
Animal Models and Molecular Pathogenesis of Arrhythmogenic Cardiomyopathy Associated with Pathogenic Variants in Intercalated Disc Genes



Abstract
1. Introduction
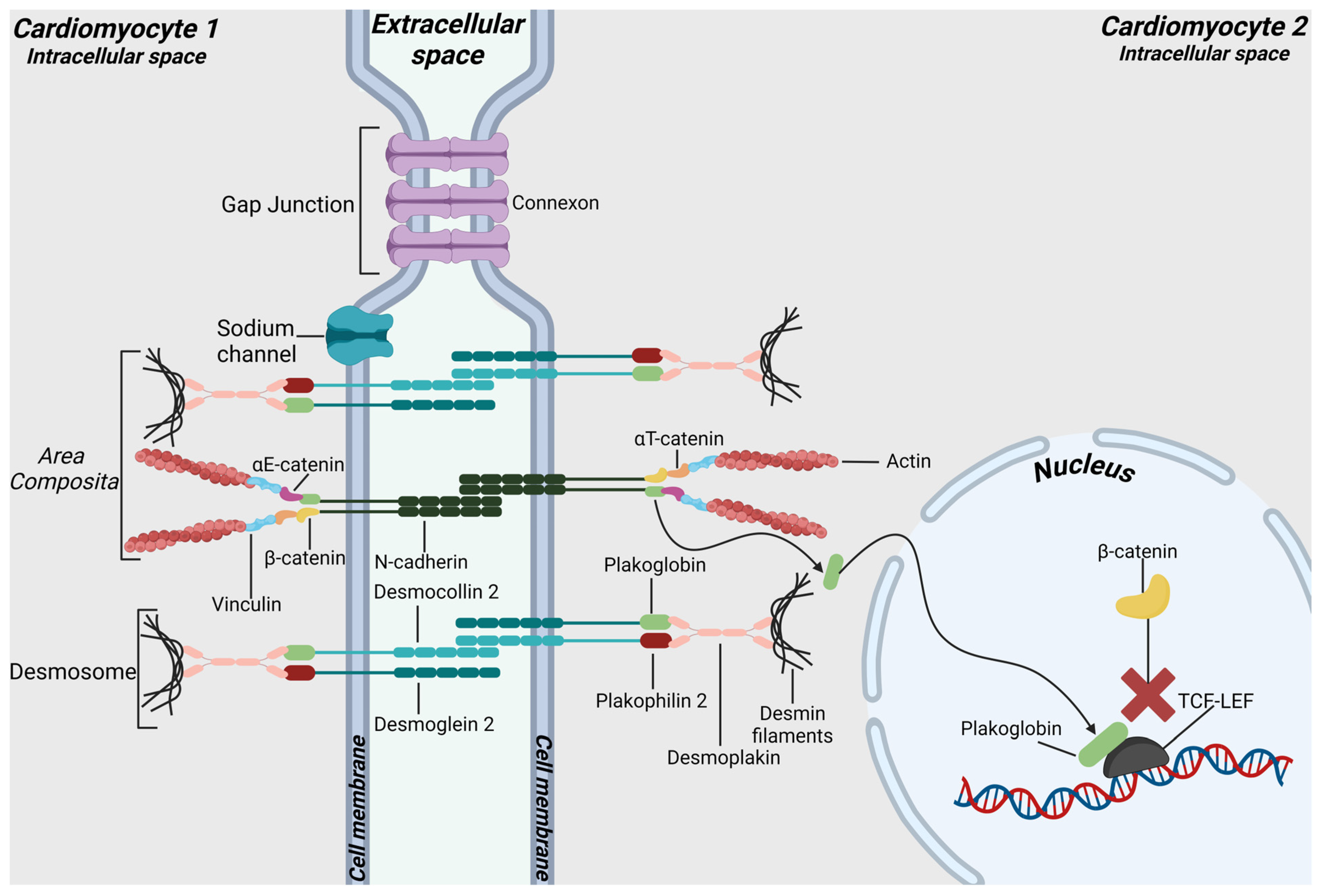
2. Intercalated Discs
3. Murine Models for ACM Associated with Plakophilin 2 Variants

{kind=link}
| Gene | Experimental Model | Alterations | Refs. |
|---|---|---|---|
| Pkp2 | Pkp2 homozygous-null embryos | Mislocation of desmosomal proteins Altered organization of cardiac junctions | [22] |
| Pkp2 heterozygous-null mice | Reduced number of mechanical junctions Reduced intercellular distances in correspondence of IDs Upregulation of ECM components ↑TGF-β1/p38 MAPK pathway Desmosomes sporadic or absent Expanded space between cardiomyocytes Ventricular arrhythmias | [23,24,25] | |
| Cardiomyocyte-specific Pkp2 conditional KO | Right ventricle dysfunction Arrhythmias Cardiac fibrosis Increased calcium concentration Enhanced frequency of spontaneous calcium release events measured in vitro | [26,27] | |
| Complete Pkp2 knock-down via shRNA | Enlarged left ventricle Interstitial fibrosis Presence of cells with lipid droplets | [36] | |
| Tg mice with cardiomyocyte-specific overexpression of PKP2 with p.R375* variation | Right ventricle dysfunction exacerbated by training Altered localization and distribution of CX43 | [29] | |
| Tg mice with cardiomyocyte-specific overexpression of PKP2 with p.S329* variation | Ventricular arrhythmias Reduced left ventricle systolic function Reduced expression of proteins CX43 and NaV1.5 Downregulation of junctional proteins expression | [30] | |
| Pkp2 heterozygous KI mice with c.1086InsT variation | Reduced right ventricle functionality Increased apoptotic cardiomyocytes Increased widening of intercalated discs | [32] | |
| Pkp2 heterozygous KI mice with c.1755delA variation | Cardiac fibrosis Reduced expression of junctional proteins Increased width of IDs | [33] | |
| Pkp2 homozygous KI mice | Alterations in sodium current Loss of Nav1.5 subunit Enlarged ventricles Cardiac fibrosis Subepicardial adiposis Reduced number of desmosomes Reduced expression of junctional proteins | [34] |
4. Murine Models for ACM Associated with Desmoplakin Variants
| Gene | Experimental Model | Alterations | Refs. |
|---|---|---|---|
| Dsp | General Dsp KO mice | Embryonic lethality | [38] |
| Cardiomyocyte-specific Dsp deficient mice | High embryonic lethality in Dsp−/− mice Molecular remodeling of IDs Fibro-fatty replacement LV dilation Reduced fractional shortening Nuclear localization of plakoglobin ↑ Adipogenesis and fibrogenesis ↓ Wnt/β-catenin signaling ↑ Hippo pathway ↑ Inflammation and EMT-related genes ↓ Oxidative phosphorylation-related genes | [39,40,42] | |
| Cardiomyocyte-specific Dsp KO mice | Structural defects in desmosomal integrity Cardiac cells death Fibro-fatty replacement Ventricular arrhythmias exacerbated with exercise or catecholamine stimulation ↓ CX40 protein level ↓CX43 protein level | [43] | |
| Mice with conditional heterozygous Dsp deletion in cFAPs | Increased fibro-adipogenesis Mild cardiac dysfunction ↓ Wnt/β-catenin signaling ↑ Adipogenesis in cFAPs | [44] | |
| Tg mice with cardiomyocyte-specific overexpression of DSP with p.R3834H variation | Increased cardiomyocytes apoptosis Cardiac fibrosis and lipid accumulations Ultrastructural alterations of IDs Cardiac hypertrophy ↑ PKP2 and β-catenin Accelerated ACM pathogenesis when exposed to endurance exercise ↓ Wnt/β-catenin signaling when exposed to endurance exercise | [45,49] | |
| Dsp KI mice | Embryonic lethality in Dsp R451G/R451G Aberrant CX43 localization Stress-induced arrhythmias Accelerated heart failure following pressure overload | [51] |
5. Murine Models for ACM Associated with Desmoglein 2 Variants
| Gene | Experimental Model | Alterations | Refs. |
|---|---|---|---|
| Dsg2 | Dsg2 KO mice lacking exons 7–8 | Embryonic lethality | [53] |
| Dsg2 KO mice lacking exons 4–6 | Cardiac fibrosis Inflammatory infiltrates Ventricular dilation Arrhythmias and cardiac insufficiency | [54,55] | |
| Cardiomyocyte-specific Dsg2 KO mice lacking exons 4–6 | Aberrant CX43 localization Cardiomyocyte necrosis Cardiac fibrosis Functional alterations | [56] | |
| Dsg2 KO mice lacking exons 4–5 | Abnormal localization of junctional proteins Cardiac fibrosis Inflammation GSK3β constitutive activation ↑ NFkB signaling pathway | [57,61,62] | |
| Dsg2 KI mice | Wide IDs Cardiac fibrosis Echocardiography and ECG abnormalities ↑ TGF-β signaling pathway | [64] | |
| Tg mice with cardiomyocyte-specific overexpression of DSG2 with p.N271S variation | Cardiac fibrosis Ventricular dilation Cardiac dysfunction Ventricular arrhythmias | [65,68] | |
| Tg mice with cardiomyocyte-specific overexpression of human DSG2 with p.Q558* variation | Decrease in size and number of desmosomes Cardiac fibrosis ↓ Wnt signaling | [66] | |
| Cardiomyocyte-specific Dsg2 null mice | Lipid accumulation Cardiac fibrosis Ventricular dilation Impaired contractile function ↓ PPARα ↓ mTOR-4EBP1 axis ↑ TGF-β signaling pathway | [71,72] |
6. Murine Models for ACM-Associated Desmocollin 2 Variants
| Gene | Experimental Model | Alterations | Refs. |
|---|---|---|---|
| Dsc2 | Dsc2 KI mice | No structural and functional defects Slight LV dilation in homozygous G790del mice Aberrant Ca2+ release in homozygous G790del mice | [75] |
| Tg mice with cardiomyocyte-specific overexpression of WT DSC2 | Myocardial necrosis Fibrotic replacement Severe cardiac dysfunction | [76] |
7. Murine Models for ACM Associated with Plakoglobin Variants
| Gene | Experimental Model | Alterations | Refs. |
|---|---|---|---|
| Jup | Homozygous Jup KO embryos | Less developed heart Thin cardiac walls Reduced number of cardiac desmosomes | [81] |
| Heterozygous Jup KO mice | Right ventricle dilatation and dysfunction exacerbated with exercise Ventricular arrhythmias exacerbated with exercise Low concentration of CX43 protein | [82,83] | |
| Cardiomyocyte-specific Jup KO mice lacking exons 3–5 | Enlarged hearts Right ventricle dilatation Hypertrophic cardiomyocytes Enhanced apoptosis of cardiomyocytes Cardiac fibrosis Absence of normal desmosomes ↑ TGF-β pathway | [84] | |
| Cardiomyocyte-specific Jup KO mice lacking exon 1 | Cardiac dilatation Cardiac fibrosis Reduced levels of DSG2 protein at intercalated discs Abrogation of positive effects of β-adrenergic signaling | [85] | |
| Cardiomyocyte-specific Jup conditional KO mice | Enlarged ventricles Focal areas of cardiomyocyte loss Inflammatory infiltrates Cardiac fibrosis Reduced junctional proteins expression at the intercalated discs Reduced CX43-containing gap junction plaques Reduction in the number and length of desmosomes ↑ β-catenin ↑ Myc and Fos ↑ c-MYC ↓ GSK3β ↑ Wnt/β-catenin signaling | [86] | |
| Cardiomyocyte-specific Jup and Ctnnb1 conditional KO mice | Enlarged ventricles Increased number of apoptotic cells Fibroblasts interspersed through myocardium Collagen deposition Reduced junctional proteins levels Decreased area occupied by gap junction plaques Loss of IDs structures | [87] | |
| Tg mice with cardiomyocyte-specific overexpression of Flag-tagged PG | Cardiac fibrosis and adiposis ↓ Wnt/β-catenin signaling | [88] | |
| Tg mice with cardiomyocyte-specific overexpression of truncated or wild-type PG | Cardiac fibrosis and adiposis Enhanced adipogenesis in cardiac progenitor cells Molecular remodeling of IDs Nuclear localization of plakoglobin ↓ Wnt/β-catenin signaling ↑ Hippo pathway Abnormal localization of junctional proteins Inflammatory infiltrates | [40,57,90] |
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gerull, B.; Brodehl, A. Insights into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 223–233. [Google Scholar] [CrossRef]
- Bauce, B.; Frigo, G.; Marcus, F.I.; Basso, C.; Rampazzo, A.; Maddalena, F.; Corrado, D.; Winnicki, M.; Daliento, L.; Rigato, I.; et al. Comparison of Clinical Features of Arrhythmogenic Right Ventricular Cardiomyopathy in Men Versus Women. Am. J. Cardiol. 2008, 102, 1252–1257. [Google Scholar] [CrossRef]
- Pruna, M.; Ehler, E. The intercalated disc: A mechanosensing signalling node in cardiomyopathy. Biophys. Rev. 2020, 12, 931–946. [Google Scholar] [CrossRef]
- Calore, M.; Lorenzon, A.; De Bortoli, M.; Poloni, G.; Rampazzo, A. Arrhythmogenic cardiomyopathy: A disease of intercalated discs. Cell. Tissue Res. 2015, 360, 491–500. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- James, C.A.; Syrris, P.; van Tintelen, J.P.; Calkins, H. The role of genetics in cardiovascular disease: Arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1393–1400. [Google Scholar] [CrossRef]
- Lie, Ø.H.; Rootwelt-Norberg, C.; Dejgaard, L.A.; Leren, I.S.; Stokke, M.K.; Edvardsen, T.; Haugaa, K.H. Prediction of Life-Threatening Ventricular Arrhythmia in Patients with Arrhythmogenic Cardiomyopathy: A Primary Prevention Cohort Study. J. Am. Coll. Cardiovasc. Imaging 2018, 11, 1377–1386. [Google Scholar] [CrossRef]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Pilichou, K.; Thiene, G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart 2011, 97, 530–539. [Google Scholar] [CrossRef]
- Nielsen, M.S.; van Opbergen, C.J.M.; van Veen, T.A.B.; Delmar, M. The intercalated disc: A unique organelle for electromechanical synchrony in cardiomyocytes. Physiol. Rev. 2023, 103, 2271–2319. [Google Scholar] [CrossRef]
- Rampazzo, A.; Calore, M.; van Hengel, J.; van Roy, F. Intercalated discs and arrhythmogenic cardiomyopathy. Circ. Cardiovasc. Genet. 2014, 7, 930–940. [Google Scholar] [CrossRef]
- Franke, W.W.; Borrmann, C.M.; Grund, C.; Pieperhoff, S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur. J. Cell Biol. 2006, 85, 69–82. [Google Scholar] [CrossRef]
- Vermij, S.H.; Abriel, H.; van Veen, T.A.B. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef]
- Zhao, G.; Qiu, Y.; Zhang, H.M.; Yang, D. Intercalated discs: Cellular adhesion and signaling in heart health and diseases. Heart Fail. Rev. 2019, 24, 115–132. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Sig. Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Manring, H.R.; Dorn, L.E.; Ex-Willey, A.; Accornero, F.; Ackermann, M.A. At the heart of inter- and intracellular signaling: The intercalated disc. Biophys. Rev. 2018, 10, 961–971. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Rasmussen, T.B.; Nissen, P.H.; Palmfeldt, J.; Gehmlich, K.; Dalager, S.; Jensen, U.B.; Kim, W.Y.; Heickendorff, L.; Baandrup, U.T.; Bross, P.; et al. Truncating plakophilin-2 mutations in arrhythmogenic cardiomyopathy are associated with protein haploinsufficiency in both myocardium and epidermis. Circ. Cardiovasc. Genet. 2014, 7, 230–240. [Google Scholar] [CrossRef]
- Kirchner, F.; Schuetz, A.; Boldt, L.H.; Martens, K.; Dittmar, G.; Haverkamp, W.; Thierfelder, L.; Heinemann, U.; Gerull, B. Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ. Cardiovasc. Genet. 2012, 5, 400–411. [Google Scholar] [CrossRef]
- Dries, A.M.; Kirillova, A.; Reuter, C.M.; Garcia, J.; Zouk, H.; Hawley, M.; Murray, B.; Tichnell, C.; Pilichou, K.; Protonotarios, A.; et al. The genetic architecture of Plakophilin 2 cardiomyopathy. Genet. Med. 2021, 10, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K.S.; Grund, C.; Huelsken, J.; Behrend, M.; Erdmann, B.; Franke, W.W.; Birchmeier, W. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J. Cell Biol. 2004, 167, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Leo-Macias, A.; Liang, F.X.; Delmar, M. Ultrastructure of the intercellular space in adult murine ventricle revealed by quantitative tomographic electron microscopy. Cardiovasc. Res. 2015, 107, 442–452. [Google Scholar] [CrossRef]
- Dubash, A.D.; Kam, C.Y.; Aguado, B.A.; Patel, D.M.; Delmar, M.; Shea, L.D.; Green, K.J. Plakophilin-2 loss promotes TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J. Cell Biol. 2016, 212, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Noorman, M.; Lin, X.; Chkourko, H.; Liang, F.X.; Van Der Nagel, R.; Hund, T.; Birchmeier, W.; Mohler, P.; Van Veen, T.A.; et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc. Res. 2012, 95, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Pérez-Hernández, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca2+i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000006755.26 (accessed on 3 January 2024).
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; García-Prieto, J.; García-Ruiz, J.M.; Pizarro, G.; Jiménez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruíz-Cabello, J.; et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll. Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef]
- Moncayo-Arlandi, J.; Guasch, E.; Sanz-de la Garza, M.; Casado, M.; Garcia, N.A.; Mont, L.; Sitges, M.; Knöll, R.; Buyandelger, B.; Campuzano, O.; et al. Molecular disturbance underlies to arrhythmogenic cardiomyopathy induced by transgene content, age and exercise in a truncated PKP2 mouse model. Hum. Mol. Genet. 2016, 25, 3676–3688. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000045015.9 (accessed on 8 February 2024).
- Camors, E.M.; Roth, A.H.; Alef, J.R.; Sullivan, R.D.; Johnson, J.N.; Purevjav, E.; Towbin, J.A. Progressive Reduction in Right Ventricular Contractile Function Attributable to Altered Actin Expression in an Aging Mouse Model of Arrhythmogenic Cardiomyopathy. Circulation 2022, 145, 1609–1624. [Google Scholar] [CrossRef]
- Tsui, H.; van Kampen, S.J.; Han, S.J.; Meraviglia, V.; van Ham, W.B.; Casini, S.; van der Kraak, P.; Vink, A.; Yin, X.; Mayr, M.; et al. Desmosomal protein degradation as an underlying cause of arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2023, 15, 4248. [Google Scholar] [CrossRef] [PubMed]
- Bradford, W.H.; Zhang, J.; Gutierrez-Lara, E.J.; Liang, Y.; Do, A.; Wang, T.; Nguyen, L.; Mataraarachchi, N.; Wang, J.; Gu, Y.; et al. Plakophilin 2 gene therapy prevents and rescues arrhythmogenic right ventricular cardiomyopathy in a mouse model harboring patient genetics. Nat. Cardiovasc. Res. 2023, 2, 1246–1261. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000006756.65 (accessed on 27 March 2024).
- Gurha, P.; Chen, X.; Lombardi, R.; Willerson, J.T.; Marian, A.J. Knockdown of Plakophilin 2 Downregulates miR-184 Through CpG Hypermethylation and Suppression of the E2F1 Pathway and Leads to Enhanced Adipogenesis In Vitro. Circ. Res. 2016, 119, 731–750. [Google Scholar] [CrossRef] [PubMed]
- Hatzfeld, M.; Keil, R.; Magin, T.M. Desmosomes and intermediate filaments: Their consequences for tissue mechanics. Cold Spring Harb. Perspect. Biol. 2017, 9, a029157. [Google Scholar] [CrossRef] [PubMed]
- Gallicano, G.I.; Kouklis, P.; Bauer, C.; Yin, M.; Vasioukhin, V.; Degenstein, L.; Fuchs, E. Desmoplakin Is Required Early in Development for Assembly of Desmosomes and Cytoskeletal Linkage. J. Cell Biol. 1998, 143, 2009–2022. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.L.; Luo, T.; Luo, M. The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct. Target. Ther. 2022, 7, 376. [Google Scholar] [CrossRef] [PubMed]
- Cheedipudi, S.M.; Hu, J.; Fan, S.; Yuan, P.; Karmouch, J.; Czernuszewicz, G.; Robertson, M.J.; Coarfa, C.; Hong, K.; Yao, Y.; et al. Exercise restores dysregulated gene expression in a mouse model of arrhythmogenic cardiomyopathy. Cardiovasc. Res. 2021, 116, 1199–1213. [Google Scholar] [CrossRef]
- Lyon, R.C.; Mezzano, V.; Wright, A.T.; Pfeiffer, E.; Chuang, J.; Banares, K.; Castaneda, A.; Ouyang, K.; Cui, L.; Contu, R.; et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 2014, 23, 1134–1150. [Google Scholar] [CrossRef]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef]
- Yang, Z.; Bowles, N.E.; Scherer, S.E.; Taylor, M.D.; Kearney, D.L.; Ge, S.; Nadvoretskiy, V.V.; DeFreitas, G.; Carabello, B.; Brandon, L.I.; et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Res. 2006, 99, 646–655. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000016846.51 (accessed on 3 January 2024).
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000163236.22 (accessed on 3 January 2024).
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000016847.2 (accessed on 3 January 2024).
- Martherus, R.; Jain, R.; Takagi, K.; Mendsaikhan, U.; Turdi, S.; Osinska, H.; James, J.F.; Kramer, K.; Purevjav, E.; Towbin, J.A. Accelerated cardiac remodeling in desmoplakin transgenic mice in response to endurance exercise is associated with perturbed Wnt/β-catenin signaling. Am. J. Physiol. Heart. Circ. Physiol. 2016, 310, 174–187. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000948761.9 (accessed on 3 January 2024).
- Stevens, T.L.; Manring, H.R.; Wallace, M.J.; Argall, A.; Dew, T.; Papaioannou, P.; Antwi-Boasiako, S.; Xu, X.; Campbell, S.G.; Akar, F.G.; et al. Humanized Dsp ACM Mouse Model Displays Stress-Induced Cardiac Electrical and Structural Phenotypes. Cells 2022, 11, 3049. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Y.; Yang, X.; Xiang, Y.; Yang, B. Molecular insight into arrhythmogenic cardiomyopathy caused by DSG2 mutations. Biomed. Pharmacother. 2023, 167, 115448. [Google Scholar] [CrossRef] [PubMed]
- Eshkind, L.; Tian, Q.; Schmidt, A.; Franke, W.W.; Windoffer, R.; Leube, R.E. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur. J. Cell Biol. 2002, 81, 592–598. [Google Scholar] [CrossRef]
- Krusche, C.A.; Holthöfer, B.; Hofe, V.; Van De Sandt, A.M.; Eshkind, L.; Bockamp, E. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic. Res. Cardiol. 2011, 106, 617–633. [Google Scholar] [CrossRef]
- Kant, S.; Krull, P.; Eisner, S.; Leube, R.E.; Krusche, C.A. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res. 2012, 348, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Holthöfer, B.; Magin, T.M.; Krusche, C.A.; Leube, R.E. Desmoglein 2-Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ. Cardiovasc. Genet. 2015, 8, 553–563. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 2016, 1, 85923. [Google Scholar] [CrossRef]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2014, 6, 240ra74. [Google Scholar] [CrossRef] [PubMed]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Goñi-Oliver, P.; Lucas, J.J.; Avila, J.; Hernández, F. N-terminal cleavage of GSK-3 by calpain: A new form of GSK-3 regulation. J. Biol. Chem. 2007, 282, 22406–22413. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.P.; Keceli, G.; Carpi, A.; Doti, N.; Agrimi, J.; Asimaki, A.; Beti Bueno, C.; Miyamoto, M.; Amat-Codina, N.; Bedja, D.; et al. Exercise triggers CAPN1-mediated AIF truncation, inducing myocyte cell death in arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2021, 13, eabf0891. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Hua, X.; Shu, S.; Xu, X.; Zhang, H.; Peng, Z.; Mo, H.; Liu, Y.; Chen, X.; Yang, Y.; et al. Single-cell RNA sequencing in donor and end-stage heart failure patients identifies NLRP3 as a therapeutic target for arrhythmogenic right ventricular cardiomyopathy. BMC. Med. 2024, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Schinner, C.; Xu, L.; Franz, H.; Zimmermann, A.; Wanuske, M.T.; Rathod, M.; Hanns, P.; Geier, F.; Pelczar, P.; Liang, Y.; et al. Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy by Involving an Integrin-αVβ6/TGF-β Signaling Cascade. Circulation 2022, 146, 1610–1626. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; Van den Hoff, M.J.B.; De Bakker, J.M.T.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Calore, M.; Lorenzon, A.; Vitiello, L.; Poloni, G.; Khan, M.A.F.; Beffagna, G.; Dazzo, E.; Sacchetto, C.; Polishchuk, R.; Sabatelli, P.; et al. A novel murine model for arrhythmogenic cardiomyopathy points to a pathogenic role of Wnt signalling and miRNA dysregulation. Cardiovasc. Res. 2019, 115, 739–751. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000016815.1 (accessed on 3 January 2024).
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated disc abnormalities, reduced Na+ current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001075931.6 (accessed on 3 January 2024).
- Soliman, H.; Paylor, B.; Scott, R.W.; Lemos, D.R.; Chang, C.K.; Arostegui, M.; Low, M.; Lee, C.; Fiore, D.; Braghetta, P.; et al. Pathogenic Potential of Hic1-Expressing Cardiac Stromal Progenitors. Cell Stem Cell 2020, 26, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, R.; Huang, Y.; Yang, Z.; Xian, J.; Huang, J.; Qiu, Z.; Lin, X.; Zhang, M.; Chen, H.; et al. Reactivation of PPARα alleviates myocardial lipid accumulation and cardiac dysfunction by improving fatty acid β-oxidation in Dsg2-deficient arrhythmogenic cardiomyopathy. Acta Pharm. Sin. B 2023, 13, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Zhao, Y.; Tao, T.; Guo, W.; Liu, R.; Huang, J.; Xu, G. Activation of PPARα Ameliorates Cardiac Fibrosis in Dsg2-Deficient Arrhythmogenic Cardiomyopathy. Cells 2022, 11, 3184. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.J.; Nassal, D.M.; Gratz, D.; Hund, T.J. Emerging therapeutic targets for cardiac arrhythmias: Role of STAT3 in regulating cardiac fibroblast function. Expert. Opin. Ther. Targets 2021, 25, 63–73. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000180319.28 (accessed on 3 January 2024).
- Hamada, Y.; Yamamoto, T.; Nakamura, Y.; Sufu-Shimizu, Y.; Nanno, T.; Fukuda, M.; Ono, M.; Oda, T.; Okuda, S.; Ueyama, T.; et al. G790del mutation in DSC2 alone is insufficient to develop the pathogenesis of ARVC in a mouse model. Biochem. Biophys. Rep. 2020, 21, 100711. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.-X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.R.; Calway, T.; Harmon, C.; Farrell, P.; Kim, G.H. MicroRNA-130a Regulation of Desmocollin 2 in a Novel Model of Arrhythmogenic Cardiomyopathy. Microrna 2017, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Osbourne, A.; Calway, T.; Broman, M.; McSharry, S.; Earley, J.; Kim, G.H. Downregulation of connexin43 by microRNA-130a in cardiomyocytes results in cardiac arrhythmias. J. Mol. Cell Cardiol. 2014, 74, 53–63. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Yeruva, S.; Waschke, J. Structure and regulation of desmosomes in intercalated discs: Lessons from epithelia. J. Anat. 2023, 242, 81–90. [Google Scholar] [CrossRef]
- Bierkamp, C.; Mclaughlin, K.J.; Schwarz, H.; Huber, O.; Kemler, R. Embryonic Heart and Skin Defects in Mice Lacking Plakoglobin. Dev. Biol. 1996, 180, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Fabritz, L.; Zwiener, M.; Witt, H.; Schäfers, M.; Zellerhoff, S.; Paul, M.; Athai, T.; Hiller, K.H.; Baba, H.A.; et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 2006, 114, 1799–1806. [Google Scholar] [CrossRef]
- Fabritz, L.; Hoogendijk, M.G.; Scicluna, B.P.; Van Amersfoorth, S.C.M.; Fortmueller, L.; Wolf, S.; Laakmann, S.; Kreienkamp, N.; Piccini, I.; Breithardt, G.; et al. Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. J. Am. Coll. Cardiol. 2011, 57, 740–750. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.; Haneline, L.S.; Field, L.J.; et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef]
- Schinner, C.; Vielmuth, F.; Rötzer, V.; Hiermaier, M.; Radeva, M.Y.; Co, T.K.; Hartlieb, E.; Schmidt, A.; Imhof, A.; Messoudi, A.; et al. Adrenergic Signaling Strengthens Cardiac Myocyte Cohesion. Circ. Res. 2017, 120, 1305–1317. [Google Scholar] [CrossRef]
- Li, J.; Swope, D.; Raess, N.; Cheng, L.; Muller, E.J.; Radice, G.L. Cardiac Tissue-Restricted Deletion of Plakoglobin Results in Progressive Cardiomyopathy and Activation of β-Catenin Signaling. Mol. Cell Biol. 2011, 31, 1134–1144. [Google Scholar] [CrossRef]
- Swope, D.; Cheng, L.; Gao, E.; Li, J.; Radice, G.L. Loss of Cadherin-Binding Proteins β-Catenin and Plakoglobin in the Heart Leads to Gap Junction Remodeling and Arrhythmogenesis. Mol. Cell Biol. 2012, 32, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Dong, J.; Rodriguez, G.; Bell, A.; Leung, T.K.; Schwartz, R.J.; Willerson, J.T.; Brugada, R.; Marian, A.J. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2009, 104, 1076–1084. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000013599.7 (accessed on 3 January 2024).
- Lombardi, R.; Da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A.J. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2011, 109, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Lu, Y.W.; Lin, Z.; Huang, Z.P.; Liu, J.; Wang, Y.; Seok, H.Y.; Hu, X.; Ma, Q.; Li, K.; et al. Intercalated disc protein Xinβ is required for Hippo-YAP signaling in the heart. Nat. Commun. 2020, 11, 4666. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Zuo, Y.; Farmer, S.R. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol. Cell. Biol. 2006, 26, 5827–5837. [Google Scholar] [CrossRef]
- Sacchetto, C.; Vitiello, L.; de Windt, L.J.; Rampazzo, A.; Calore, M. Modeling Cardiovascular Diseases with hiPSC-Derived Cardiomyocytes in 2D and 3D Cultures. Int. J. Mol. Sci. 2020, 21, 3404. [Google Scholar] [CrossRef]
- Kyriakopoulou, E.; Versteeg, D.; de Ruiter, H.; Perini, I.; Seibertz, F.; Döring, Y.; Zentilin, L.; Tsui, H.; van Kampen, S.J.; Tiburcy, M.; et al. Therapeutic efficacy of AAV-mediated restoration of PKP2 in arrhythmogenic cardiomyopathy. Nat. Cardiovasc. Res. 2023, 2, 1262–1276. [Google Scholar] [CrossRef]
- van Opbergen, C.J.M.; Narayanan, B.; Sacramento, C.B.; Stiles, K.M.; Mishra, V.; Frenk, E.; Ricks, D.; Chen, G.; Zhang, M.; Yarabe, P.; et al. AAV-Mediated Delivery of Plakophilin-2a Arrests Progression of Arrhythmogenic Right Ventricular Cardiomyopathy in Murine Hearts: Preclinical Evidence Supporting Gene Therapy in Humans. Circ. Genom. Precis. Med. 2024, 17, e004305. [Google Scholar] [CrossRef]
- Wu, I.; Zeng, A.; Greer-Short, A.; Aycinena, J.A.; Tefera, A.E.; Shenwai, R.; Farshidfar, F.; Van Pell, M.; Xu, E.; Reid, C.; et al. AAV9:PKP2 improves heart function and survival in a Pkp2-deficient mouse model of arrhythmogenic right ventricular cardiomyopathy. Commun. Med. 2024, 4, 38. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vencato, S.; Romanato, C.; Rampazzo, A.; Calore, M. Animal Models and Molecular Pathogenesis of Arrhythmogenic Cardiomyopathy Associated with Pathogenic Variants in Intercalated Disc Genes. Int. J. Mol. Sci. 2024, 25, 6208. https://doi.org/10.3390/ijms25116208
Vencato S, Romanato C, Rampazzo A, Calore M. Animal Models and Molecular Pathogenesis of Arrhythmogenic Cardiomyopathy Associated with Pathogenic Variants in Intercalated Disc Genes. International Journal of Molecular Sciences. 2024; 25(11):6208. https://doi.org/10.3390/ijms25116208
Chicago/Turabian StyleVencato, Sara, Chiara Romanato, Alessandra Rampazzo, and Martina Calore. 2024. "Animal Models and Molecular Pathogenesis of Arrhythmogenic Cardiomyopathy Associated with Pathogenic Variants in Intercalated Disc Genes" International Journal of Molecular Sciences 25, no. 11: 6208. https://doi.org/10.3390/ijms25116208
APA StyleVencato, S., Romanato, C., Rampazzo, A., & Calore, M. (2024). Animal Models and Molecular Pathogenesis of Arrhythmogenic Cardiomyopathy Associated with Pathogenic Variants in Intercalated Disc Genes. International Journal of Molecular Sciences, 25(11), 6208. https://doi.org/10.3390/ijms25116208