV and Pigmentation to Behaviour Modulation. Int. J. Mol. Sci. 2024, 25, 6199. https://doi.org/10.3390/ijms25116199
Ascsillán AA, Kemény LV. The Skin–Brain Axis: From UV and Pigmentation to Behaviour Modulation. International Journal of Molecular Sciences. 2024; 25(11):6199. https://doi.org/10.3390/ijms25116199
Chicago/Turabian StyleAscsillán, Anna A., and Lajos V. Kemény. 2024. "The Skin–Brain Axis: From UV and Pigmentation to Behaviour Modulation" International Journal of Molecular Sciences 25, no. 11: 6199. https://doi.org/10.3390/ijms25116199
APA StyleAscsillán, A. A., & Kemény, L. V. (2024). The Skin–Brain Axis: From UV and Pigmentation to Behaviour Modulation. International Journal of Molecular Sciences, 25(11), 6199. https://doi.org/10.3390/ijms25116199