

New Molecules in Type 2 Diabetes: Advancements, Challenges and Future Directions

 , ,
, , 
Abstract

1. Introduction
2. GLP-1 Receptor Agonists
3. Cardiovascular Effects of GLP-1 Receptor Agonists: Preclinical Studies
4. Cardiovascular Effects of GLP-1 Receptor Agonists: Clinical Studies
5. SGLT2 Inhibitors
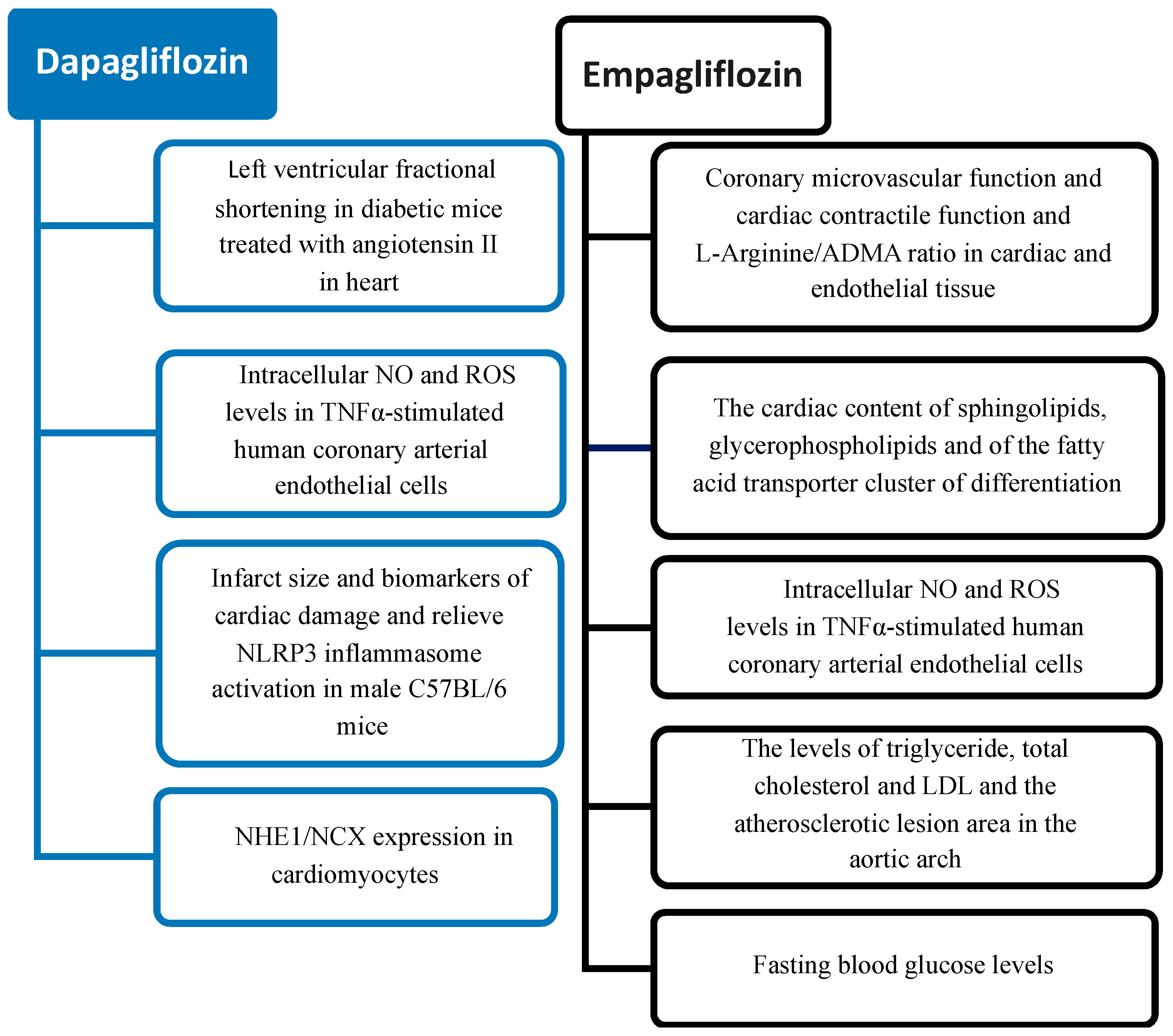
6. Cardiovascular Effects of SGLT2 Inhibitors: Preclinical Studies
7. Cardiovascular Effects of SGLT2 Inhibitors: Clinical Studies
8. Innovative Therapeutic Developments in the T2D Scenario
8.1. β-Cells Senescence in Type 2 Diabetes
8.2. Mitochondrial Dynamics in T2D
8.3. Epigenetic Modifications in T2D
8.4. Gut Microbiota Profile in T2D
9. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Diabetes Is “a Pandemic of Unprecedented Magnitude” Now Affecting One in 10 Adults Worldwide. Diabetes Res. Clin. Pact. 2021, 181, 109133. [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Gyldenkerne, C.; Mortensen, M.B.; Kahlert, J.; Thrane, P.G.; Olesen, K.K.W.; Sørensen, H.T.; Thomsen, R.W.; Maeng, M. 10-Year Cardiovascular Risk in Patients With Newly Diagnosed Type 2 Diabetes Mellitus. J. Am. Coll. Cardiol. 2023, 82, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on Diabetes, Pre-Diabetes, and Cardiovascular Diseases Developed in Collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [PubMed]
- Fan, W. Epidemiology in Diabetes Mellitus and Cardiovascular Disease. Cardiovasc. Endocrinol. 2017, 6, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M. Cardiovascular Disease in Type 2 Diabetes: Challenge for Treatment and Prevention. J. Intern. Med. 2001, 249, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The Cardiovascular Biology of Glucagon-like Peptide-1. Cell Metab. 2016, 24, 15–30. [Google Scholar] [CrossRef]
- Defronzo, R.A. From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed]
- Lovshin, J.A. Glucagon-like Peptide-1 Receptor Agonists: A Class Update for Treating Type 2 Diabetes. Can. J. Diabetes 2017, 41, 524–535. [Google Scholar] [CrossRef]
- Premji, R.; Nylen, E.S.; Naser, N.; Gandhi, S.; Burman, K.D.; Sen, S. Lipid Profile Changes Associated with SGLT-2 Inhibitors and GLP-1 Agonists in Diabetes and Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2022, 20, 321–328. [Google Scholar] [CrossRef]
- Diallo, A.; Carlos-Bolumbu, M.; Galtier, F. Blood Pressure-Lowering Effects of SGLT2 Inhibitors and GLP-1 Receptor Agonists for Preventing of Cardiovascular Events and Death in Type 2 Diabetes: A Systematic Review and Meta-Analysis. Acta Diabetol. 2023, 60, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Ansari, H.U.H.; Qazi, S.U.; Sajid, F.; Altaf, Z.; Ghazanfar, S.; Naveed, N.; Ashfaq, A.S.; Siddiqui, A.H.; Iqbal, H.; Qazi, S. Efficacy and Safety of Glucagon-Like Peptide-1 Receptor Agonists on Body Weight and Cardiometabolic Parameters in Individuals With Obesity and Without Diabetes: A Systematic Review and Meta-Analysis. Endocr. Pract. 2024, 30, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Abildstrøm, S.Z.; Borlaug, B.A.; Davies, M.J.; Kitzman, D.W.; Petrie, M.C.; Shah, S.J.; Verma, S.; Abhayaratna, W.P.; Chopra, V.; et al. Semaglutide in Patients With Obesity and Heart Failure Across Mildly Reduced or Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2023, 82, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.-J.; Deswal, A. In Patients with HFpEF and Obesity, Semaglutide Increased Weight Loss and Reduced Symptoms and Physical Limitations at 52 Wk. Ann. Intern. Med. 2023, 176, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Cimino, G.; Vaduganathan, M.; Lombardi, C.M.; Pagnesi, M.; Vizzardi, E.; Tomasoni, D.; Adamo, M.; Metra, M.; Inciardi, R.M. Obesity, Heart Failure with Preserved Ejection Fraction, and the Role of Glucagon-like Peptide-1 Receptor Agonists. ESC Heart Fail. 2023, 11, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Tahrani, A.A.; Barnett, A.H. Future Glucose-Lowering Drugs for Type 2 Diabetes. Lancet Diabetes Endocrinol. 2016, 4, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.; Costello, R.A. Glucagon-Like Peptide-1 Receptor Agonists; StatPearls Publishing LLC.: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Aslam, B.; Zafar, B.; Changez, M.I.K.; Abdullah, M.; Safwan, M.; Qamar, B.; Shinwari, A.; RAI, S. Exploring the Potential Impact of GLP-1 Receptor Agonists in Cancer Therapy. Minerva Endocrinol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, A.; Oyama, J.-i.; Komoda, H.; Asaka, M.; Komatsu, A.; Sakuma, M.; Kodama, K.; Sakamoto, Y.; Kotooka, N.; Hirase, T.; et al. The Glucagon-like Peptide 1 Analog Liraglutide Reduces TNF-α-Induced Oxidative Stress and Inflammation in Endothelial Cells. Atherosclerosis 2012, 221, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Noyan-Ashraf, M.H.; Shikatani, E.A.; Schuiki, I.; Mukovozov, I.; Wu, J.; Li, R.K.; Volchuk, A.; Robinson, L.A.; Billia, F.; Drucker, D.J.; et al. A Glucagon-like Peptide-1 Analog Reverses the Molecular Pathology and Cardiac Dysfunction of a Mouse Model of Obesity. Circulation 2013, 127, 74–85. [Google Scholar] [CrossRef]
- Chen, J.; Wang, D.; Wang, F.; Shi, S.; Chen, Y.; Yang, B.; Tang, Y.; Huang, C. Exendin-4 Inhibits Structural Remodeling and Improves Ca2+ Homeostasis in Rats with Heart Failure via the GLP-1 Receptor through the ENOS/CGMP/PKG Pathway. Peptides 2017, 90, 69–77. [Google Scholar] [CrossRef]
- McLean, B.A.; Wong, C.K.; Kabir, M.G.; Drucker, D.J. Glucagon-like Peptide-1 Receptor Tie2+ Cells Are Essential for the Cardioprotective Actions of Liraglutide in Mice with Experimental Myocardial Infarction. Mol. Metab. 2022, 66, 101641. [Google Scholar] [CrossRef]
- Dimsdale, J.E. Psychological Stress and Cardiovascular Disease. J. Am. Coll. Cardiol. 2008, 51, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Lei, Y.; Inoue, A.; Piao, L.; Hu, L.; Jiang, H.; Sasaki, T.; Wu, H.; Xu, W.; Yu, C.; et al. Exenatide Mitigated Diet-Induced Vascular Aging and Atherosclerotic Plaque Growth in ApoE-Deficient Mice under Chronic Stress. Atherosclerosis 2017, 264, 1–10. [Google Scholar] [CrossRef]
- Bose, A.K.; Mocanu, M.M.; Carr, R.D.; Brand, C.L.; Yellon, D.M. Glucagon-like Peptide 1 Can Directly Protect the Heart against Ischemia/Reperfusion Injury. Diabetes 2005, 54, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Køber, L.V.; Lawson, F.C.; Ping, L.; Wei, X.; Lewis, E.F.; et al. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Jones, B. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. Ann. Clin. Biochem. 2016, 53, 712. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef]
- Green, J.B.; Hernandez, A.F.; D’Agostino, R.B.; Granger, C.B.; Janmohamed, S.; Jones, N.P.; Leiter, L.A.; Noronha, D.; Russell, R.; Sigmon, K.; et al. Harmony Outcomes: A Randomized, Double-Blind, Placebo-Controlled Trial of the Effect of Albiglutide on Major Cardiovascular Events in Patients with Type 2 Diabetes Mellitus—Rationale, Design, and Baseline Characteristics. Am. Heart J. 2018, 203, 30–38. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.D.; Lam, C.S.P.; Khurmi, N.S.; Heenan, L.; Del Prato, S.; et al. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 896–907. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and Cardiovascular Outcomes in Type 2 Diabetes (REWIND): A Double-Blind, Randomised Placebo-Controlled Trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Marsenic, O. Glucose Control by the Kidney: An Emerging Target in Diabetes. Am. J. Kidney Dis. 2009, 53, 875–883. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V. SGLT2 Inhibitors and Mechanisms of Cardiovascular Benefit: A State-of-the-Art Review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 Inhibitors for Primary and Secondary Prevention of Cardiovascular and Renal Outcomes in Type 2 Diabetes: A Systematic Review and Meta-Analysis of Cardiovascular Outcome Trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Williams, D.M.; Nawaz, A.; Evans, M. Renal Outcomes in Type 2 Diabetes: A Review of Cardiovascular and Renal Outcome Trials. Diabetes Ther. 2020, 11, 369–386. [Google Scholar] [CrossRef]
- Bolinder, J.; Ljunggren, Ö.; Kullberg, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sugg, J.; Parikh, S. Effects of Dapagliflozin on Body Weight, Total Fat Mass, and Regional Adipose Tissue Distribution in Patients with Type 2 Diabetes Mellitus with Inadequate Glycemic Control on Metformin. J. Clin. Endocrinol. Metab. 2012, 97, 1020–1031. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Sjöström, C.D.; Greasley, P.J.; Cain, V.; Boulton, D.W.; Heerspink, H.J.L. Effects of the Sodium-Glucose Co-Transporter-2 Inhibitor Dapagliflozin on Estimated Plasma Volume in Patients with Type 2 Diabetes. Diabetes Obes. Metab. 2019, 21, 2667–2673. [Google Scholar] [CrossRef]
- Packer, M. SGLT2 Inhibitors Produce Cardiorenal Benefits by Promoting Adaptive Cellular Reprogramming to Induce a State of Fasting Mimicry: A Paradigm Shift in Understanding Their Mechanism of Action. Diabetes Care 2020, 43, 508–511. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Zannad, F. Effects of Sodium-Glucose Cotransporter 2 Inhibitors for the Treatment of Patients with Heart Failure—Proposal of a Novel Mechanism of Action. JAMA Cardiol. 2017, 2, 1025–1029. [Google Scholar] [CrossRef]
- Szekeres, Z.; Toth, K.; Szabados, E. The Effects of Sglt2 Inhibitors on Lipid Metabolism. Metabolites 2021, 11, 87. [Google Scholar] [CrossRef]
- Dobner, S.; Bernhard, B.; Asatryan, B.; Windecker, S.; Stortecky, S.; Pilgrim, T.; Gräni, C.; Hunziker, L. SGLT2 Inhibitor Therapy for Transthyretin Amyloid Cardiomyopathy: Early Tolerance and Clinical Response to Dapagliflozin. ESC Heart Fail. 2023, 10, 397–404. [Google Scholar] [CrossRef]
- Monami, M.; Nardini, C.; Mannucci, E. Efficacy and Safety of Sodium Glucose Co-Transport-2 Inhibitors in Type 2 Diabetes: A Meta-Analysis of Randomized Clinical Trials. Diabetes Obes. Metab. 2014, 16, 457–466. [Google Scholar] [CrossRef]
- Monica Reddy, R.P.; Inzucchi, S.E. SGLT2 Inhibitors in the Management of Type 2 Diabetes. Endocrine 2016, 53, 364–372. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of Hyperglycemia in Type 2 Diabetes: A Patient-Centered Approach. Diabetes Care 2012, 35, 1364–1379. [Google Scholar] [CrossRef]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-Glucose Cotransporter 2 Inhibitors for Type 2 Diabetes: A Systematic Review and Meta-Analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef]
- Arow, M.; Waldman, M.; Yadin, D.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Freimark, D.; Kornowski, R.; Aravot, D.; Hochhauser, E.; et al. Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Attenuates Diabetic Cardiomyopathy. Cardiovasc. Diabetol. 2020, 19, 7. [Google Scholar] [CrossRef]
- Adingupu, D.D.; Göpel, S.O.; Grönros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L.M.; Jönsson-Rylander, A.C. SGLT2 Inhibition with Empagliflozin Improves Coronary Microvascular Function and Cardiac Contractility in Prediabetic Ob/Ob−/− Mice. Cardiovasc. Diabetol. 2019, 18, 16. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 Inhibitor Empagliflozin Improves the Primary Diabetic Complications in ZDF Rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef]
- Aragón-Herrera, A.; Feijóo-Bandín, S.; Otero Santiago, M.; Barral, L.; Campos-Toimil, M.; Gil-Longo, J.; Costa Pereira, T.M.; García-Caballero, T.; Rodríguez-Segade, S.; Rodríguez, J.; et al. Empagliflozin Reduces the Levels of CD36 and Cardiotoxic Lipids While Improving Autophagy in the Hearts of Zucker Diabetic Fatty Rats. Biochem. Pharmacol. 2019, 170, 113677. [Google Scholar] [CrossRef]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmanna Benedikt Preckel, M.W.; Koolwijk, P.; Van Hinsbergh, V.W.M.; Zuurbier, C.J.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore No Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell. Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, J.; Wu, M.; Xu, B.; Kang, L. Empagliflozin Protects against Atherosclerosis Progression by Modulating Lipid Profiles and Sympathetic Activity. Lipids Health Dis. 2021, 20, 5. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.W.; Que, J.Q.; Liu, S.; Huang, K.Y.; Qian, L.; Weng, Y.B.; Rong, F.N.; Wang, L.; Zhou, Y.Y.; Xue, Y.J.; et al. Sodium-Glucose Co-Transporter-2 Inhibitor of Dapagliflozin Attenuates Myocardial Ischemia/Reperfusion Injury by Limiting NLRP3 Inflammasome Activation and Modulating Autophagy. Front. Cardiovasc. Med. 2021, 8, 768214. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.D.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Rocca, H.-P.B.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Iwasaki, K.; Abarca, C.; Aguayo-Mazzucato, C. Regulation of Cellular Senescence in Type 2 Diabetes Mellitus: From Mechanisms to Clinical Applications. Diabetes Metab. J. 2023, 47, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin Suppresses Gluconeogenesis by Inhibiting Mitochondrial Glycerophosphate Dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Metformin Enhances B Cell Function and Antibody Responses of Elderly Individuals With Type-2 Diabetes Mellitus. Front. Aging 2021, 2, 715981. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, I.Y.; Agostinucci, K.; Sadaf, B.; Grant, M.K.O.; Zordoky, B.N. Metformin Mitigates SASP Secretion and LPS-Triggered Hyper-Inflammation in Doxorubicin-Induced Senescent Endothelial Cells. Front. Aging 2023, 4, 1170434. [Google Scholar] [CrossRef] [PubMed]
- Hooten, N.N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-Mediated Increase in DICER1 Regulates MicroRNA Expression and Cellular Senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Forouzandeh, F.; Salazar, G.; Patrushev, N.; Xiong, S.; Hilenski, L.; Fei, B.; Alexander, R.W. Metformin beyond Diabetes: Pleiotropic Benefits of Metformin in Attenuation of Atherosclerosis. J. Am. Heart Assoc. 2014, 3, e001202. [Google Scholar] [CrossRef] [PubMed]
- Oeseburg, H.; De Boer, R.A.; Buikema, H.; van der Harst, P.; Van Gilst, W.H.; Silljé, H.H.W. Glucagon-like Peptide 1 Prevents Reactive Oxygen Species-Induced Endothelial Cell Senescence through the Activation of Protein Kinase A. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.E.; Rakipovski, G.; Raun, K.; Lykkesfeldt, J. Does Glucagon-like Peptide-1 Ameliorate Oxidative Stress in Diabetes? Evidence Based on Experimental and Clinical Studies. Curr. Diabetes Rev. 2016, 12, 331–358. [Google Scholar] [CrossRef]
- Bode-Böger, S.M.; Martens-Lobenhoffer, J.; Täger, M.; Schröder, H.; Scalera, F. Aspirin Reduces Endothelial Cell Senescence. Biochem. Biophys. Res. Commun. 2005, 334, 1226–1232. [Google Scholar] [CrossRef]
- Li, F.; Guo, Y.; Jiang, X.; Zhong, J.; Li, G.; Sun, S. Aspirin Inhibits Human Telomerase Activation in Unstable Carotid Plaques. Exp. Ther. Med. 2013, 6, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Brouilette, S.W.; Moore, J.S.; McMahon, A.D.; Thompson, J.R.; Ford, I.; Shepherd, J.; Packard, C.J.; Samani, N.J. Telomere Length, Risk of Coronary Heart Disease, and Statin Treatment in the West of Scotland Primary Prevention Study: A Nested Case-Control Study. Lancet 2007, 369, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, L.; Li, Y. Change of Telomere Length in Angiotensin II-Induced Human Glomerular Mesangial Cell Senescence and the Protective Role of Losartan. Mol. Med. Rep. 2011, 4, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Lodovici, M.; Bigagli, E.; Tarantini, F.; Di Serio, C.; Raimondi, L. Losartan Reduces Oxidative Damage to Renal DNA and Conserves Plasma Antioxidant Capacity in Diabetic Rats. Exp. Biol. Med. 2015, 240, 1500–1504. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, B.; Xiao, F.Q.; Wu, Y.T.; Wang, R.H.; Sun, Z.W.; Fu, G.S.; Mou, Y.; Tao, W.; Hu, X.S.; et al. Autophagy Protects against Senescence and Apoptosis via the Ras-Mitochondria in High-Glucose-Induced Endothelial Cells. Cell. Physiol. Biochem. 2014, 33, 1058–1074. [Google Scholar] [CrossRef] [PubMed]
- Chrienova, Z.; Nepovimova, E.; Kuca, K. The Role of MTOR in Age-Related Diseases. J. Enzyme Inhib. Med. Chem. 2021, 36, 1679–1693. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Inagaki, N.; Kondoh, H. Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells. Front. Endocrinol. 2022, 13, 869414. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef]
- Miura, Y.; Endo, K.; Komori, K.; Sekiya, I. Clearance of Senescent Cells with ABT-263 Improves Biological Functions of Synovial Mesenchymal Stem Cells from Osteoarthritis Patients. Stem Cell Res. Ther. 2022, 13, 222. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed Elimination of Senescent Cells by Inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Rysanek, D.; Vasicova, P.; Kolla, J.N.; Sedlak, D.; Andera, L.; Bartek, J.; Hodny, Z. Synergism of BCL-2 Family Inhibitors Facilitates Selective Elimination of Senescent Cells. Aging 2022, 14, 6381–6414. [Google Scholar] [CrossRef] [PubMed]
- Nerstedt, A.; Smith, U. The Impact of Cellular Senescence in Human Adipose Tissue. J. Cell Commun. Signal. 2023, 17, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Tavana, O.; Zhu, C. Too Many Breaks (Brakes): Pancreatic β-Cell Senescence Leads to Diabetes. Cell Cycle 2011, 10, 2471–2484. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Alexander, P.B.; Wang, X.F. Cellular Senescence: From Anti-Cancer Weapon to Anti-Aging Target. Sci. China Life Sci. 2020, 63, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.K.; Hughes, C.M.; Gu, X.; Rozenblatt-Rosen, O.; McLean, G.W.; Xiong, Y.; Meyerson, M.; Kim, S.K. Menin Regulates Pancreatic Islet Growth by Promoting Histone Methylation and Expression of Genes Encoding P27Kip1 and P18INK4c. Proc. Natl. Acad. Sci. USA 2005, 102, 14659–14664. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xing, B.; Cao, Y.; He, X.; Bennett, K.E.; Tong, C.; An, C.; Hojnacki, T.; Feng, Z.; Deng, S.; et al. Menin-regulated Pbk Controls High Fat Diet-induced Compensatory Beta Cell Proliferation. EMBO Mol. Med. 2021, 13, e13524. [Google Scholar] [CrossRef] [PubMed]
- Armata, H.L.; Chamberland, S.; Watts, L.; Ko, H.J.; Lee, Y.; Jung, D.Y.; Kim, J.K.; Sluss, H.K. Deficiency of the Tumor Promoter Gene Wip1 Induces Insulin Resistance. Mol. Endocrinol. 2015, 29, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Le Guezennec, X.; Bulavin, D.V. WIP1 Phosphatase at the Crossroads of Cancer and Aging. Trends Biochem. Sci. 2010, 35, 109–114. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor Cell Escape from Therapy-Induced Senescence. Biochem. Pharmacol. 2019, 162, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Geng, Q.; Jin, S.; Teng, X.; Xiao, L.; Wu, Y.; Tian, D. Exercise Protects Vascular Function by Countering Senescent Cells in Older Adults. Front. Physiol. 2023, 14, 1138162. [Google Scholar] [CrossRef] [PubMed]
- de França, N.A.G.; Rolland, Y.; Guyonnet, S.; de Souto Barreto, P. The Role of Dietary Strategies in the Modulation of Hallmarks of Aging. Ageing Res. Rev. 2023, 87, 101908. [Google Scholar] [CrossRef] [PubMed]
- Canudas, S.; Becerra-Tomas, N.; Hernandez-Alonso, P.; Galie, S.; Leung, C.; Crous-Bou, M.; De Vivo, I.; Gao, Y.; Gu, Y.; Meinila, J.; et al. Mediterranean Diet and Telomere Length: A Systematic Review and Meta-Analysis. Adv. Nutr. 2020, 11, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, K.; Seibert, D.; Wysocki, K.; Seibert, D. Genomics of Aging: Genes, Adducts, and Telomeres. J. Am. Assoc. Nurse Pract. 2020, 32, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; Spiga, R.; Mancuso, E.; La Sala, L.; Antonicelli, R.; Testa, R.; Procopio, A.D.; Olivieri, F.; Ceriello, A. Inflammageing and Metaflammation: The Yin and Yang of Type 2 Diabetes. Ageing Res. Rev. 2018, 41, 1–17. [Google Scholar] [CrossRef]
- Gaggini, M.; Fenizia, S.; Vassalle, C. Sphingolipid Levels and Signaling via Resveratrol and Antioxidant Actions in Cardiometabolic Risk and Disease. Antioxidants 2023, 12, 1102. [Google Scholar] [CrossRef]
- Truong, V.L.; Jun, M.; Jeong, W.S. Role of Resveratrol in Regulation of Cellular Defense Systems against Oxidative Stress. BioFactors 2018, 44, 36–49. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, Y.; Wang, C. Impacts of an Exercise Intervention on the Health of Pancreatic Beta-Cells: A Review. Int. J. Environ. Res. Public Health 2022, 19, 7229. [Google Scholar] [CrossRef]
- Werner, C.; Fürster, T.; Widmann, T.; Pöss, J.; Roggia, C.; Hanhoun, M.; Scharhag, J.; Büchner, N.; Meyer, T.; Kindermann, W.; et al. Physical Exercise Prevents Cellular Senescence in Circulating Leukocytes and in the Vessel Wall. Circulation 2009, 120, 2438–2447. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial Fusion and Fission: The Fine-Tune Balance for Cellular Homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tan, J.; Miao, Y.; Zhang, Q. Mitochondrial Dynamics, Mitophagy, and Mitochondria–Endoplasmic Reticulum Contact Sites Crosstalk Under Hypoxia. Front. Cell Dev. Biol. 2022, 10, 848214. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, H.; Lin, S.; Lv, Y.; Lin, Y.; Liu, Y.; Peng, R.; Jin, H. New Therapeutic Directions in Type II Diabetes and Its Complications: Mitochondrial Dynamics. Front. Endocrinol. 2023, 14, 1230168. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Zhuravlev, A.D.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. Mitochondria-Mediated Cardiovascular Benefits of Sodium-Glucose Co-Transporter 2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 5371. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xu, C.; Liu, X.; Li, X.; Li, T.; Yu, X.; Xue, M.; Yang, J.; Kosmas, C.E.; Moris, D.; et al. Empagliflozin Induces White Adipocyte Browning and Modulates Mitochondrial Dynamics in KK Cg-Ay/J Mice and Mouse Adipocytes. Front. Physiol. 2021, 12, 745058. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, T.; Watanabe, M.; Yokota, T.; Tsuda, M.; Handa, H.; Koya, J.; Nishino, K.; Tatsuta, D.; Natsui, H.; Kadosaka, T.; et al. Empagliflozin Suppresses Mitochondrial Reactive Oxygen Species Generation and Mitigates the Inducibility of Atrial Fibrillation in Diabetic Rats. Front. Cardiovasc. Med. 2023, 10, 1005408. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a Sodium Glucose Co-Transporter-2 Inhibitor, Alleviates Atrial Remodeling and Improves Mitochondrial Function in High-Fat Diet/Streptozotocin-Induced Diabetic Rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin Rescues Diabetic Myocardial Microvascular Injury via AMPK-Mediated Inhibition of Mitochondrial Fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef]
- Yang, W.; Li, X.; He, L.; Zhu, S.; Lai, S.; Zhang, X.; Huang, Z.; Yu, B.; Cui, C.; Wang, Q. Empagliflozin Improves Renal Ischemia–Reperfusion Injury by Reducing Inflammation and Enhancing Mitochondrial Fusion through AMPK–OPA1 Pathway Promotion. Cell. Mol. Biol. Lett. 2023, 28, 42. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, Y.; Chen, X.; Huang, S.; Li, Y.; Ye, G.; Cao, X.; Su, W.; Zhuo, Y. Empagliflozin Targets Mfn1 and Opa1 to Attenuate Microglia-Mediated Neuroinflammation in Retinal Ischemia and Reperfusion Injury. J. Neuroinflamm. 2023, 20, 296. [Google Scholar] [CrossRef]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin Normalizes the Size and Number of Mitochondria and Prevents Reduction in Mitochondrial Size after Myocardial Infarction in Diabetic Hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.T.; Huo, J.Y.; Jiang, W.Y.; Yang, W.; Wang, S.C.; Zhang, S.G.; Cheng, Y.D.; Jiang, Z.X.; Shan, Q.J. Empagliflozin Ameliorates Cardiac Dysfunction in Heart Failure Mice via Regulating Mitochondrial Dynamics. Eur. J. Pharmacol. 2023, 942, 175531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, H.; Yang, X.; Shi, J.; Sheng, X.; Wang, L.; Li, T.; Quan, H.; Zhai, X.; Li, W. Effects of Dapagliflozin Monotherapy and Combined Aerobic Exercise on Skeletal Muscle Mitochondrial Quality Control and Insulin Resistance in Type 2 Diabetes Mellitus Rats. Biomed. Pharmacother. 2023, 169, 115852. [Google Scholar] [CrossRef] [PubMed]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 Inhibitor Dapagliflozin Suppresses Prolonged Ventricular-Repolarization through Augmentation of Mitochondrial Function in Insulin-Resistant Metabolic Syndrome Rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.; Semmler, L.; Wakula, P.; Hegemann, N.; Primessnig, U.; Beindorff, N.; Powell, D.; Dahmen, R.; Ruetten, H.; Oeing, C.; et al. Dual SGLT-1 and SGLT-2 Inhibition Improves Left Atrial Dysfunction in HFpEF. Cardiovasc. Diabetol. 2021, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Li, J.; Takagaki, Y.; Kitada, M.; Nitta, K.; Takasu, T.; Kanasaki, K.; Koya, D. Ipragliflozin Improves Mitochondrial Abnormalities in Renal Tubules Induced by a High-Fat Diet. J. Diabetes Investig. 2018, 9, 1025–1032. [Google Scholar] [CrossRef]
- Wu, Y. Metformin Inhibits Mitochondrial Dysfunction and Apoptosis in Cardiomyocytes Induced by High Glucose via Upregulating AMPK Activity. Exp. Biol. Med. 2023, 248, 1556–1565. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, T.; Sun, G.F.; Xiao, J.X.; Jiang, L.P.; Tou, F.F.; Qu, X.H.; Han, X.J. Metformin Protects against Retinal Ischemia/Reperfusion Injury through AMPK-Mediated Mitochondrial Fusion. Free Radic. Biol. Med. 2023, 205, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Kim, J.; Lee, J.Y.; Kim, J.; Oh, C.M. Mitochondrial Quality Control: Its Role in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD). J. Obes. Metab. Syndr. 2023, 32, 289–302. [Google Scholar] [CrossRef]
- Mima, A. Mitochondria-Targeted Drugs for Diabetic Kidney Disease. Heliyon 2022, 8, e08878. [Google Scholar] [CrossRef]
- Nakajima, R.; Sekiya, M.; Furuta, Y.; Miyamoto, T.; Sato, M.; Fukuda, K.; Hattori, K.; Suehara, Y.; Sakata-Yanagimoto, M.; Chiba, S.; et al. A Case of NASH with Genetic Predisposition Successfully Treated with an SGLT2 Inhibitor: A Possible Involvement of Mitochondrial Dysfunction. Endocrinol. Diabetes Metab. Case Rep. 2022, 2022, 0368. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Ferreira, J.P.; Butler, J.; Filippatos, G.; Januzzi, J.L.; Sumin, M.; Zwick, M.; Saadati, M.; Pocock, S.J.; Sattar, N.; et al. Effect of Empagliflozin on Circulating Proteomics in Heart Failure: Mechanistic Insights into the EMPEROR Programme. Eur. Heart J. 2022, 43, 4991–5002. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and Insulin Resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Gao, Z.; Chen, K.; Zhang, Q.; Hu, S.; Zhao, L. Inflammation-Related Epigenetic Modification: The Bridge Between Immune and Metabolism in Type 2 Diabetes. Front. Immunol. 2022, 13, 883410. [Google Scholar] [CrossRef] [PubMed]
- Remely, M.; Aumueller, E.; Jahn, D.; Hippe, B.; Brath, H.; Haslberger, A.G. Microbiota and Epigenetic Regulation of Inflammatory Mediators in Type 2 Diabetes and Obesity. Benef. Microbes 2014, 5, 33–43. [Google Scholar] [CrossRef]
- Na, Y.K.; Hong, H.S.; Lee, W.K.; Kim, Y.H.; Kim, D.S. Increased Methylation of Interleukin 6 Gene Is Associated with Obesity in Korean Women. Mol. Cells 2015, 38, 452–456. [Google Scholar] [CrossRef]
- Mohamed, I.N.; Li, L.; Ismael, S.; Ishrat, T.; El-Remessy, A.B. Thioredoxin Interacting Protein, a Key Molecular Switch between Oxidative Stress and Sterile Inflammation in Cellular Response. World J. Diabetes 2021, 12, 1979–1999. [Google Scholar] [CrossRef] [PubMed]
- Pedroso, J.A.B.; Ramos-Lobo, A.M.; Donato, J. SOCS3 as a Future Target to Treat Metabolic Disorders. Hormones 2019, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, P.; Landes, R.D.; Graw, S.; Byrum, S.D.; Bennuri, S.; Delhey, L.; Randolph, C.; MacLeod, S.; Reis, A.; Børsheim, E.; et al. Effect of Excess Weight and Insulin Resistance on DNA Methylation in Prepubertal Children. Sci. Rep. 2022, 12, 8430. [Google Scholar] [CrossRef]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef]
- Gharipour, M.; Mani, A.; Baghbahadorani, M.A.; de Souza Cardoso, C.K.; Jahanfar, S.; Sarrafzadegan, N.; de Oliveira, C.; Silveira, E.A. How Are Epigenetic Modifications Related to Cardiovascular Disease in Older Adults? Int. J. Mol. Sci. 2021, 22, 9949. [Google Scholar] [CrossRef] [PubMed]
- Kaimala, S.; Kumar, C.A.; Allouh, M.Z.; Ansari, S.A.; Emerald, B.S. Epigenetic Modifications in Pancreas Development, Diabetes, and Therapeutics. Med. Res. Rev. 2022, 42, 1343–1371. [Google Scholar] [CrossRef] [PubMed]
- Scisciola, L.; Taktaz, F.; Fontanella, R.A.; Pesapane, A.; Surina; Cataldo, V.; Ghosh, P.; Franzese, M.; Puocci, A.; Paolisso, P.; et al. Targeting High Glucose-Induced Epigenetic Modifications at Cardiac Level: The Role of SGLT2 and SGLT2 Inhibitors. Cardiovasc. Diabetol. 2023, 22, 24. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, S.; Fukuhara, A.; Shin, J.; Okuno, Y.; Otsuki, M.; Shimomura, I. Metabolomic and Microarray Analyses of Adipose Tissue of Dapagliflozin-Treated Mice, and Effects of 3-Hydroxybutyrate on Induction of Adiponectin in Adipocytes. Sci. Rep. 2018, 8, 8805. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current Understanding of the Human Microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Wang, X.; Li, L. Human Gut Microbiome: The Second Genome of Human Body. Protein Cell 2010, 1, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-Z.; Stirling, K.; Yang, J.-J.; Zhang, L. Gut Microbiota and Diabetes: From Correlation to Causality and Mechanism. World J. Diabetes 2020, 11, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Sircana, A.; Framarin, L.; Leone, N.; Berrutti, M.; Castellino, F.; Parente, R.; De Michieli, F.; Paschetta, E.; Musso, G. Altered Gut Microbiota in Type 2 Diabetes: Just a Coincidence? Curr. Diabetes Rep. 2018, 18, 98. [Google Scholar] [CrossRef]
- Patra, D.; Banerjee, D.; Ramprasad, P.; Roy, S.; Pal, D.; Dasgupta, S. Recent Insights of Obesity-Induced Gut and Adipose Tissue Dysbiosis in Type 2 Diabetes. Front. Mol. Biosci. 2023, 10, 1224982. [Google Scholar] [CrossRef]
- Redondo-Useros, N.; Nova, E.; González-Zancada, N.; Díaz, L.E.; Gómez-Martínez, S.; Marcos, A. Microbiota and Lifestyle: A Special Focus on Diet. Nutrients 2020, 12, 1776. [Google Scholar] [CrossRef] [PubMed]
- Breton, J.; Galmiche, M.; Déchelotte, P. Dysbiotic Gut Bacteria in Obesity: An Overview of the Metabolic Mechanisms and Therapeutic Perspectives of Next-Generation Probiotics. Microorganisms 2022, 10, 452. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chang, Y.; Zhang, K.; Chen, H.; Tao, S.; Zhang, Z. Implication of the Gut Microbiome Composition of Type 2 Diabetic Patients from Northern China. Sci. Rep. 2020, 10, 5450. [Google Scholar] [CrossRef] [PubMed]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short Chain Fatty Acids in Human Gut and Metabolic Health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef] [PubMed]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of Gut Microbiota in Type 2 Diabetes Pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut Metagenome in European Women with Normal, Impaired and Diabetic Glucose Control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Kaul, R.; Harfouche, M.; Arabi, M.; Al-Najjar, Y.; Sarkar, A.; Saliba, R.; Chaari, A. The Effect of Microbiome-Modulating Probiotics, Prebiotics and Synbiotics on Glucose Homeostasis in Type 2 Diabetes: A Systematic Review, Meta-Analysis, and Meta-Regression of Clinical Trials. Pharmacol. Res. 2022, 185, 106520. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Luo, Z.; Zhou, J.; Sun, B. Gut Microbiota and Antidiabetic Drugs: Perspectives of Personalized Treatment in Type 2 Diabetes Mellitus. Front. Cell. Infect. Microbiol. 2022, 12, 853771. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, N. Effects of Metformin on the Gut Microbiota in Obesity and Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 5003–5014. [Google Scholar] [CrossRef]
- Huda, M.N.; Kim, M.; Bennett, B.J. Modulating the Microbiota as a Therapeutic Intervention for Type 2 Diabetes. Front. Endocrinol. (Lausanne) 2021, 12, 632335. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Y. Mechanism and Application of Lactobacillus in Type 2 Diabetes-Associated Periodontitis. Front. Public Health 2023, 11, 1248518. [Google Scholar] [CrossRef] [PubMed]
- Seferović, P.M.; Fragasso, G.; Petrie, M.; Mullens, W.; Ferrari, R.; Thum, T.; Bauersachs, J.; Anker, S.D.; Ray, R.; Çavuşoğlu, Y.; et al. Sodium–Glucose Co-Transporter 2 Inhibitors in Heart Failure: Beyond Glycaemic Control. A Position Paper of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2020, 22, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Das, S.R.; Everett, B.M.; Birtcher, K.K.; Brown, J.M.; Cefalu, W.T.; Januzzi, J.L.; Kalyani, R.R.; Kosiborod, M.; Magwire, M.L.; Morris, P.B.; et al. 2018 ACC Expert Consensus Decision Pathway on Novel Therapies for Cardiovascular Risk Reduction in Patients With Type 2 Diabetes and Atherosclerotic Cardiovascular Disease: A Report of the American College of Cardiology Task Force on Expert Consensus Deci. J. Am. Coll. Cardiol. 2018, 72, 3200–3223. [Google Scholar] [CrossRef] [PubMed]
- Dave, C.V.; Schneeweiss, S.; Wexler, D.J.; Brill, G.; Patorno, E. Trends in Clinical Characteristics and Prescribing Preferences for SGLT2 Inhibitors and GLP-1 Receptor Agonists, 2013–2018. Diabetes Care 2020, 43, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, R.; Jha, K.; Dardari, Z.; Heyward, J.; Blumenthal, R.S.; Eckel, R.H.; Alexander, G.C.; Blaha, M.J. National Trends in Use of Sodium-Glucose Cotransporter-2 Inhibitors and Glucagon-like Peptide-1 Receptor Agonists by Cardiologists and Other Specialties, 2015 to 2020. J. Am. Heart Assoc. 2022, 11, e023811. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, M.C.; Chang, L.S.; McGuire, D.K.; Plutzky, J.; Aroda, V.R.; Vaduganathan, M. Use of Glucagon-Like Peptide-1 Receptor Agonists in Patients with Type 2 Diabetes and Cardiovascular Disease: A Review. JAMA Cardiol. 2020, 5, 1182–1190. [Google Scholar] [CrossRef]
- Scheen, A.J. Bridging the Gap in Cardiovascular Care in Diabetic Patients: Are Cardioprotective Antihyperglycemic Agents Underutilized? Expert Rev. Clin. Pharmacol. 2023, 16, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Fatima, H.; Rangwala, H.S.; Mustafa, M.S.; Shafique, M.A.; Abbas, S.R.; Rizwan, A.; Fadlalla Ahmed, T.K.; Arshad, A. Evaluating Glycemic Control Efficacy and Safety of the Oral Small Molecule Glucagon-Like Peptide 1 Receptor Agonist Danuglipron in Type 2 Diabetes Patients: A Systemic Review and Meta-Analysis. Diabetes Metab Syndr Obes. 2023, 16, 3567–3578. [Google Scholar] [CrossRef]
- Frias, J.P.; Hsia, S.; Eyde, S.; Liu, R.; Ma, X.; Konig, M.; Kazda, C.; Mather, K.J.; Haupt, A.; Pratt, E.; et al. Efficacy and safety of oral orforglipron in patients with type 2 diabetes: A multicenter, randomized, dose-response, phase 2 study. Lancet 2023, 402, 472–483. [Google Scholar] [CrossRef]
- Kaore, S.; Bhavya, B.; Khasbage, S.; Atal, S. Evaluating the Efficacy and Safety of Tirzepatide on Glycaemic and Non-glycaemic Outcomes in Diabetes: A Systematic Review of Meta-Analyses. Cureus 2024, 16, e56939. [Google Scholar] [CrossRef] [PubMed]
- Jakubowska, A.; Roux, C.W.L.; Viljoen, A. The Road towards Triple Agonists: Glucagon-Like Peptide 1, Glucose-Dependent Insulinotropic Polypeptide and Glucagon Receptor—An Update. Endocrinol. Metab. 2024, 39, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Forzano, I.; Varzideh, F.; Avvisato, R.; Jankauskas, S.S.; Mone, P.; Santulli, G. Tirzepatide: A Systematic Update. Int. J. Mol. Sci. 2022, 23, 14631. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Wysham, C.; Frías, J.P.; Kaneko, S.; Lee, C.J.; Fernández Landó, L.; Mao, H.; Cui, X.; Karanikas, C.A.; Thieu, V.T. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): A double-blind, randomised, phase 3 trial. Lancet 2021, 398, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Pérez Manghi, F.C.; Fernández Landó, L.; Bergman, B.K.; Liu, B.; Cui, X.; Brown, K. SURPASS-2 Investigators. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Ludvik, B.; Giorgino, F.; Jódar, E.; Frias, J.P.; Fernández Landó, L.; Brown, K.; Bray, R.; Rodríguez, Á. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): A randomised, open-label, parallel-group, phase 3 trial. Lancet 2021, 398, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S.; Kahn, S.E.; Pavo, I.; Weerakkody, G.J.; Yang, Z.; Doupis, J.; Aizenberg, D.; Wynne, A.G.; Riesmeyer, J.S.; Heine, R.J.; et al. SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): A randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet 2021, 398, 1811–1824. [Google Scholar] [CrossRef] [PubMed]
- Dahl, D.; Onishi, Y.; Norwood, P.; Huh, R.; Bray, R.; Patel, H.; Rodríguez, Á. Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial. JAMA 2022, 327, 534–545. [Google Scholar] [CrossRef]
- Tall Bull, S.; Nuffer, W.; Trujillo, J.M. Tirzepatide: A novel, first-in-class, dual GIP/GLP-1 receptor agonist. J. Diabetes Complicat. 2022, 36, 108332. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| 1. reduced insulin secretion by β-cells (pancreas) |
| 2. increased glucagon secretion by ⲁ-cells (pancreas) |
| 3.increased glucose production (liver) |
| 4. increase lipolysis (adipose tissue) |
| 5. reduced appetite (brain) |
| 6. decreased incretin effect (bowel) |
| 7. increased glucose (kidney) |
| 8. decrease in glucose uptake (muscle) |
| GLP-1RA | Frequency of Administration | Structural Class | Administration Route | Delivery | Dose | Contraindications | Adverse Effects |
|---|---|---|---|---|---|---|---|
| Liraglutide | Once/day | GLP-1 | SCI | Multidose pen | 0.6–1.8 mg | History of pancreatitis and pancreatitis development while taking these medications. Severe gastrointestinal diseases, hypersensitivity and pregnancy. Personal or family history significant for multiple endocrine neoplasia 2A, multiple endocrine neoplasia 2B, or medullary thyroid cancer | Nausea, vomiting, diarrhea; dizziness, mild tachycardia, infections, headaches, and dyspepsia may also occur. Low risk of minor hypoglycemic episodes. Combination with dipeptidyl peptidase-4 inhibitors not currently recommended due to not significant glycemic improvement but enhanced hypoglycemic effects. Still unclear possible interactions with other oral anti-diabetic medications. |
| Exenatide immediate release | twice/day | Exendin-4 | SCI | Multidose pen | 5–10 µg | ||
| Exenatide long acting | once/week | Exendin-4 | SCI | Single dose/powder and diluent supplied | 2 mg | ||
| Dulaglutide | once/week | GLP-1 | SCI | Multidose pen/syringe | 0.75–1.5 mg | ||
| Semaglutide | once/week | GLP-1 | SCI | Multidose pen | 2.25–1 mg | ||
| Oral semaglutide | once/day | GLP-1 | O | Capsule | 3–14 mg | ||
| Lixisenatide | once/day | Exendin-4 | SCI | Multidose pen | 10–20 µg |
| improved glucose control |
| minimal hypoglycemic events |
| improved endothelial function |
| body weight reduction |
| reduced blood pressure |
| improved lipid profile; reduced triglycerides |
| improved recovery after ischemia |
| a reduction in MACE events and CV mortality in high CV risk T2D patients |
| improved hemodynamics in patients with left ventricular dysfunction or heart failure |
| improved glucose control |
| natriuretic effect (reduced risk of hospitalizations for heart failure and the progression of the renal damage) |
| minimal hypoglycemic events |
| improved endothelial function |
| body weight reduction |
| cardioprotection |
| reduced blood pressure |
| improved lipid profile |
| improvement of inflammation and oxidative stress |
| possible use for transthyretin amyloid cardiomyopathy |
| a reduction in MACE events and cardiovascular mortality in high cardiovascular risk T2D patients |
| a reduction in infarct size and the occurrence of ischemia-reperfusion-induced arrhythmias |
| reduction in heart failure risk |
| SGLT 2 I | Frequency of Administration | Structural Class | Administration Route | Dose | Contraindications | Adverse Effects |
|---|---|---|---|---|---|---|
| Empagliflozin | Once/day | SGLT2 inhibitor | oral | 10 mg and 25 mg |
|
|
| Dapagliflozin | Once/day | SGLT2 inhibitor | oral | 10 mg | ||
| Canagliflozin | Once/day | SGLT2 inhibitor | oral | 100 mg and 300 mg | ||
| Ertugliflozin | Once/day | SGLT2 inhibitor | oral | 5 mg and 15 mg |
| Trial Name | Trial Type | Trial Target | Trial Primary Endpoint | Trial Results | Reference |
|---|---|---|---|---|---|
| Surpass-1 | randomized, double-blind, parallel-group, phase 3 trial | Effectiveness of tirzepatide (5, 10 or 15 mg; subcutaneous injections once weekly) compared to placebo in patients with T2D inadequately controlled by diet and exercise | Mean change in HbA1c from baseline at 40 weeks | Effectiveness of tirzepatide compared with placebo in glycemic control and weight loss, without increased risk of hypoglycaemia; safety profile comparable with GLP-1 receptor agonists | [165] |
| Surpass-2 | randomized, parallel-group, open-label, phase 3 trial | Comparison of tirzepatide with the GLP-1R agonist Semaglutide | Mean change in HbA1c from baseline at 40 weeks | Tirzepatide at all doses noninferior and superior to semaglutide; loss weight greater with tirzepatide than with semaglutide. Most common adverse events were gastrointestinal (nausea, diarrhea, vomiting). | [166] |
| Surpass-3 | randomized, open-label, parallel-group, phase 3 trial | Comparison of tirzepatide (once-weekly) with insulin degludec (once-daily) as an add-on to metformin with or without SGLT2 inhibitors | Mean change from baseline in HbA1c at week 52. | Tirzepatide at all doses superior to titrated insulin degludec, with greater reductions in HbA1c and bodyweight (week 52) and lower hypoglycaemic risk; safety profile comparable with GLP-1 receptor agonists | [167] |
| Surpass-4 | randomized, open-label, parallel-group, phase 3 trial | Comparison of tirzepatide (10 or 15 mg, or both), with insulin glargine in patients with a high cardiovascular risk, inadequately controlled by oral glucose-lowering drugs | HbA1c change from baseline to 52 weeks. | Superior HbA1c reduction with a lower incidence of hypoglycaemia at week 52. Tirzepatide was not associated with excess cardiovascular risk. | [168] |
| Surpass-5 | randomized, double-blind, phase 3 trial | Addition of subcutaneous tirzepatide, compared with placebo, to titrated insulin glargine in T2D patients inadequately controlled with basal insulin, with or without metformin. | HbA1c change from baseline to 40 weeks. | Improved glycemic control in terms of HbA1c reduction at week 40, with higher % of patients treated with tirzepatide vs those treated with placebo who showed HbA1c less than 7%; more weight reduction. | [169] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatzianagnostou, K.; Gaggini, M.; Suman Florentin, A.; Simonini, L.; Vassalle, C. New Molecules in Type 2 Diabetes: Advancements, Challenges and Future Directions. Int. J. Mol. Sci. 2024, 25, 6218. https://doi.org/10.3390/ijms25116218
Chatzianagnostou K, Gaggini M, Suman Florentin A, Simonini L, Vassalle C. New Molecules in Type 2 Diabetes: Advancements, Challenges and Future Directions. International Journal of Molecular Sciences. 2024; 25(11):6218. https://doi.org/10.3390/ijms25116218
Chicago/Turabian StyleChatzianagnostou, Kyriazoula, Melania Gaggini, Adrian Suman Florentin, Ludovica Simonini, and Cristina Vassalle. 2024. "New Molecules in Type 2 Diabetes: Advancements, Challenges and Future Directions" International Journal of Molecular Sciences 25, no. 11: 6218. https://doi.org/10.3390/ijms25116218
APA StyleChatzianagnostou, K., Gaggini, M., Suman Florentin, A., Simonini, L., & Vassalle, C. (2024). New Molecules in Type 2 Diabetes: Advancements, Challenges and Future Directions. International Journal of Molecular Sciences, 25(11), 6218. https://doi.org/10.3390/ijms25116218